

Abstract
Very intriguing questions arise with our advancing knowledge on gut microbiota composition and the relationship with health, particularly relating to the factors that contribute to maintaining the population balance. However, there are limited available methodologies to evaluate these factors. Bacteriocins are antimicrobial peptides produced by many bacteria that may confer a competitive advantage for food acquisition and/or niche establishment. Many probiotic lactic acid bacteria (LAB) strains have great potential to promote human and animal health by preventing the growth of pathogens. They can also be used for immuno-modulation, as they produce bacteriocins. However, the antagonistic activity of bacteriocins is normally determined by laboratory bioassays under well-defined but over-simplified conditions compared to the complex gut environment in humans and animals, where bacteria face multifactorial influences from the host and hundreds of microbial species sharing the same niche. This work describes a complete and efficient procedure to assess the effectof a variety of bacteriocins with different target specificities in a murine system. Changes in the microbiota composition during the bacteriocin treatment are monitored using compositional 16S rDNA sequencing. Our approach uses both the bacteriocin producers and their isogenic non-bacteriocin-producing mutants, the latter giving the ability to distinguish bacteriocin-related from non-bacteriocin-related modifications of the microbiota. The fecal DNA extraction and 16S rDNA sequencing methods are consistent and, together with the bioinformatics, constitute a powerful procedure to find faint changes in the bacterial profiles and to establish correlations, in terms of cholesterol and triglyceride concentration, between bacterial populations and health markers. Our protocol is generic and can thus be used to study other compounds or nutrients with the potential to alter the host microbiota composition, either when studying toxicity or beneficial effects.
Keywords: Cellular Biology, Issue 125, Mouse model, fecal samples, gut microbiota, bacteriocin, massive sequencing, Bioinformatic analysis, bacterial populations, cholesterol, triglycerides
Introduction
Bacteriocins are antimicrobial peptides produced by a wide range of bacterial species1,2. These compounds and their producers, especially LAB, have been explored and exploited worldwide for decades for their potential applications in food preservation and medicine3. Several bacteriocins are known to kill important pathogens, including species of Listeria, Enterococcus, Staphylococcus, and Bacillus. Some bacteriocins even have the ability to modulate the immune response4. Many bacteriocins have relatively narrow spectra, a property that is much appreciated in some applications. For example, some narrow-spectrum bacteriocins can be used to direct specific activity against selected groups of problematic bacteria, without much disturbance on the commensal or beneficial flora sharing the same niche; this is especially essential in the gut environment, where numerous beneficial microbes thrive in an interactive and dynamic manner5. Bacteriocins are also very attractive for prophylactic or probiotic use, as they can suppress the (out)growth of pathogens, pathobionts, or opportunistic bacteria that may unbalance the gut homeostasis6,7.
In terms of their nature and physicochemical properties, bacteriocins are very diverse, as they have different structures, target specificities, modes of action, etc. Most bacteriocins have been studied in great detail in in vitro settings, but very few have been tested in food matrices8,9 or in vivo, such as in an animal gut6,10. The in vitro properties can differ to a great extent when assessed in vivo due to the complexity of the gut environment and also to putative unintended effects on beneficial bacteria. Most probiotics are LAB. They produce an array of other metabolites, including short-chain fatty acids, which are known to influence the physiology of the host, as well as to demonstrate antimicrobial properties toward certain bacteria. Therefore, in the case of probiotic strains that produce bacteriocins, it is best to establish realistic assays, such as healthy animals with normal microbiota.
In the present study, we provide a strategy that allows for the assessment of the effect of different bacteriocin-producing strains, whose bacteriocins have different inhibitory spectra, on healthy mice. Our strategy includes feeding mice with isogenic non-bacteriocin mutants, which enables the differentiation of bacteriocin-mediated effects from non-bacteriocin-mediated effects. Sequencing 16S rDNA allows for following the dynamic changes of the bacterial population in the gut. Subsequent statistical analysis deciphers correlations between bacterial species and also between bacterial species and measured physiological parameters (e.g., bodyweight, serum biochemical parameters, etc.). We believe that the protocol presented in this study is also applicable to other probiotic or prebiotic applications beyond the study of bacteriocins in live animals.
Protocol
Care and handling must be carried out at a specialized animal care unit. Procedures described here were approved by the corresponding Ethics Committee of the University of Valencia and local authorities, following the principles of laboratory animal care mandatory by European Union Law and 2010/63/EU and the Spanish Government RD 53/2013 on the protection of animals used for scientific purposes, in order to respect the 3R principle in animal experimentation (Replacement, Reduction, Refinement).
1. Frozen Bacterial Cultures Used to Inoculate Mice
Inoculate individual strains of LAB in 5 mL of brain-heart infusion (BHI) medium and grow them for 20 - 24 h (overnight) at 30 °C.
Dilute each overnight bacterial culture 100-fold in BHI by transferring 2 mL of overnight culture to 200 mL of BHI. Grow them at 30 °C for 20 - 24 h. NOTE: Strains used in this study are listed in Table 1.
Harvest cells by centrifugation at 5,000 x g for 20 min at 4 °C.
Remove the supernatant and wash the cells twice by suspending each cell pellet in 100 mL of ice-cold phosphate-buffered saline (PBS) and collecting the cells as in step 1.3.
After the second wash, suspend each cell pellet in 20 mL of ice-cold 15% glycerol in PBS.
Aliquot the cell suspension into 1-mL volumes in tubes and then store them at -80 °C until use. NOTE: Cells should be used within 1-2 months after this point.
- Determine the cell number before storage and prior to administration so that the number of live cells given to mice is known.
- For cell counting, make ten-fold serial dilutions of cells in ice-cold 0.9% sodium chloride (NaCl) solution. Spread 100 µL of each dilution onto a BHI agar plate. Incubate overnight (20 - 24 h) at 30 °C for cell growth and colony formation, the latter to determine colony-forming units (cfu)/mL.
To make bacteria-containing drinking water, dilute cells of each bacterial stock culture in 100 mL of sterile, filtered drinking water, with a final average cfu of 109/mL of water. For bacterial survival assessment, count live cells just after dilution and after 24 h once a week during the first two weeks of bacteria treatment.
2. Mice Assay and Experimental Design
Note: Specific pathogen-free (SPF) BALB/c young female mice (6 - 8 weeks) are needed for this experiment; here, a total of 100 animals were purchased.
Provide the mice with free access to pelleted food and water throughout the experiment.
- Design the experiment and calculate the power.
- If each treatment has its own control, apply a paired case-control design (t- test). Use a preliminary assay to calculate the mean and standard error of the most important effect(s) to be determined.
- Optimize the power of the assay and the number of animals needed per group using different methods and freely available programs11.
- To test for the effects of bacteriocin producer versus non-producer strains, use 9 animals per experimental condition. NOTE: This number was determined based on the experimental standard error and provided a satisfactory power (0.7 - 1.0) and a significance level below 0.05. NOTE: In the given example, 11 groups of mice were made, one per cage. Ten cages corresponded to the treatments with 5 bateriocin-producing strains and the corresponding 5 isogenic non-producing strains; the 11th cage had 10 mice and constituted the untreated control.
After the arrival of the mice to the husbandry facilities, distribute them in cages and ear-label the animals to allow for individual tracking. Acclimatize the mice to the husbandry facilities and conditions for a week before proceeding.
Prepare bacteria-containing drinking water, as described in step 1.8, and place drinking water bottles in the corresponding independent cages that are clearly identified so that the mice in each cage share the same water bottle. NOTE: The control group must be kept in a separate cage, with no contact with the tested bacteria.
Prepare a new bottle with fresh drinking water (100 mL) every day and add freshly defrosted bacterial suspensions. Do this for 15 consecutive days.
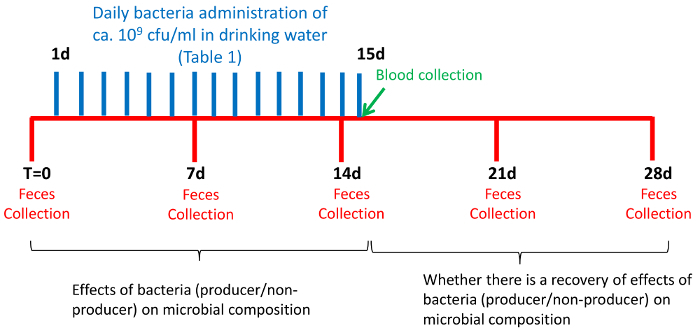
Follow the bacteria treatment with a period of two weeks, during which time mice drink bacteria-free water (Figure 1).
3. Collecting Samples
- Collect fecal samples.
- Prepare autoclaved cages (empty) and sterile forceps and label sterile 1.5-mL tubes for each mouse.
- Collect samples at the same time of day to avoid variations in microbiota composition during the course of a day. For optimal results, collect fresh samples separately. NOTE: Preferably, collect feces early in the morning, when mice tend to defecate spontaneously.
- Clean an empty cage with 70% alcohol and place a mouse inside. Allow it to defecate, and collect 2 - 4 fecal pellets using sterile forceps, placing them into 1.5 mL tubes. NOTE: Sometimes it is enough to hold the mouse tail upwards; the stool can then be directly collected from the anus into a tube.
- Collect fecal samples from each mouse once a week during the four-week experiment (Figure 1). Collect the first samples on day one (T0, time zero), just before the exposure of the mice to bacteria. Collect the following fecal samples on days 7, 14, 21, and 28.
- Refrigerate the samples at 4 °C until their transport to the laboratory, where they are frozen at -80 °C. Plan in advance and provide enough storage boxes, as a large number of fecal samples will be collected during the experiment.
Weigh all mice every week on fecal collection day.
Collect blood samples from the facial vein from all mice on 15th day, the last day of bacteria administration, and determine the levels of triglycerides, total cholesterol, high-density lipoprotein (HDL), and low-density lipoprotein (LDL) in the blood serum. NOTE: Experienced personnel should collect the blood to reduce stress in the animals.
4. LAB Counting and Bacteriocin Activity
Weigh one fecal pellet of each sample in a 1.5 mL tube and add an adequate volume of ice-cold 0.9% NaCl to achieve a 10% (w/v) solution. Use sterile micropestles for 1.5 mL tubes to homogenize the fecal suspension and prepare ten-fold serial dilutions in ice-cold 0.9% NaCl.
- Count the total number of LAB cells.
- Transfer diluted cells (100 µL) to 4 mL of prewarmed Man Rogosa Sharpe (MRS) soft agar (50 °C, 0.8% agar). Mix by vortexing before pouring the cells onto a solid MRS agar plate (1.5% agar).
- Dry the plate by taking the lid off for 5 - 10 min in the sterile hood before incubating the cells for 20 - 24 h at 30 °C for cell growth and colony formation.
- Count bacteriocin-producing cells.
- Transfer diluted cells (100 µL) of the bacteriocin producer to 4 mL of prewarmed MRS soft agar (50 °C, 0.8% agar), mix by vortexing, and pour the cells onto an MRS agar plate (1.5% agar); this layer is referred to as the first layer.
- Transfer another 4 mL of cell-free MRS soft agar (0.8% agar) onto the first layer to embed all cells within the soft agar. NOTE: This layer is meant to prevent cells from growing as colonies on the surface of the agar plates. Cells growing on the surface are easily displaced when pouring indicator cells on the top (step 4.3.4) and will therefore complicate the interpretation of results. This layer is referred to as the second layer.
- Dry the plate by taking the lid off for 5 - 10 min in the sterile hood before incubating for 20 - 24 h at 30 °C for cell growth and colony formation.
- To identify bacteriocin-producing colonies, mix 40 µL of an overnight culture of an appropriate indicator strain, as shown in Table 1, with 4 mL of prewarmed soft agar. Pour the mixture onto the plate; this layer is referred to as the third layer.
- Dry the plate by taking the lid off for 5 - 10 min in the sterile hood before incubating for 20 - 24 h at 30 °C for cell growth and colony formation. NOTE: Bacteriocin-producing colonies have clear (inhibition) zones around the colonies (Figure 2).

5. DNA Extraction, 16S rDNA Amplification, and Sequencing
NOTE: Steps for DNA extraction are described for the use of a commercial kit (e.g., Realpure "SSS" Kit).
Suspend 1 - 2 fecal mice pellets (~200 mg) in 300 µL of lysis solution.
Transfer the sample to a 2 mL microtube in which about 0.5 g of glass beads (diameter: 0.1 mm) have previously been placed.
Add 1 µL of mutanolysin at 10 U/µL and 2 µL of lysozyme at 20 mg/mL to each sample and incubate at 37 °C for 40 - 60 min. Proceed with one of the standard protocols for DNA extraction from fecal samples12.
Apply two cycles of bead-beater steps, 1 min each.
Add 2 µL of proteinase K and mix thoroughly.
Incubate for 20 min and vortex every 5 min.
Cool the samples to 37 °C and add 1 µL of 5 µg/mL RNase A to the sample. Mix thoroughly.
Incubate at 37 °C for 30 - 60 min.
Cool the samples to room temperature and add 180 µL of protein precipitation solution. Vortex vigorously at maximum speed for 20 - 30 s.
Centrifuge at 16,000 x g for 5 min. If there are any particles floating in the supernatant, incubate the samples in ice for 5 min and then centrifuge again.
Transfer the supernatant containing the DNA to a new 1.5-mL microtube containing 300 µL of isopropanol.
Mix by inverting the tubes 20-30 times and incubate for 1 h at -20 °C. Alternatively, incubate the tubes for longer a period (e.g., overnight).
Centrifuge at 16,000 x g for 2 min.
Remove the supernatant and dry the tube on absorbent paper. Add 300 µL of 70% ethanol to wash the DNA pellet.
Centrifuge at 16,000 x g for 1 min, remove the supernatant, and leave the pellet to dry for about 15 min.
Once the pellet is dry, add 100 µL of sterile water or Tris (10 mM)/EDTA (1 mM) buffer and resuspend the pellet.
To eliminate any possible inhibitory compounds during subsequent steps, clean the DNA. Mix extracted DNA with 2 volumes (200 µL) of buffer NTI to adjust the binding conditions for step 5.15.
Place a silica membrane containing PCR clean-up column into a collection tube (2 mL) and load the sample.
Centrifuge for 30 s at 11,000 x g. Discard the flow-through.
Wash the silica membrane with 700 µL of buffer NT3 and repeat step 5.16.
Dry the silica membrane by centrifugation for 1 min at 11,000 x g to remove any residual ethanol containing wash buffer NT3.
Transfer the column to a new 1.5-mL microtube and incubate for 2 - 5 min at 70 °C for the total removal of the ethanol.
Add 30 µL of PCR-grade water and incubate for 5 min at 70 °C. Elute by centrifugation for 1 min at 11,000 x g.
Quantify the DNA using a UV/Vis spectrophotometer or a fluorometric method.
Normalize and pool the DNA samples from mice sharing same cage at each time point.
To observe individual variations during time, sequence DNA samples from three randomly selected mice from the control and two other random cages on days 0, 14, and 28.
6. 16S rRNA Gene Amplification and Sequencing
Prepare a library of amplified 16S rRNA genes. Amplify the V3-V4 region of the bacterial 16S rRNA gene5 using forward and reverse primers with appropriate overhang adaptors: 5'-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG-3' 5'-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA CTA CHV GGG TAT CTA ATC C-3'
Check the amplified bands using electrophoresis with 1% agarose gel.
Clean up the polymerase chain reaction (PCR) products using magnetic beads for amplicon purification.
Proceed with the following steps as indicated by the manufacturer according to the massive sequencing platform used6.
7. Data Analysis and Statistics
- Quality filtering (usually performed at sequencing facilities).
- Filter the raw reads data and de-multiplex using the software supplied with the equipment.
- Cluster the sequences into operational taxonomic units (OTUs) on a 97% sequence identity and use UCHIME15 to filter chimeric sequences.
- OTU picking, taxonomic assignment and phylogenetic reconstruction, and diversity analyses and visualizations.
- Process the OTUs using Quantitative Insights Into Microbial Ecology (QIIME)16.
- Assign taxonomy to the aligned sequences using the Ribosomal Database Project (RDP) classifier program19 with a confidence of 0.8.
- Normalize the OTU table to the sequence count of the sample with the lowest sequence depth.
- Build a phylogenic tree from aligned sequences using Fast Tree20 after the filtration step in order to remove highly variable regions and positions that are all gaps. Use this tree to calculate alpha and beta diversities and to calculate rarefaction curves and Shannon indexes.
- For the comparison of Shannon indexes of diversity between the different treatments, use an ANCOVA, considering time as a continuous dependent variable in R.
- Compare the distances between treatments in PCoA in QIIME using a two-sided Student's t-test and calculate the nonparametric p-values with 1,000 Monte Carlo permutations using the Bonferroni correction.
- Analyze for correlations between the relative abundance of specific OTUs and other measured parameters, such as serum levels of cholesterol, HDL, LDL, etc., using Pearson correlation analysis. To this end, use CoNet23 with the subsequent visualization of correlation networks using Cytoscape 3.1.124.
Representative Results
The production of bacteriocins has been considered a positive probiotic feature in LAB, as it was assumed to prevent the growth of opportunistic bacteria and pathogens. The aim of this work was to show the capacity of bacteriocins to modulate gut microbiota populations in a mouse model. For this purpose, a procedure was developed to compare the effect of the intake of bacteriocin-producing strains and their isogenic non-producing strains. The procedure for the inoculation and the time extension of the assay are shown in Figure 1. Mice were grouped into eleven cages: a control without any treatment, five cages treated with bacteriocin-producer strains, and five cages treated with the corresponding isogenic mutants that have reduced or no bacteriocin production. The tested bacteriocin-producing LAB, as well as the respective counts (cfu/mL) in drinking water and counts in feces, are summarized in Table 1. The determination of bacteriocin production by LAB recovered from feces is illustrated in Figure 2. There were no significant differences between the bacteriocin treatments and the isogenic strains when all samples and all bacterial groups were considered together (Figure 3). However, there were likely differences when particular bacterial genera were studied independently, as shown in Figure 4. Furthermore, Pearson's correlation analysis revealed significant linkage between different bacterial genera and a correlation between bacterial genera and triglycerides and LDL in serum, as shown by the network built in CoNet in Figure 5.
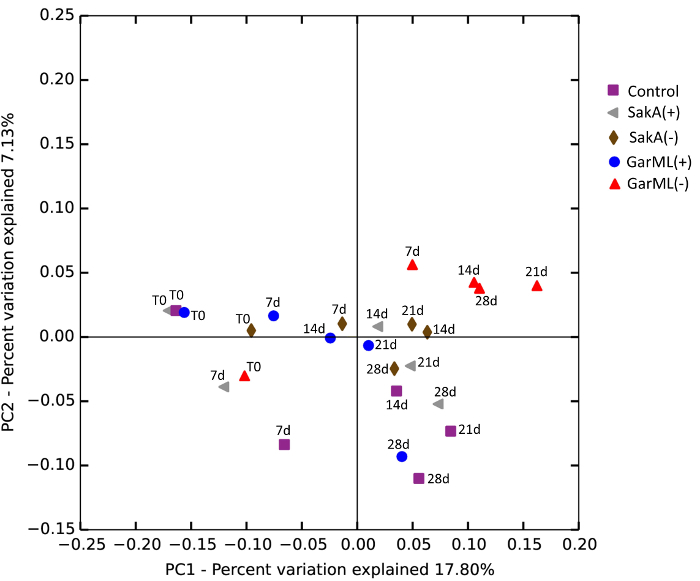

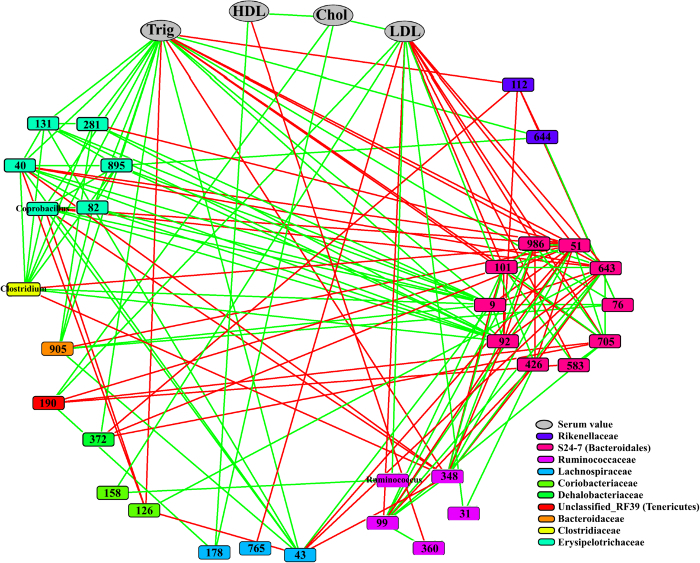
Figure 1: Scheme Showing the Time Course Design of the Experiment. Please click here to view a larger version of this figure.
Figure 2: Detection of Bacteriocin Production in LAB Recovered from Feces by a Three-layer Protocol (as Described in the Procedure). Please click here to view a larger version of this figure.
Figure 3: Comparison of the Composition of Bacteria Treatments. A PCoA plot was generated based on the calculated distances in an unweighted UniFrac matrix. Different time points from the same sample (Figure 1) have the same color, and each color identifies a treatment group. For clarity of the illustration, only four strains are shown: garvicin and sakacin producer and non-producer strains (see the legend in the figure to relate color and treatment). The time of the samples is annotated as T0 (before inoculation), 7d (7 days), 14d (14 days), 21d (21 days), and 28d (28 days). For statistics, see the Procedure. Adapted from Umu et al., 201612. Please click here to view a larger version of this figure.
Figure 4: Changes in the Relative Abundance of LAB and Bacteriocin-targeted Bacterial Groups at the Genus Level during the Treatment and Post-treatment Periods. Changes in the relative abundances of genera in treatments, obtained with respect to time 0, were compared to those in the control group (CON). For clarity of the illustration, only four strains are shown: garvicin and sakacin producer and non-producer strains (see the legend in the figure to relate color and treatment). Error bars represent the standard deviation. Significance degree is represented as follows: P <0.1 with dot (.); P <0.05 with one star (*); P <0.01 with two stars (**). Adapted from Umu et al., 201612. Please click here to view a larger version of this figure.
Figure 5: Correlation Network of Relative Abundances of OTUs on Day 14 and Serum Levels. The correlations were calculated using Pearson's correlation in CoNet. Only the significant ones (P <0.05) are shown on the network. Parameters determined in serum – total cholesterol (Chol), high-density lipoprotein (HDL), low-density lipoprotein (LDL), and triglycerides (Trig – are shown by one color (gray), while OTUs belonging to different families are represented by different colors (see the legend). Positive correlations are displayed with green edges, and negative correlations with red edges. OTUs on the nodes are represented with OTU numbers or the genus to which they belong. Adapted from Umu et al., 201612. Please click here to view a larger version of this figure.
| Strain | Inoculated in drinking water | Lactic acid bacteria counts in feces | Indicator strains | |
| CFU/ml (log10) | CFU/g (mean log10) | |||
| CONTROL | ---- | 10 mice | ND | |
| Lactobacillus sakei B1500 (sakA +) | 1.6 | 9 mice | 8.53 ± 0.18 | Enterococcus faecium P21 (LMG 2783) |
| Lactobacillus sakei B1501 (sakA -) | 2.0 | 9 mice | 8.22 ± 0.05 | |
| Pediocccus acidilactici B1502 (ped +) | 26 | 9 mice | 8.73 ± 0.12 | Enterococcus faecium P21 (LMG 2783) |
| Pediocccus acidilactici B1503 (ped -) | 25 | 9 mice | 8.75 ± 0.09 | |
| Enterococcus faecium B1504 (L50 wt +) | 6 | 9 mice | 8.83 ± 0.14 | Pediococcus damnosus (LMG 3397) |
| Enterococcus faecium B1505 (L50 cured -) | 10 | 9 mice | 8.72 ± 0.14 | |
| Lactobacillus plantarum B1507 (planta +) | 26 | 9 mice | 9.14 ± 0.14 | Lactobacillus spp. 965 (LMG 2003) |
| Lactobacillus plantarum B1508 (planta -) | 23 | 9 mice | 8.60 ± 0.18 | |
| Lactococcus garviae B1515 (GarML +) | 17 | 9 mice | 8.41 ± 0.08 | Lactococcus lactis IL1403 (LMG2705) |
| Lactococcus garviae B1516 (GarML -) | 17 | 9 mice | 8.05 ± 0.18 | |
| Enterococcus faecium P21 (LMG 2783) for SakA and PedPA-1, Pediococcus damnosus (LMG 3397) for enterocins, Lactobacillus spp. 965 (LMG 2003) for plantaricins, and Lactococcus lactis IL1403 (LMG2705) for GarML. |
Table 1: Inoculated Strains and the Counts in Fecal Samples.
Discussion
The procedure described here has been used to determine whether changes in the microbiota are bound to health or age. Different parts of the protocol are important, but among them, sampling the feces, choosing the DNA fragment to be sequenced and analyzed, and performing the DNA extraction and bioinformatic analysis could certainly be the most critical points. Sampling is crucial because, for ethical reasons, mice should not be stressed and because it is known to change the proportion of bacteria in the gut. Samples must be processed as soon as possible or should be frozen until use, as some bacteria are very sensitive to dehydration and/or to oxygen. The selection of the DNA fragment to be sequenced from the amplified 16S rRNA genes is a crucial point. In this work, the variable regions V3 - V4 were selected25. The importance of the DNA extraction method must not be underestimated, as bacterial cells present in the samples are not all equally sensitive to different lysis protocols, and biases in the estimation of the bacterial populations can result from incomplete lysis. Therefore, the introduction of a bead beater step is required. Isogenic strains can be constructed by genetic knockdown or by plasmid curation, although in the case of strains, refractory to transformation chromosome-encoded bacteriocins may represent a problem. This procedure can be adapted using different specific culturing techniques, such as anaerobic methods26.
The procedure can be modified in most steps, depending upon the type of factor changes to be studied. The inoculation procedure could be shortened, but changes in serum parameters may need some time to develop. A possible limitation of the method could be the study of bacterial strains (nutrients or compounds) that cannot survive several hours in water or that may clump (or precipitate for compounds). Hence, this is a factor that must also be considered.
Until now, bacteriocin activity was generally determined in vitro27,28,29 or in mixed cultures30. This method has allowed for the in vivo monitoring of the activities of five bacteriocins from the whole-gut microbiota of standard laboratory mice. Small variations in populations could be determined, as well as their influence on other bacterial groups or physiological factors, like serum parameters12. Bacteriocins have a relatively narrow activity spectrum, inhibiting the growth of taxonomically related bacteria. However, those used in this assay are among the most effective, as some of them are active against Staphylococcus aureus, Listeria monocytogenes31, and even some strains of Escherichia coli, as in the case of sakacin A32. When inoculated in mice, LAB producing bacteriocins did not give rise to remarkable changes in the global microbiota composition, but rather to a reduced number of bacterial taxa. Nevertheless, global correlation network analysis revealed interesting ecological compatibilities and antagonism between different bacteria. This method could be adapted to analyze the effect of food constituents, drugs, or other compounds, although the expected outcome may be deliberately restricted to preserve healthy conditions.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors wish to thank the EEA Grant NILS Science and Sustainability Coordinated Mobility of Researchers (reference 017-ABEL-CM-2013). C.B. and G.P.-M. were supported by the AGL2015-70487-P grant from the Spanish Ministry of Economy and Competitiveness. O.C.O.U. and D.B.D. were supported by a strategic scholarship program for food science research from the Norwegian University of Life Sciences (NMBU) (project 1205051025). We would also like to thank Inmaculada Noguera for her assistance with animal care and sampling and Jesus Dehesa for his help with ensuring the availability of laboratory materials in the animal facility. We also appreciate Professor Lars-Gustav Snipen for his advice on statistics.
References
- Papagianni M. Ribosomally synthesized peptides with antimicrobial properties: biosynthesis, structure, function, and applications. Biotechnol Adv. 2003;21(6):465–499. doi: 10.1016/s0734-9750(03)00077-6. [DOI] [PubMed] [Google Scholar]
- Gillor O, Kirkup BC, Riley MA. Advances in Applied Microbiology. Vol. 54. Academic Press; 2004. pp. 129–146. [DOI] [PubMed] [Google Scholar]
- Dzung BD, Ingolf FN. Ribosomally Synthesized Antibacterial Peptides in Gram Positive Bacteria. Curr Drug Targets. 2002;3(2):107–122. doi: 10.2174/1389450024605409. [DOI] [PubMed] [Google Scholar]
- Dobson A, Cotter PD, Ross RP, Hill C. Bacteriocin Production: a Probiotic Trait? Appl Environ Microb. 2012;78(1):1–6. doi: 10.1128/AEM.05576-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Wang P, Wang P, Hu X, Chen F. The gut microbiota: A treasure for human health. Biotechnol Adv. 2016;34(7):1210–1224. doi: 10.1016/j.biotechadv.2016.08.003. [DOI] [PubMed] [Google Scholar]
- Millette M, et al. Capacity of Human Nisin- and Pediocin-Producing Lactic Acid Bacteria To Reduce Intestinal Colonization by Vancomycin-Resistant Enterococci. Appl Environ Microb. 2008;74(7):1997–2003. doi: 10.1128/AEM.02150-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter PD, Ross RP, Hill C. Bacteriocins [mdash] a viable alternative to antibiotics? Nat Rev Microb. 2013;11(2):95–105. doi: 10.1038/nrmicro2937. [DOI] [PubMed] [Google Scholar]
- Leroy F, Foulquié Moreno MR, De Vuyst L. Enterococcus faecium RZS C5, an interesting bacteriocin producer to be used as a co-culture in food fermentation. Int J Food Microb. 2003;88(2-3):235–240. doi: 10.1016/s0168-1605(03)00185-5. [DOI] [PubMed] [Google Scholar]
- Ananou S, et al. Combined effect of enterocin AS-48 and high hydrostatic pressure to control food-borne pathogens inoculated in low acid fermented sausages. Meat Sci. 2010;84(4):594–600. doi: 10.1016/j.meatsci.2009.10.017. [DOI] [PubMed] [Google Scholar]
- Riboulet-Bisson E, et al. Effect of Lactobacillus salivarius Bacteriocin Abp118 on the Mouse and Pig Intestinal Microbiota. PLoS One. 2012;7(2):e31113. doi: 10.1371/journal.pone.0031113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charan J, Kantharia ND. How to calculate sample size in animal studies? J Pharmacol Pharmacother. 2013;4(4):303–306. doi: 10.4103/0976-500X.119726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umu ÖCO, et al. The Potential of Class II Bacteriocins to Modify Gut Microbiota to Improve Host Health. PLoS One. 2016;11(10):e0164036. doi: 10.1371/journal.pone.0164036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Meth. 2013;10(10):996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Meth. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, et al. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl Environ Microb. 2006;72(7):5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26(2):266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl Environ Microb. 2007;73(16):5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastTree 2 - Approximately Maximum-Likelihood Trees for Large Alignments. PLoS One. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, Knight R. UniFrac: a New Phylogenetic Method for Comparing Microbial Communities. Appl Environ Microb. 2005;71(12):8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R Foundation for Statistical Computing. Vienna, Austria: 2014. Available from: http://www.R-project.org. [Google Scholar]
- Faust K, et al. Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol. 2012;8(7):e1002606. doi: 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, et al. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klindworth A, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41(1):e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AN, Suárez JM, McBride SM. Culturing and Maintaining Clostridium difficile in an Anaerobic Environment. J Vis Exp. 2013. p. e50787. [DOI] [PMC free article] [PubMed]
- Delves-Broughton J, Blackburn P, Evans RJ, Hugenholtz J. Applications of the bacteriocin, nisin. Antonie Van Leeuwenhoek. 1996;69(2):193–202. doi: 10.1007/BF00399424. [DOI] [PubMed] [Google Scholar]
- Allende A, et al. Growth and bacteriocin production by lactic acid bacteria in vegetable broth and their effectiveness at reducing Listeria monocytogenes in vitro and in fresh-cut lettuce. Food Microb. 2007;24(7-8):759–766. doi: 10.1016/j.fm.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Strompfová V, Lauková A. In vitro study on bacteriocin production of Enterococci associated with chickens. Anaerobe. 2007;13(5-6):228–237. doi: 10.1016/j.anaerobe.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Caballero-Guerrero B, Jiménez Díaz R, Maldonado-Barragán A, Ruiz-Barba JL. Coculture with specific bacteria enhances survival of Lactobacillus plantarum NC8, an autoinducer-regulated bacteriocin producer, in olive fermentations. Food Microb. 2010;27(3):413–417. doi: 10.1016/j.fm.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Drider D, Fimland G, Héchard Y, McMullen LM, Prévost H. The Continuing Story of Class IIa Bacteriocins. Microb. Mol Biol Rev. 2006;70(2):564–582. doi: 10.1128/MMBR.00016-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Jia S, Gao Q, Tan Z. A novel bacteriocin with a broad inhibitory spectrum produced by Lactobacillus sake C2, isolated from traditional Chinese fermented cabbage. Food Control. 2010;21(1):76–81. [Google Scholar]