

Abstract
LapA is the biggest protein in Pseudomonas putida and a key factor for biofilm formation. Its importance and posttranslational regulation is rather thoroughly studied but less is known about the transcriptional regulation. Here we give evidence that transcription of lapA in LB-grown bacteria is initiated from six promoters, three of which display moderate RpoS-dependence. The global transcription regulator Fis binds to the lapA promoter area at six positions in vitro, and Fis activates the transcription of lapA while overexpressed in cells. Two of the six Fis binding sites, Fis-A7 and Fis-A5, are necessary for the positive effect of Fis on the transcription of lapA in vivo. Our results indicate that Fis binding to the Fis-A7 site increases the level of transcription from the most distal promoter of lapA, whereas Fis binding to the Fis-A5 site could be important for modifying the promoter area topology.
Introduction
Bacteria, including plant-associated species from the genus Pseudomonas, may display two distinct lifestyles. Firstly, they can exist as freely moving planktonic cells. Secondly, given appropriate environmental conditions, they may adhere to surfaces and form complex sessile communities called biofilms. The structure of the bacterial biofilm, as well as its development, differs from species to species due to the variability of the environment and the ability of the specific bacterium to respond to various biotic and abiotic signals [1,2]. Therefore, the regulation of biofilm development is sophisticated and comprises the involvement of several global and specific regulators [2,3]. The biofilm of Pseudomonas putida seems to be positively regulated by availability of nutrients in LB medium. We have shown that P. putida biofilm is the strongest after 4 hours of inoculation in LB medium and it decays about threefold within the next 4 hours [4]. While exact numbers vary, similar trends of P. putida biofilm development are in agreement with earlier works [5–7].
The matrix of P. putida biofilm is proteinaceous and the two largest proteins of the bacterium, LapA and LapF, are known to play an important role in biofilm formation [4,5,8]. LapA provides cell-surface interactions [9,10] and is necessary for both for initial attachment and mature biofilm [7], whereas LapF rather provides cell-cell interactions contributing to mature biofilm formation [9]. LapA seems to be the most important biofilm factor [7,11] as no conditions efficiently rescuing the lapA mutant’s biofilm formation defect have been reported [5,11,12]. The second large extracellular protein, LapF, is the cell surface hydrophobicity factor [13] and probably contributes to cell-cell attachment by regulating cell hydrophobicity [9]. However, LapF seems to be an important factor for biofilm formation only when P. putida is grown in glucose minimal medium, but not in a rich medium like LB [4,9].
Although the post-translational regulation of lapA expression is well-described [12,14,15], the regulation of transcription has been rather briefly studied. LapA is transcribed from early exponential to late stationary phase and the transcription is about twice as active in the stationary phase compared to the exponential phase [16]. FleQ, c-di-GMP, Fis and the GacS-GacA two-component signal system have been shown to positively influence the expression of lapA [4,16–18]. The global regulator FleQ activates lapA transcription directly by DNA binding but its exact binding sites are yet to be determined. The activating effect of FleQ varies from 2 to 10 times between different authors and methods [16–18]. The effector molecule c-di-GMP only affects lapA transcription through FleQ and their effect is synergistic [17,18]. The two component system GacS-GacA regulates lapA mRNA levels, but whether directly or indirectly remains unknown [16]. We have shown that the global regulator Fis increases the amount of LapA 1.6 times when it is overexpressed in stationary phase P. putida [4]. However, the number and location of the lapA promoters remains unknown and the in vitro binding of regulators had not been tested prior to this work.
In P. putida, the Fis mRNA levels are highest in exponentially growing planktonic cells and drop approximately three times in stationary phase cells [19]. The pattern of Fis amount follows a surprisingly similar trend to the development of P. putida biofilm. Moreover, we have reported that artificially overexpressing Fis in the stationary phase, when the native amount of Fis is lowest, enhances the mature biofilm of P. putida 3 times in LB medium [4,20].
Fis has been shown to activate and repress the transcription of genes involved in biofilm development [21–23]. In general, Fis can affect transcription by modulating DNA supercoiling and topology (e. g., [24–26]) or directly by binding to the upstream region of genes (e. g., [22,27–29]. For example, Fis directly represses the transcription of lapF 4.2 times in P. putida by binding on its RpoS-dependent promoter [22]. Additionally, it has been shown that Fis can activate biofilm formation indirectly by repressing signal transduction in the Vibrio cholerae quorum sensing regulatory pathway [30].
Fis seems to be an essential protein for P. putida [20,31]. Fis is well described in E. coli, where the Fis-deficient strains are viable, although with a prolonged lag-phase [28]. Yet, we have been unable to construct Fis-deficient or fis under-expression strains in P. putida [20,31]. Therefore, a fis overexpression strain was used to elucidate its effects in P. putida.
The main goal of this study was to locate the promoter(s) of lapA and ascertain the impact of Fis on the transcription of lapA. In the present report, we show that the transcription of lapA is initiated from six promoters in LB medium, three of which display moderate RpoS-dependence. We identified six Fis binding sites in front of lapA by DNase I footprint and gel-shift assays. Two of the six binding sites are necessary for the Fis-enhanced expression of lapA in vivo. Surprisingly, these two binding sites are located 575 bp and 755 bp upstream of the lapA start codon. Fis activates the transcription of lapA from the most distal promoter PlapA8 and can additionally regulate lapA transcription via modification of lapA upstream DNA topology.
Materials and methods
Bacterial strains, plasmids, oligonucleotides and media
The bacterial strains and plasmids used in this study are described in S1 Table and oligonucleotides in S2 Table. E. coli was incubated at 37°C and P. putida at 30°C. E. coli strain DH5αλpir [32] was used for cloning suicide vector pEMG constructs and P. putida PSm for routine cloning pBLKT and p9TTBlacZ constructs [22,33]. Bacteria were grown in LB medium [34]. Solid media contained 1.5% Difco agar. Antibiotics were added at the following concentrations: gentamicin, 10 μg ml-1; kanamycin, 50 μg ml-1; benzylpenicillin, 1.5 mg ml-1; streptomycin, 200 μg ml-1. Bacteria were electrotransformed as described by Sharma & Schimke [35].
DNA manipulations and strains construction
Two types of promoter probe vectors were used to clone lapA promoter fragments in front of the lacZ gene and thereafter measure β-galactosidase activity. A medium-copy number pBRR1-based promoter probe vector pBLKT [22,36] was used for the functionality assessment of potential promoters (S1 Table). A low-copy-number RK2-based promoter probe vector p9TTBlacZ [33,37] was used to ascertain the influence of Fis to the transcription of lapA (S1 Table). The lapA promoter regions were amplified by PCR, and all fragments were cloned into the pBLKT or p9TTBlacZ BamHI site except for PlapA8, which was cloned into blunted BamHI site (Fig 1). The oligonucleotides, the length of amplified lapA promoter regions and content of DNA used for construction of plasmids are specified in the S1 and S2 Tables.
Fig 1. Scheme of the lapA promoter region, promoter region fragments cloned into pBLKT (or p9TTBlacZ) plasmid and the names of yielding constructs.

The first nucleotides of the mRNA 5ʹ ends determined by 5ʹ RACE are written in bold and designated as A-I to A-VIII. Potential promoters PlapA1 to PlapA8 are highlighted as white boxes in the black DNA. The black arrows show the beginnings of lapA and lapB genes.
For the construction of plasmids carrying substitutions in lapA promoter region (mutated Fis binding sites and mutated -10 box of potential promoters), site-directed mutagenesis of wild-type lapA promoter region was performed using two sequential PCRs and the P. putida PSm chromosomal DNA or different plasmids as a template. In the first PCR, the substitutions were introduced into the target site using a pair of oligonucleotides, one of which contained substituted nucleotides in the target site. In the second PCR, the needed lapA promoter region was amplified by using the second pair of oligonucleotides, one of which was the product of the first PCR. All mutated PCR fragments were cloned into the BamHI site of pBLKT or p9TTBlacZ plasmids [22,33].
For construction of pBPlapA1-8_F1mut and F2mut, respective mutational primers LapA-1mut-uus or LapA-2mut and LapA-I-rev were used for first PCR. The products of first PCR and PP0167-down were used as oligonucleotides for the second PCR to amplify a 951 bp long lapA promoter area. For construction of pBPlapA1-8_F4mut, F5mut, F6mut and F7mut, the respective mutational primers LapA-4mut-uus, FisA5-mut, FisA6-mut or FisA7-mut and PP0167 down were used for first PCR and the products of the first PCR and LapA-I-rev for second PCR. These amplifications resulted in fragments with substituted nucleotides in the different Fis binding sites but are otherwise identical to p9_PlapA1-8. The number of substitutions made in every mutated Fis binding site is specified in S1 Table. For the construction of p9_PlapA1-8_F1,2mut, the plasmid p9_PlapA1-8_F2mut was used as the template and mutations of Fis-A1 binding site were introduced as described previously via two sequential PCRs. For the construction of p9_PlapA1-8_F4,5,6,7mut, plasmids p9_PlapA1-8_F4,5mut and p9_PlapA1-8_F6,7mut were constructed first. The construct p9_PlapA1-8_F4mut was used as the template and mutations of the Fis-A5 were introduced to make p9_PlapA1-8_F4,5mut. Additionally, p9_PlapA1-8_F6mut was used as the template and Fis-A7 was mutated via two sequential PCRs to make p9_PlapA1-8_F6,7mut. Thereafter, p9_PlapA1-8_F6,7mut was amplified with FisA6-mut and PP0167-down primers. NheI (Thermo Scientific) digestion was used to cut the template p9_PlapA1-8_6,7m in the mixture of first PCR to hinder its use as a template in the second PCR. The product of first PCR, oligonucleotide lapA-I-rev and template p9_PlapA1-8_4,5m were used in the second PCR.
For construction pB_PlapA1-3 variant carrying the lapA promoter area with substitutions in the potential -10 box of PlapA3, two sequential PCR-s were used. The substitutions in the -10 box of the potential promoter PlapA3 was introduced by an oligonucleotide LapA-IIImut-piken in the first PCR. LapA-IV was used as the second oligonucleotide in the PCR mixture and pB_PlapA3mut as a template. The PCR product carrying the mutated -10 box of PlapA3 was obtained. The second PCR was carried out with a pair of oligonucleotides, one of which was the product of the first PCR and the second was LapA-I-rev and p9_PlapA1-8 as template (S1 and S2 Tables).
For construction of p9TTBlacZ variants carrying the individual promoters PlapA6, PlapA7, PlapA8 and functional Fis binding site(s) or mutated Fis binding site(s), the lapA promoter region was amplified by PCR using oligonucleotides shown in S1 Table and different templates. The pB_PlapA1-8 was used as a PCR template for the construction of p9_PlapA6B, p9_ PlapA7 and p9_ PlapA8B; the p9_A1-8_F4mut for construction of p9_PlapA6B_F4mut; the p9_PlapA1-8_F5mut for construction of p9_PlapA7_F5mut; and the p9_PlapA1-8 _F6mut, p9_PlapA1-8_F7mut and p9_PlapA1-8_F4567mut for construction of p9_PlapA8B _F6mut, p9_PlapA8B_F7mut and p9_PlapA8B_F67mut, respectively. Additionally, the parallel constructs pB_PlapA6B and pB_PlapA6B_4mut were created similarly to p9TTBlacZ variants.
For construction of PSm ΔrpoS, the DNA regions which flanked the rpoS were cloned into the suicide vector pEMG using the protocol described by Martinez-Garcia et al. [32]. The 457 bp long region located upstream of rpoS (Up) was amplified by primers RpoS-I-fw and RpoS-I-rev and the 468 bp long region downstream of rpoS (Down) was amplified by primers RpoS-2-fw and RpoS-2-rev. The Up and Down rpoS flanking regions were joined together by overlap extension (SOE)-PCR [38]. Thereafter the 925-bp PCR fragment (Up+Down) was purified and cloned into pEMG using the EcoRI and BamHI sites, resulting in pEMG-ΔrpoS (S1 Table). The rpoS deletion mutant of P. putida strain PSm (S1 Table) was constructed by a previously described protocol [32]. The pEMG-ΔrpoS was delivered to P. putida PSm by electroporation [39] to obtain a cointegrate between P. putida chromosome and pEMG-ΔrpoS carrying the recombination targets. A mutant with desired deletions was obtained after electroporating bacteria with plasmid pSW and expression of the Sce-I homing restrictase from the pSW plasmid [40].
All designed constructs were sequenced in order to exclude PCR-generated errors in the cloned DNA fragments. The accuracy of recombination was checked by sequencing the relevant regions of P. putida’s chromosome.
Identification of 5ʹ ends of mRNA by RACE
The mRNA 5ʹ ends of the lapA gene were identified by RACE (rapid amplification of cDNA ends) as described by Sambrook and Russell [41]. The cells were grown in LB medium for 18 hours at 30°C and total RNA was purified using the Thermo Scientific GeneJET RNA Purification kit. To obtain RNA from exponentially growing bacteria, cells were pre-grown for 18 hours, diluted 50 times and then grown to the optical density of 0.5. The cells were thereafter diluted 1:1 and grown for 30 minutes. This step was repeated 3 times. After the third time the cells were grown to the optical density of 0.5. 1.5 μg of purified total RNA and the LapA-RACE1 primer were used for the synthesize the first strand of cDNA. The second strands of cDNA were synthesized by primers Adapt-pikkC or Adapt-pikkT, with 5ʹ ends binding accordingly to poly-G or poly-A, synthesised by terminal deoxynucleotidyltransferase (TdT) to the 3ʹ ends of the first strand of cDNA. To amplify the second strand of cDNA, the Adapt-lyh and LapA-RACE2, LapA-rev, LapA-IV-rev or LapA-VI-rev primers were used. Zymo Research DNA Clean & ConcentratorTM-5 kit was used for DNA purification between RACE steps.
Prediction of Fis binding sites on the promoter region of the lapA gene
Putative Fis binding sequences on the promoter regions of the lapA gene were predicted using the E. coli Fis binding sites matrix [42] and the matrix-scan program available at the Regulatory Sequence Analysis Tools homepage [43]. The -1000 bp to -1 bp DNA region of the lapA gene was used for the prediction of potential Fis binding sites. The Markov model of one order, organism-specific probability of nucleotides in the upstream region of genes in P. putida KT2440 and a P-value upper threshold of 0.001 were selected for the conditions of the background model. The rest of the parameters were left at the program’s default values.
DNase I footprint analyses
DNase I footprint assays were performed for the identification of P. putida Fis binding sequences on the lapA promoter region. PCR-amplified fragments were used for DNase I footprint assay and were generated as follows. To study Fis binding to Fis-A1 and Fis-A2 sites, 207 bp-long DNA fragments containing the two sites were amplified by primers LapAdown and LapA2up. Depending on the template (p9_PlapA1-8, p9_PlapA1-8_F1mut or p9_PlapA1-8_F2mut), the fragments contained the wild-type Fis-A1 and Fis-A2 sites or either the mutated Fis-A1mut site or the mutated Fis-A2mut site. The 220 bp-long Fis-A4 fragment was amplified with LapAdown2 and LapA-fw using the full-length lapA promoter construct with or without mutated Fis-A4 binding site as template. To study Fis binding to Fis-A5 and Fis-A6 sites, the 235 bp-long DNA fragment containing the two sites was amplified using PP0167-I-fw and PP0168-I-fw primers. p9_PlapA1-8, p9_PlapA1-8_F5mut or p9_PlapA1-8_F6mut were used as templates. The 238 bp-long Fis-A7 fragment was amplified with LapBCup and LapBCdown using the full-length lapA promoter construct with or without mutated Fis-A7 binding site as template. Primers used for amplifications are listed in S2 Table. The following procedures: labelling PCR products with [γ-32P]-ATP, preparing reaction mixtures and gel electrophoresis were carried out as described by Teras et al. in 2009.
Gel mobility shift assay
The same radiolabelled PCR products that were used for DNase I footprint assays were used for the gel mobility assay. Additionally, the non-labelled PCR product containing the Fis binding site LF2 [44] and a PCR product without Fis binding site RF1 [44] were used in out-competition experiments. The unlabelled DNA fragment LF2 was amplified using the oligonucleotides TnLsisse and SIDD-2. Oligonucleotides PRH8 and Tnots were used for the amplification of unlabelled DNA RF1. Plasmids pLA1-12 and pRA1-12 [44] were used as templates for amplifying LF2 and RF1, respectively. Amounts of competing DNA in the reaction mixes were calculated in molecules. Binding reactions with purified P. putida His-tagged Fis were carried out with 2 × 1010 molecules (750–1000 c.p.m.) of labelled DNA fragment in a reaction buffer (24 mM Tris/HCl pH 7.5, 50 mM KCl, 10 mM MgCl2, 1 mM CaCl2, 0.1 mM EDTA, 5% glycerol, 0.05 μg BSA μl-1 and 0.05 μg salmon sperm DNA μl-1) in a final volume of 20 μl. The mixtures were preincubated for 20 min at room temperature. After incubation, reaction mixtures were applied to a 5% non-denaturing polyacrylamide gel buffered with TBE (50 mM Tris, 60 mM boric acid, 5 mM EDTA; pH 7.5). Electrophoresis was carried out at 4°C at 10 V cm-1 for 3 hours. Gels were vacuum dried and exposed to a Typhoon Trio screen (GE Healthcare).
Measurement of β-galactosidase activity
To measure β-galactosidase activities, the pBLKT or p9TTBlacZ constructs containing the lapA promoter region in front of lacZ gene were electroporated into P. putida wild-type strain PSm or IPTG-inducible fis overexpression strain F15. The resulting colonies were streaked onto LB agar plates, grown overnight at 30°C and incubated at 4°C for 3 days. Incubation at 4°C reduced the variability between biological replicates. The cells were thereafter grown in LB medium with or without 1 mM IPTG supplementation for 18 hours at 30°C. 18 hours of incubation was chosen as the cells are in stationary phase after 18 hours of growth and the growth rate difference between PSm and F15 does not play a role [20]. Also the native amount of Fis has dropped at that timepoint [19]. For exponential phase measurements, cells were pre-grown for 18 hours, diluted 50 times and then grown to the optical density of 0.5. The method of serial dilutions for growing exponential phase bacteria [16] enabled similar results of lapA transcription activity compared to a single dilution (data not shown), indicating that one dilution is enough for exponential phase measurements. The β-galactosidase measurements from cell suspension were performed according to the protocol of Miller [34]. At least five independent measurements were performed.
Statistical analysis
Factorial analysis of variance (ANOVA) and post-hoc Bonferroni test at a significance level of 0.05 were used to assess the variability of experimental data. The calculations were performed using Statistica 13 software.
Results
Mapping lapA promoters
In order to investigate where the promoters of lapA are located, we mapped the 5ʹ ends of lapA mRNA obtained from exponential and stationary phase LB-grown P. putida by RACE. Eight 5ʹ ends for the lapA mRNA located at 27, 71, 158, 200, 262/3, 387, 532/3 and 597 bp upstream of the lapA start codon were identified from stationary phase cells (Figs 1 and 2). The ninth PCR product, approximately 900 bp long (Fig 2B, above A-VIII), was probably the result of a nonspecific amplification since it was not verified as a 5ʹ end of lapA mRNA by cDNA sequencing. The same 5ʹ ends of lapA mRNA were identified from exponentially growing bacteria, except for the 5ʹ ends located at 27, 262 and 597 bp upstream of the lapA start codon. Using the consensus sequence of E. coli sigma70-dependent promoters [45] we predicted the -10 boxes of the eight putative promoters PlapA1 to PlapA8 (Figs 1 and 2).
Fig 2. Mapped mRNA 5ʹ ends, promoters and Fis binding sites at the lapA promoter region.

(A) Sequence of the lapA promoter region and downstream DNA. The start codons of the lapA and lapB genes are shown in bold. The first nucleotides of the mRNA 5ʹ ends determined by 5ʹ RACE are written in bold and designated as A-I to A-VIII. The potential –10 and –35 elements of the lapA promoters are shown in grey boxes with solid and dotted outlines, respectively. The Fis binding sites Fis-A1, Fis-A2 and Fis-A4 to Fis-A7 are shown in black brackets. The E. coli Fis binding consensus according to Finkel and Johnson (1992) and Shao et al. (2008) is shown above every Fis binding sequence [46,47]. The most important nucleotides in the E. coli Fis binding consensus are shown in bold. The point mutations in the Fis binding sequences are indicated by arrows on the antisense strand. The substituted nucleotides in the -10 boxes of potential promoters are shown in bold on the sense strand. In silico predicted FleQ binding sequences [48] are indicated by grey boxes. (B) Agarose gel electrophoresis of cDNA amplified by the RACE method for the identification of the lapA mRNA 5ʹ ends. The arrows point to the PCR products used to determine the mRNA 5ʹ ends. The primer used is shown under the gel images.
To confirm that the identified 5ʹ mRNA ends correspond to transcription start sites, lapA promoter region fragments were cloned into the promoter probe vector pBLKT containing the lacZ reporter gene (Fig 1). All of the cloned DNA fragments shared a common 3ʹ end at the position -27 from the lapA gene start codon. The first construct pB_PlapA1 contained one potential promoter PlapA1 (Fig 1). The second construct pB_PlapA1-2 contained two potential promoters PlapA1 and PlapA2 and so on; so that each subsequent construct contained one additional potential promoter compared to the previous one. The β-galactosidase activity was measured in stationary-phase cells of the P. putida wild-type strain PSm (Table 1). By extending the lapA promoter region from the 5ʹ end, we expected to see an increase in β-galactosidase activity every time a functional promoter was added.
Table 1. B-galactosidase activity (Miller units) expressed from different length lapA promoter constructs.
| Construct | 4 h | 18 h | ||
|---|---|---|---|---|
| PSm | PSmΔrpoS | PSm | PSmΔrpoS | |
| pBLKT* | 0.36 (0.05) a | 0.58 (0.30) a | 0.98 (0.05) b | 1.09 (0.13) b |
| pB_PlapA1 | - | - | 0.8 (0.2) a | - |
| pB_PlapA1-2 | - | - | 9.9 (2.5) a | - |
| pB_PlapA1-3 | - | - | 570.6 (46.4) b | - |
| pB_PlapA1-4 | - | - | 501.5 (95.7) bc | - |
| pB_PlapA1-5 | - | - | 567.7 (21.4) bc | - |
| pB_PlapA1-6 | - | - | 861.7 (53.0) d | - |
| pB_PlapA1-7 | - | - | 457.5 (95.4) c | - |
| pB_PlapA1-8 | - | - | 516.7 (37.7) bc | - |
| pB_PlapA2 | 9.7 (1.6) a | 7.7 (1.0) a | 18.4 (3.3) b | 16.9 (2.4) b |
| pB_PlapA2mut | 7.4 (1.2) a | - | 15.1 (3.0) b | - |
| pB_PlapA3 | 398.4 (31.2) b | 480.2 (44.0) b | 1217.8 (106.5) c | 1443.3 (90.0) d |
| pB_PlapA3mut | 28.7 (2.6) a | - | 71.0 (4.4) a | - |
| pB_PlapA4 | 70.4 (9.0) b | 67.7 (3.2) b | 374.5 (74.2) c | 385.5 (113.8) c |
| pB_PlapA4mut | 2.4 (0.6) a | - | 10.8 (2.1) ab | - |
| pB_PlapA5 | 20.9 (2.3) b | 22.6 (4.1) b | 91.2 (5.5) d | 83.9 (9.0) d |
| pB_PlapA5mut | 10.2 (0.8) a | - | 38.8 (2.9) c | - |
| pB_PlapA6 | 8.1 (1.1) b | 8.7 (1.3) b | 34.4 (1.5) d | 19.7 (1.5) c |
| pB_PlapA6mut | 0.4 (0.1) a | - | 2.4 (0.3) a | - |
| pB_PlapA7 | 8.8 (1.5) b | 7.6 (0.3) b | 37.6 (4.0) d | 24.0 (1.5) c |
| pB_PlapA7mut | 1.1 (0.1) a | - | 2.8 (0.3) ab | - |
| pB_PlapA8 | 32.6 (2.2) b | 35.9 (2.4) b | 163.2 (7.5) d | 123.0 (11.5) c |
| pB_PlapA8mut | 12.0 (1.4) a | - | 33.1 (7.4) b | - |
*The promoterless pBLKT does not contain lapA upstream DNA.
Data from at least 5 independent measurements is shown. 95% confidence intervals are shown in parentheses. Not determined activities are marked with the “-”symbol. Groups of data analysed separately are divided by horizontal lines. Letters a-e depict different homogeneity groups according to ANOVA post hoc Bonferroni test. Identical letters denote non-significant differences (P>0.05) between averages of β-galactosidase activity.
All constructed plasmids ensured β-galactosidase activity in P. putida wild-type strain PSm except pB_PlapA1, which β-galactosidase activity was comparable to the activity of a promoterless pBLKT vector which was approximately 1 Miller unit (Table 1). This indicated that there is no σ70 type promoter in pB_PlapA1. The wild-type cells harbouring pB_PlapA1-2 showed a β-galactosidase activity of 10 Miller units. The increase in β-galactosidase activity compared to pB_PlapA1 was not statistically significant, suggesting that this upstream lapA region also lacks a functional σ70 type promoter or contains only a very weak one. Only two constructs, pB_PlapA1-3 and pB_PlapA1-6, displayed a statistically significant increase in β-galactosidase activity compared to the previous shorter version, suggesting that PlapA3 and PlapA6 are lapA’s promoters (Table 1). Adding subsequent hypothetical promoters PlapA4, PlapA5, PlapA7 and PlapA8 did not increase the β-galactosidase activity, which suggested that these may not contribute to transcriptional activation of lapA. However, cumulative extension of the regulatory region may add potential regulator (repressor) binding sites in addition to promoters. Thus, it might be possible that additional regulators that bind longer fragments may mask the effect of weaker promoters in these constructs.
Therefore, to examine this possibility, we decided to assess the effect of each individual potential promoter to the transcription of the reporter gene (Table 1). The β-galactosidase activity of hypothetical promoters PlapA2, PlapA3, PlapA4, PlapA5, PlapA6, PlapA7 and PlapA8 cloned into the promoter probe vector pBLKT were measured in both exponential and stationary-phase cells of the P. putida wild-type strain PSm (Table 1). All assessed pBLKT constructs (except for previously measured pB_PlapA1) revealed β-galactosidase activity in PSm (Table 1). To verify the existence of promoters in cloned lapA upstream region, we used the same individual promoter regions but with mutated -10 boxes (Table 1). Although the number of substituted nucleotides varied, in general, the A-T nucleotides of the σ70 type promoter consensus were substituted with G-C nucleotides. Disrupting putative -10 boxes decreased the activity of promoters PlapA3, PlapA4, PlapA6 and PlapA7 between 8 and 35 times in both stationary phase and exponentially growing cells (Table 1), confirming that these are functional promoters. Disrupting -10 boxes of putative promoters PlapA5 and PlapA8 reduced the LacZ activity in both growth phases between 2 and 5 times (Table 1), showing that these are probably functional promoters as well. At the same time, mutating the potential -10 box of PlapA2 had no effect on β-galactosidase activity in stationary phase nor in exponentially growing PSm (Table 1). This indicated that PlapA2 is either not a σ70 promoter or not a functional promoter at all.
To examine the possibility of the presence of any unidentified promoters in the proximal upstream region of lapA gene, we constructed pB_PlapA1-3_PlapA3mut (S1 Table). This construct is otherwise identical to pB_PlapA1-3 except for the mutated -10 box of PlapA3, which was identified as the strongest lapA promoter (Table 1). P. putida PSm harbouring pB_PlapA1-3 with mutated PlapA3 showed a LacZ activity of 14 Miller units in exponential growth phase and 62 Miller units in stationary phase, which is respectively 12.6 and 7.1 times less than wild-type pB_PlapA1-3 (Fig 3). The LacZ activity measured with pB_PlapA1-3_PlapA3mut was probably a residual promoter activity left after mutating PlapA3 because the mutated PlapA3 promoter revealed a similar pattern in the study of individual promoters (Table 1). Thus, it seems that PlapA3 is the most important promoter for lapA transcription in LB medium and the proximal region of lapA up to PlapA3 does not carry any additional promoters that would be active in LB medium, at least not at a considerable level.
Fig 3. The effect of mutating PlapA3 on the transcription from the proximal upstream DNA of lapA.

B-galactosidase (β-Gal) activity expressed from the lapA promoter-lacZ reporter constructs containing upstream DNA at position -27 to -200 was measured in P. putida wild-type strain PSm grown in LB medium to optical density of 0.5 (exponential phase) and for 18 hours (stationary phase). Schemes of lapA proximal upstream DNA carrying the functional PlapA3 promoter and predicted promoters PlapA1 and PlapA2 are shown above the diagrams. The Dotted box denotes the mutated promoter PlapA3, lacZ reporter gene is shown as a black arrow and location of sequences previously described as PlapA1 and PlapA2 in white boxes. Vertical bars denote 95% confidence intervals of means. Data of at least 9 independent measurements is shown. Letters a–c depict homogeneity groups according to ANOVA post hoc Bonferroni test. Identical letters denote non-significant differences (P>0.05) between averages of β-galactosidase activity.
RpoS-dependency of lapA promoters
We were interested in the RpoS-dependency of all identified promoters. Considering that lapA has six promoters unequally contributing to the expression of lapA, we decided to test the RpoS-dependency of all promoters individually to avoid the masking effects of strong promoters like PlapA3. We measured β-galactosidase activity in stationary phase PSmΔrpoS carrying the individual promoter constructs (Table 1). This experiment revealed that PlapA6, PlapA7 and PlapA8 show a moderate RpoS-dependence in stationary phase as the plasmids with the corresponding promoters ensured decreased β-galactosidase activity in rpoS deletion strain compared to the β-galactosidase activity in wild-type strain (Table 1). PlapA4 and PlapA5 are not RpoS-dependent and PlapA3 showed an approximately 1.2 times higher activity in the rpoS deletion strain than in the wild-type PSm (Table 1).
To verify the RpoS-dependence of promoters, we measured the β-galactosidase activity also in exponentially growing PSm and PSmΔrpoS (Table 1). The transcription and translation of rpoS in P. putida is downregulated in exponentially growing cells [19,49,50] and therefore RpoS should not affect promoters in exponentially growing cells [9,51,52]. As expected, rpoS deletion has no statistically significant effect on the activities of the measured promoters PlapA3, PlapA4, PlapA5, PlapA6, PlapA7 and PlapA8 in exponentially growing cells. However, it seems that RpoS did not affect the activity of PlapA3, since similarly to stationary phase cells, exponentially grown cells carrying pB_PlapA3 exhibited an approximately 1.2-times higher β-galactosidase activity in the absence of RpoS. Moreover, Martinez-Gil et al. have reported in 2014 that the transcription from promoter probe vector carrying lapA proximal promoters identified in this work as PlapA3, PlapA4 and PlapA5 is RpoS-independent. Thus, the statistically significant activation of PlapA3 promoter in PSmΔrpoS strain can be a type I error of statistical hypothesis testing without biological importance.
Altogether, we have identified six lapA promoters with recognisable -10 elements: PlapA3, PlapA4, PlapA5, PlapA6, PlapA7 and PlapA8. All of them are active in both exponential and stationary phase. PlapA3 seems to be the most important for the expression of lapA as it shows the highest activity in both β-galactosidase experiments: cumulative extensions of the 5ʹ end of lapA promoter area and studying individual promoters. PlapA6, PlapA7 and PlapA8 display RpoS-dependence. As the effects are moderate, these promoters are probably recognized by both sigma factors RpoS and RpoD.
Mapping Fis binding sites in the lapA promoter region
We have previously reported that fis-overexpression increases the amount of LapA in P. putida about 1.6 times [4]. However, it was yet unclear if Fis regulates the expression of lapA directly by binding lapA promoter area and controlling its transcription. To elucidate where Fis binding sites are located, we used in silico prediction and thereafter determined Fis binding by DNase I footprint and gel-shift analysis.
Eight Fis binding sequences, Fis-A1 to Fis-A8, were predicted for the lapA promoter region in silico (Table 2). DNase I footprint analysis verified six of the Fis binding sites: Fis-A1 at approximately -95 bp relative to the lapA gene, Fis-A2 at -145 bp (Figs 2A and 4A); Fis-A4 at -400 bp (Figs 2A and 5A) Fis-A5 at -575 bp, Fis-A6 at -615 bp (Figs 2A and 6A) and Fis-A7 at -755 bp relative to the lapA gene (Figs 2A and 7A). We could not verify Fis binding to the predicted Fis-A3 and Fis-A8 sequences by DNase I footprint analysis (data not shown).
Table 2. Sequences of in silico predicted Fis binding sites in the upstream region of lapA gene.
| Name of site | Sequence | Strand | Positions from start codon | Weight scorea | P-value |
|---|---|---|---|---|---|
| Fis-A1 | GACGTCAATATTCCGTCTAT | Antisense | -117…-98 | 5.7 | 4.7 × 10−4 |
| Fis-A2 | AAGGTCAATAGTTTGGCAGT | Sense | -156…-137 | 7.5 | 6.4 × 10−5 |
| CTGCCAAACTATTGACCTTA | Antisense | -157…-138 | 6.5 | 2.0 × 10−4 | |
| Fis-A3 | ATGTAAGACATTTGTCCTCA | Sense | -245…-226 | 5.7 | 4.7 × 10−4 |
| Fis-A4 | GTGCTTATTTGGCAATCAGT | Sense | -404…-385 | 5.4 | 6.3× 10−4 |
| CTGATTGCCAAATAAGCACA | Antisense | -405…-386 | 5.6 | 5.1 × 10−4 | |
| Fis-A5 | TTGCATTCAATATCCGCAAG | Sense | -584…-565 | 5.5 | 5.7 × 10−4 |
| TTGCGGATATTGAATGCAAG | Antisense | -585…-566 | 5.4 | 6.3 × 10−4 | |
| Fis-A6 | GTTAGTCAAAATTCATCTAT | Sense | -625…-606 | 6.6 | 1.8 × 10−4 |
| TAGATGAATTTTGACTAACA | Antiense | -626…-607 | 5.6 | 5.1 × 10−4 | |
| Fis-A7 | TTGTTTCATTTTTGATCCAG | Sense | -765…-746 | 7.3 | 8.1 × 10−5 |
| TGGATCAAAAATGAAACAAT | Antisense | -766…-747 | 7.4 | 7.2 × 10−5 | |
| Fis-A8 | ACGTAGAATTGTTTACTCAA | Antisense | -818…-799 | 5.3 | 6.9 × 10−4 |
a The maximum possible weight score for the Fis matrix was 12.5.
Fig 4. Fis binding to Fis-A1 and Fis-A2 sites upstream of the lapA gene.

(A) Protection of the lapA upstream DNA against DNase I cleavage by Fis binding on the sense and the antisense strands. Lines at the right side of the panels indicate the regions protected by Fis from DNase I cleavage at the positions -110 to -85 on the sense strand and -109 to -84 on the antisense strand corresponding to Fis-A1; and -158 to -132 on the sense strand and -159 to -138 on the antisense strand corresponding to Fis-A2. (B) Gel shift assay of the Fis binding to the lapA promoter DNA containing the wild-type Fis binding site Fis-A1 and Fis-A2 and one or the other mutated site. 2 × 1010 molecules of radioactively labelled PCR products containing both Fis binding sites Fis-A1 and FisA2 or one mutated binding site FisA1mut-Fis-A2, and Fis-A1-FisA2mut were used in Fis binding assay. Fis was outcompeted from Fis-DNA complex with unlabelled PCR product containing the Fis binding site (LF2) or a PCR product without Fis binding site (RF1). Arrows point to different dissociation of Fis from radioactively labelled DNA in favour of binding unlabelled Fis-specific DNA. Added unlabelled DNA was calculated in molecules. 0.46 μM Fis was used in each reaction mixture except mixtures without Fis in lanes 2, 12 and 22.
Fig 5. Fis binding to Fis-A4 site upstream of the lapA gene.

(A) Protection of the lapA upstream DNA against DNase I cleavage by Fis binding on the sense and the antisense strands. Lines at the right side of the panels indicate the regions protected by Fis from DNase I cleavage at the positions -417 to -381 on the sense strand and -409 to -385 on the antisense strand corresponding to Fis-A4. (B) Gel shift assay of the Fis binding to the lapA promoter DNA containing the wild-type Fis binding site Fis-A4 and mutated site Fis-A4mut. 2 × 1010 molecules of radioactively labelled PCR products containing Fis-A4 and FisA4-mut sites were used for Fis binding. Fis was outcompeted from Fis-DNA complex with unlabelled PCR product containing the Fis binding site (LF2) or a PCR product without Fis binding site (RF1). Arrows point to different dissociation of Fis from radioactively labelled DNA in favour of binding unlabelled Fis-specific DNA. Added unlabelled DNA was calculated in molecules. 0.46 μM Fis was used in each reaction mixture except mixtures without Fis in lanes 2 and 12.
Fig 6. Fis binding to Fis-A5 and Fis-A6 sites upstream of the lapA gene.

(A) Protection of the lapA upstream DNA against DNase I cleavage by Fis binding on the sense and the antisense strands. Lines at the right side of the panels indicate the regions protected by Fis from DNase I cleavage at the positions -586 to -566 on the sense strand and -586 to -564 on the antisense strand corresponding to Fis-A5; and -627 to -603 on the sense strand and -626 to -604 on the antisnse strand corresponding to Fis-A6. (B) Gel shift assay of the Fis binding to the lapA promoter DNA containing the wild-type Fis binding site Fis-A5 and Fis-A6 and one or other mutated site. 2 × 1010 molecules of radioactively labelled PCR products containing both Fis binding sites Fis-A5 and FisA6 or one mutated binding site FisA5mut-Fis-A6, and Fis-A5-FisA6mut were used in the assay. Fis was outcompeted from Fis-DNA complex with unlabelled PCR product containing the Fis binding site (LF2) or a PCR product without Fis binding site (RF1). Arrows point to different dissociation of Fis from radioactively labelled DNA in favour of binding unlabelled Fis-specific DNA. Added unlabelled DNA was calculated in molecules. 0.46 μM Fis was used in each reaction mixture except mixtures without Fis in lanes 2, 12 and 22.
Fig 7. Fis binding to Fis-A7 site upstream of the lapA gene.

(A) Protection of the lapA upstream DNA against DNase I cleavage by Fis binding on the sense and the antisense strands. Lines at the right side of the panels indicate the regions protected by Fis from DNase I cleavage at the positions -744 to -768 on the sense strand and -745 to -769 on the antisense strand corresponding to Fis-A7. (B) Gel shift assay of the Fis binding to the lapA promoter DNA containing the wild-type Fis binding site Fis-A7 and mutated site Fis-A7mut. 2 × 1010 molecules of radioactively labelled PCR products containing Fis-A7 and Fis-A7mut sites were used for Fis binding. Fis was outcompeted from Fis-DNA complex with unlabelled PCR product containing the Fis binding site (LF2) or a PCR product without Fis binding site (RF1). Arrows point to different dissociation of Fis from radioactively labelled DNA in favour of binding unlabelled Fis-specific DNA. Added unlabelled DNA was calculated in molecules. 0.46 μM Fis was used in each reaction mixture except mixtures without Fis in lanes 2 and 12.
Thereafter, all verified Fis binding sites were mutated considering the most important nucleotides in E. coli Fis binding sites [46,47]. Enough positions were chosen for nucleotide substitutions to avert in silico binding [43] to the sites. Mutated Fis binding sites (Fig 2A) were used to confirm direct Fis binding to the lapA promoter region. The DNase I footprint analysis revealed that unlike the wild-type Fis binding sites, Fis did not prevent DNase I cleavage of the Fis-A1mut, A2mut, A5mut, A6mut or A7mut sequences (Figs 4A, 6A and 7A). Fis-A4 was an exception because it required more substitutions in the Fis binding site than were predicted in silico. First, five substitutions were made in Fis-A4 that averted in silico binding of Fis, but maintained the functionality of the promoter PlapA6. However, the five substitutions in the PlapA6−10 box flanking sequence did not hinder Fis protection of Fis-A4 against DNase I cleavage (data not shown). More extensive mutation of Fis-A4 that included substitutions in the -10 box of PlapA6 (Fig 2A) abolished Fis-A4mut protection by Fis against DNase I (Fig 5A).
Additionally, Fis binding to the six determined binding sequences was assessed by gel mobility shift analysis. To assess Fis-specific binding, unlabelled DNA containing the LF2 Fis binding site from the left end of Tn4652 [44] was used to outcompete Fis from the lapA promoter DNA-Fis complex (Figs 4B, 5B, 6B and 7B). Although Fis bound relatively similarly to the wild-type and mutated DNA fragments, LF2 outcompeted Fis from all of the complexes with mutated DNA fragments more easily than those with wild-type Fis binding sites (Figs 4B, 5B, 6B and 7B). Mutating Fis-A4 (Fig 5B, compare lanes 5 and 15) and Fis-A7 (Fig 7B, compare lanes 5 and 15) enabled out competition by LF2 the most. Mutating Fis-A1, Fis-A2, Fis-A5 or Fis-A6 had smaller effects. This can be due to the presence of Fis-A1 and Fis-A2 binding sites within the same DNA fragment. Mutating one site still left the other one unaffected and able to bind Fis, thereby hindering the out-competition by LF2. The same applied to Fis-A5 or Fis-A6, which are also together in one PCR fragment.
Altogether, DNase I footprint and gel mobility shift verified Fis binding to six binding sites (Fig 2) upstream of the lapA gene in vitro. Mutating Fis binding sites averted Fis binding and enabled easier outcompetition by Fis-specific DNA.
The positive effect of Fis on lapA transcription depends on Fis-A5 and Fis-A7
To investigate the possible effect of Fis on the transcription of the lapA gene, the 951 bp DNA fragment containing all of the investigated potential lapA promoters was cloned in front of the lacZ reporter gene in the low-copy-number promoter probe vector p9TTBlacZ (S1 Table). B-galactosidase activity was measured in stationary-phase PSm (wild-type) and F15 (IPTG-inducible fis overexpression strain, [20]) cells (Fig 8 and S3 Table). The fis overexpression strain was used as fis deletion is lethal to Pseudomonas species [20,31,53] and conditional expression strains were unstable (data not shown).
Fig 8. The effect of mutated Fis binding sites in the lapA promoter region on the level of reporter gene lacZ expression.

B-galactosidase (β-Gal) activity expressed from the lapA promoter-lacZ reporter constructs was measured in P. putida wild-type strain PSm and fis overexpression strain F15 grown in LB medium with or without 1 mM IPTG for 18 hours. Schemes of Fis binding sites (shown as grey boxes) are shown below the diagrams. Dotted lines denote mutated Fis binding sites and the lacZ reporter gene is shown as a black arrow. The scheme is not to scale. Vertical bars denote 95% confidence intervals of means. Data of at least 5 independent measurements is shown. Letters a–c depict homogeneity groups according to ANOVA post hoc Bonferroni test. Within subfigures, identical letters denote non-significant differences (P>0.05) between averages of β-galactosidase activity.
The P. putida strains PSm and F15 harbouring promoterless p9TTBlacZ showed a β-galactosidase activity of 0.21 to 0.45 Miller units (S3 Table). The β-galactosidase activity of PSm did not depend on added IPTG as the investigated vectors ensured a similar LacZ activity in cells grown with or without IPTG (Figs 8 and 9). This demonstrated that IPTG itself has no effect on the expression of the lacZ reporter gene. IPTG-induced fis overexpression in the F15 strain harbouring an additional fis gene copy under the Ptac promoter [20] increased the activity of the lapA promoter region 1.4 times compared to no IPTG supplementation, indicating that Fis activates the transcription of lapA in stationary phase (Fig 8A). No effect of fis overexpression was observed in exponentially growing P. putida (Fig 9). This result was expected because we have seen the impact of fis overexpression on the amount of LapA in stationary phase cells and on biofilm after 24 hours of inoculation but not in exponentially growing bacteria or 4-hours-old biofilm of P. putida [4].
Fig 9. The effect of fis overexpression to the transcription of lapA promoter region in exponential phase P. putida.
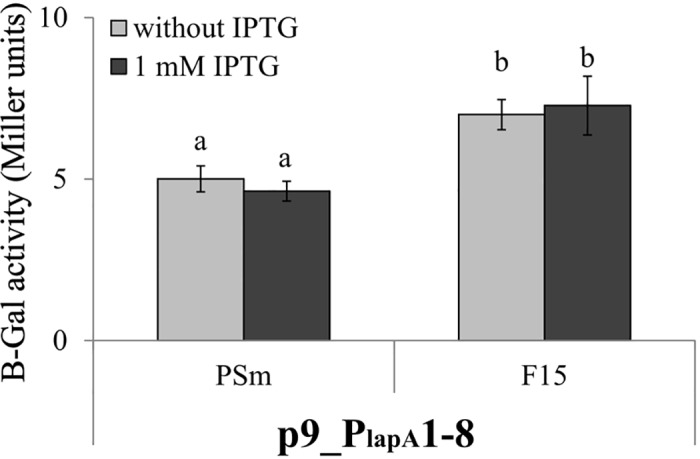
Β-galactosidase (β-Gal) activity expressed from the lapA promoter-lacZ reporter constructs was measured in P. putida wild-type strain PSm and fis overexpression strain F15 grown in LB medium with or without 1 mM IPTG to the optical density of 0.5. Vertical bars denote 95% confidence intervals of means. Data of at least 10 independent measurements is shown. Letters a–b depict homogeneity groups according to ANOVA post hoc Bonferroni test. Identical letters denote non-significant differences (P>0.05) between averages of β-galactosidase activity.
To investigate which Fis binding sites are used for the activation of lapA transcription in the stationary phase, we used mutated Fis binding sites. Substitutions in the sites Fis-A1 and/or Fis-A2 did not abolish the positive effect of Fis-overexpression on the activity of LacZ (Fig 8B–8D). Therefore, these sites alone in the lapA promoter area did not ensure the Fis’ positive effect on transcription. Constructs p9_PlapA1-8_F4mut and p9_PlapA1-8_F6mut also displayed Fis-induced activation (Fig 8E and 8G), showing that Fis-A4 and Fis-A6 are not individually responsible for the Fis-induced transcription activation of lapA. However, Fis-A5 and Fis-A7 seem to be important for the Fis induced lapA expression as mutating either one diminished the Fis-induced increase in β-galactosidase activity (Fig 8F and 8H) characteristic to the native lapA promoter region. As a control, we also measured the β-galactosidase activity of the construct p9_PlapA1-8_F4,5,6,7mut with all distal binding sites mutated together and unsurprisingly it also had no Fis-induction effect (Fig 8I).
Fis has a direct effect only to PlapA8 among the distal individual promoters
We were interested in Fis’ influence on transcription of three promoters PlapA6, PlapA7 and PlapA8 as Fis binding sites from Fis-A4 to Fis-A7 were located in positions that could influence the transcription from these promoters directly. Near the promoter PlapA6 is one Fis binding site, Fis-A4, which overlaps the -10 box of the promoter (Fig 2). Regulator binding sites in such positions generally repress transcription. The Fis binding site Fis-A5 is located upstream of the promoter PlapA7 (Fig 2). There are two Fis binding sites near PlapA8: Fis-A6 overlaps the -10 box of PlapA8 and Fis-A7 is located upstream of the promoter in position -155 from the 5ʹ end A-VIII (Fig 2).
Fis-A7 is necessary for the Fis enhanced transcription activation from the most distal lapA promoter—PlapA8. Fis overexpression induced by IPTG in F15 increased the activity of PlapA8 1.8 times compared to no IPTG supplementation. Mutating the Fis-A7 binding site abolished the positive effect of fis overexpression on the activity of PlapA8 (Fig 10). Mutating the Fis-A6 binding site overlapping the same promoter did decrease the overall transcription, but the positive effect of fis overexpression was still present (Fig 10). This indicated that Fis does not affect PlapA8 via binding to Fis-A6.
Fig 10. The effect of mutated Fis binding sites Fis-A6 and Fis-A7 to PlapA8 promoter.

B-galactosidase (β-Gal) activity expressed from the lapA promoter-lacZ reporter constructs p9_PlapA8B, p9_PlapA8B_F6mut, p9_PlapA8B_F7mut and p9_PlapA8B_F6,7mut were measured in P. putida wild-type strain PSm and fis overexpression strain F15 grown in LB medium with or without 1 mM IPTG for 18 hours. Schemes of Fis binding sites (shown as grey boxes) are shown below the diagrams. Dotted lines denote mutated Fis binding sites, lacZ reporter gene is shown as a black arrow and promoter PlapA8 in a small white box. The scheme is not to scale. Vertical bars denote 95% confidence intervals of means. Data of at least 9 independent measurements is shown. Letters a–i depict homogeneity groups according to ANOVA post hoc Bonferroni test. Identical letters denote non-significant differences (P>0.05) between averages of β-galactosidase activity.
Fis overexpression seemed to repress the activity of the individual promoter PlapA7 (Fig 11C), but mutations in Fis-A5 had no effect on the PlapA7 activity. However, in the above-described experiment with the 951 bp long lapA promoter area, we observed the positive effect of fis overexpression on lapA transcription, which depended on the functionality of the Fis-A5 binding site (Fig 8). Thus, Fis is likely still able to weakly bind the mutated Fis-binding site Fis-A5mut in vivo. The binding may be sufficient to repress the transcription from PlapA7, but not to activate the transcription of lapA via DNA topology modification. Considering the fact that PlapA7 is a weak promoter (Table 1 and Fig 11C), Fis binding to Fis-A5 affects lapA expression mostly by modifying lapA upstream DNA topology rather than regulating transcription from the individual promoter PlapA7.
Fig 11. The effect of mutated Fis binding sites to individual promoters PlapA6 and PlapA7.

B-galactosidase (β-Gal) activity expressed from the lapA promoter-lacZ reporter constructs. (A) PlapA6 promoter construct with or without mutated Fis-A4 binding site was cloned into medium-copy plasmid pBLKT and low-copy plasmid p9TTBlacZ. p9_PlapA6B, p9_PlapA6B_F4mut, (B) pB_PlapA6B, pB_PlapA6B_F4mut, (C) p9_PlapA7 and p9_PlapA7_F5mut were measured in P. putida wild-type strain PSm and fis overexpression strain F15 grown in LB medium with or without 1 mM IPTG for 18 hours. Schemes of Fis binding sites (shown as grey boxes) are shown below the diagrams. Dotted lines denote mutated Fis binding sites and mutated promoter PlapA6, lacZ reporter gene is shown as a black arrow and promoters PlapA6 and PlapA7 as white boxes. The scheme is not to scale. Vertical bars denote 95% confidence intervals of means. Data of at least 9 independent measurements is shown. Letters a–e depict homogeneity groups according to ANOVA post hoc Bonferroni test. Within subfigures, identical letters denote non-significant differences (P>0.05) between averages of β-galactosidase activity.
The impact of Fis-A4 binding site to the transcriptional activity of PlapA6 promoter remained unclear. To assess the impact of Fis to the transcription from PlapA6, we cloned the whole Fis binding site Fis-A4 mapped by DNAse I footprint analysis into p9TTBlacZ, resulting in p9_PlapA6B (S1 Table). This construct contains 28 extra nucleotides of the lapA promoter area in the 3ʹ end compared to pB_PlapA6 (Fig 1). Binding of Fis to Fis-A4 repressed the activity of PlapA6, but in a moderate manner, 1.53 times (Fig 11A and 11B). Mutating Fis-A4 also disrupted the PlapA6 promoter as we were unable to hinder Fis binding without damaging the -10 box (data not shown). Therefore, we were unable to distinguish the effects of mutating Fis binding sites from the effects of mutating the promoter.
The loss of transcription from the promoter PlapA6 with mutated -10 box depended on the copy number of the vector. Plasmid pB_PlapA6mut used in the promoter verification experiment (Table 1) and p9_PlapA6B_F4mut used to study the impact of Fis binding to the site Fis-A4 (Fig 11A) both contained identically mutated -10 boxes of PlapA6 promoter but the extent of negative effect of mutated -10 boxes differed in these constructs. Compared to the wild-type variant, the LacZ activites were 1.28 and 14.33 times lower in PSm harbouring the respective high and low copy number constructs with mutated PlapA6 (Table 1, Fig 11A and 11B). Compared to pB_PlapA6mut, the construct p9_PlapA6B_F4mut had 5 additional substitutions in the DNA flanking the -10 box due to mutation in the Fis-A4 binding site (Fig 2). To assess whether the mutation of Fis-A4 binding-site present in plasmid p9_PlapA6B_F4mut could affect the transcription from PlapA6, the DNA fragments contaning PlapA6B and PlapA6B_F4mut sequences were cloned to the medium-copy-number plasmid pBLKT, resulting in pB_PlapA6B and pB_PlapA6B_F4mut (S1 Table). Indeed, the loss of transcription from the PlapA6 in the medium-copy-number plasmid pBLKT was independent from the number of substitutions in promoter area (Table 1 and Fig 11B).
Altogether, our results indicate that the positive effect of fis overexpression on the transcription of the lapA gene depends on the Fis binding sites Fis-A5 and Fis-A7, where Fis-A7 influences the transcription from the PlapA8 promoter and Fis-A5 is probably involved in transcriptional regulation of lapA by modification of DNA topology of whole promoter area.
Discussion
LapA is the largest protein of P. putida and it is a key factor for biofilm formation in this bacterium [4,11]. LapA’s posttranslational regulation is well described but relatively little is known about the transcriptional regulation. As the excact number and location of lapA promoters was unknown, our first aim was to identify the promoters of lapA. We determined six σ70 type (RpoD) promoters upstream of lapA by RACE and by mutating potential -10 boxes of these promoters (Table 1, Figs 1 and 2). All the identified promoters of lapA were negatively affected by the substitutions in potential -10 boxes (Table 1) and were active in exponentially growing bacteria as well as in stationary phase cells. Thus, the inability to identify the 5ʹ ends of lapA mRNA corresponding to promoters PlapA5 and PlapA8 in exponential phase may have been due to technical reasons. However six promoters is an unusually high number, as most tested E. coli genes are proposed to have one or two promoters [54,55] indicating the complexity of transcriptional regulation. Indeed, some of the promoters (for example PlapA6 and PlapA7) provide only low transcription in LB-grown P. putida (Table 1) and thereby the contribution of these promoters to the expression of lapA seems insignificant. Nonetheless, the activity of promoters can depend on factors found in specific environments that may be absent in classically used growth media. Therefore, in laboratory conditions the transcription from such promoters may be downregulated. For instance, the algD transcription in P. putida KT2440 is strongly activated in the rhizosphere of maize roots and undetectable or at basal level in M9-citrate medium [56]. AlgD is the first gene in the algD-8-44KEGXLIJFA operon, which is responsible for the biosynthesis of alginate, the exopolysaccharide that is an important component of mucoid P. aeruginosa biofilm [57,58]. As the substitutions in the potential -10 boxes of PlapA6 and PlapA7 decreased LacZ activity 8 to 20 times (Table 1), it seems that PlapA6 and PlapA7 are essentially functional promoters that are weak in LB medium, yet maybe differently regulated in the nature.
Out of the identified promoters, PlapA3 seems to be the most important and the most proximal promoter for lapA transcription in LB medium (Fig 2A, Table 1). LacZ activities measured in LB-grown P. putida harbouring pB_PlapA1 or pB_PlapA1-2, which respectively contained one or two most proximal hypothetical lapA promoters, were insignificant (Table 1). Similarly to the previous two plasmids, insignificant LacZ activity was measured in LB-grown P. putida wild-type strain PSm carrying pB_PlapA1-3_PlapA3mut, which carries a mutated PlapA3 promoter.
Our results indicate that the stationary phase sigma factor RpoS can be involved in the regulation of the three distal lapA promoters, PlapA6, PlapA7 and PlapA8 (Table 1). The effects of rpoS deletion were moderate, indicating partial σS-dependence of these promoters. As RpoS and RpoD can recognize a similar promoter consensus [59,60] these promoters are probably recognized by both sigma factors, RpoD and RpoS.
The transcription regulation of lapA seems to be under the control of many regulators. In search for the lapA promoters, we extended the upstream region of lapA in the promoter probe vector, adding one hypothetical promoter at a time and expected to see an increase in activity every time a promoter is added (Table 1 and Fig 1). However, adding potential promoters to the construct did not always increase the activity of the promoter construct and adding one particular promoter PlapA7 even decreased the activity. This indicates that the extension of upstream DNA in front of the reporter gene has not added only promoters, but other regulatory areas as well.
So far, only activators have been described for lapA: FleQ, c-di-GMP, GacS and Fis have been shown to regulate lapA expression [4,16–18]. Indeed, we have previously shown that overexpressing the global transcription regulator Fis increases the amount of the LapA protein 1.6 times compared to the wild-type cells [4]. Our current work elucidates the mechanisms by which Fis activates lapA expression. We observed that fis overexpression increases the transcription of lapA 1.4 times (Fig 8A). Fis binds to the lapA promoter region in six specific sites in vitro and mutating these sites hinders binding (Figs 4–7). Fis’ positive effect on lapA transcription in vivo depends on the two distal Fis binding sites, Fis-A5 and Fis-A7 (Fig 8A, 8F and 8H). The rest of the Fis binding sites: Fis-A1, Fis-A2, Fis-A4 and Fis-A6 have a redundant impact, if any, to lapA transcription. However, the importance of the Fis-A4 binding site to the regulation of lapA transcription stays unclear, as we were unable to separate the effects of mutating the Fis-A4 binding site and the PlapA6 promoter that overlaps it (Table 1 and Fig 11A). Two mechanisms could explain the positive effect of Fis on the transcription of lapA. Firstly, Fis can enhance lapA transcription only from one individual promoter, PlapA8, which expression depends on Fis binding to Fis-A7 (Fig 10). Secondly, Fis can regulate lapA transcription by modifying the topology of upstream DNA. Indeed, the Fis binding site Fis-A5 is not important for the transcriptional activation from its nearest promoter PlapA7 (Fig 11C) but it affects transcription in the presence of all promoters (Fig 8F). Also, we cannot exclude the possibility that Fis-A1, Fis-A2, Fis-A4 and/or Fis-A6 binding sites could contribute to the lapA promoter area topology. It is likely that the two mechanisms work synergistically: Fis activates transcription of the PlapA8 promoter directly and also changes the DNA topology of the whole lapA promoter area.
Mutating potential Fis binding sites Fis-A1 and/or Fis-A2 does not abolish fis overexpression’s activating effect on lapA transcription but the overall activity of the lapA 951-bp-long promoter region decreases (Fig 8), indicating the importance of this region. Jimenez-Fernandez and other have in silico predicted three FleQ binding sites positioned at nucleotides -101 to -114, -140 to -153 and -655 to -668 [18]. Surprisingly, two of the proximal sites overlap with Fis binding sites Fis-A1 and Fis-A2 (Fig 2). The third predicted FleQ binding site is located between sites Fis-A6 and Fis-A7 (Fig 2). However, the exact FleQ binding positions in lapA upstream DNA have not been specified because FleQ binding has only been shown by gel mobility shift [18]. Mutating the Fis-A2 site also substituted three nucleotides in the predicted overlapping FleQ binding site and mutating the Fis-A1 site changed 6 nucleotides adjacent to the predicted FleQ binding site. This means these substitutions may disrupt FleQ binding and thereby decrease the total level of transcription from lapA promoter region.
This raises the question, how could FleQ binding to these two sites regulate lapA transcription in LB-grown P. putida when these sites are located downstream of all the proven promoters active in LB-grown bacteria. Activators binding downstream of promoters are uncommon in bacteria. However, FleQ seems to be an exception as Pseudomonas aeruginosa FleQ can activate transcription of flhA by binding downstream of the promoter [61]. Therefore, it is possible that FleQ activates lapA via binding sites located downstream of the promoters. The third in silico predicted FleQ binding site is located between PlapA8 and the Fis binding site Fis-A7. It is possible that Fis and FleQ regulate lapA transcription from PlapA8 promoter co-operatively. However, as the exact binding sites for FleQ remain unknown, it is impossible to propose an exact mechanism.
Biofilm development and fis expression follow similar trends in LB-grown P. putida. Both are up-regulated in a nutrient-rich environment and down-regulated or hindered by nutrient depletion. We have shown that P. putida biofilm is the strongest after 4 hours of inoculation in LB medium and it decays about threefold within the next 4 hours [4]. While exact numbers vary, others have described similar trends [5–7]. Fis mRNA levels are highest in exponentially growing planktonic P. putida and drop approximately three times in stationary phase cells [19]. It is not surprising that fis overexpression activates lapA transcription (Fig 8A) and increases the amount of LapA protein [4] resulting in enhanced mature biofilm [4,20]. At the same time, fis overexpression does not affect the 4-hours-old biofilm [4] or the expression of lapA in exponentially growing P. putida (Fig 9) [4]. Considering the increased expression of fis in exponentially growing wild-type cells [19], the native amount of Fis in fast growing P. putida could be enough to saturate the Fis binding sites Fis-A5 and Fis-A7 and enhance lapA expression. As the native fis expression decreases in stationary phase, artificial overexpression of Fis allows the detectable positive regulation of lapA.
Conclusion
Six promoters and Fis are involved in the complex regulation of lapA transcription in LB medium. Two of the six Fis binding sites are necessary for the Fis-enhanced expression of lapA in vivo. Fis activates the transcription of lapA from the most distal promoter PlapA8 and can additionally regulate lapA transcription via modification of lapA upstream DNA topology.
Supporting information
(DOCX)
a restrictases are shown in brackets. b sequences recognized by endonucleases are underlined, the nucleotides mutated in Fis binding sites or -10 boxes are shown in bold and sequences complementary to another oligonucleotide are indicated in bold and italics.
(DOCX)
Data from at least 5 independent measurements is shown. 95% confidence intervals are shown in parentheses. Letters a-c depict different homogeneity groups according to ANOVA post hoc Bonferroni test. Identical letters denote non-significant differences (P>0.05) between averages of β-galactosidase activity.
(DOCX)
Acknowledgments
We thank Andres Ainelo and Signe Saumaa for their comments on the manuscript.
Data Availability
All relevant data are included within the paper and its Supporting Information files.
Funding Statement
This work was supported by the funding of the Targeted Financing Project SF0180031s08; the Institutional Research Funding IUT20-19 from Estonian Research Council. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Wolska KI, Grudniak AM, Rudnicka Z, Markowska K (2015) Genetic control of bacterial biofilms. J Appl Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fazli M, Almblad H, Rybtke ML, Givskov M, Eberl L, Tolker-Nielsen T (2014) Regulation of biofilm formation in Pseudomonas and Burkholderia species. Environ Microbiol 16: 1961–1981. doi: 10.1111/1462-2920.12448 [DOI] [PubMed] [Google Scholar]
- 3.Waite RD, Paccanaro A, Papakonstantinopoulou A, Hurst JM, Saqi M, Littler E, et al. (2006) Clustering of Pseudomonas aeruginosa transcriptomes from planktonic cultures, developing and mature biofilms reveals distinct expression profiles. BMC Genomics 7: 162 doi: 10.1186/1471-2164-7-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moor H, Teppo A, Lahesaare A, Kivisaar M, Teras R (2014) Fis overexpression enhances Pseudomonas putida biofilm formation by regulating the ratio of LapA and LapF. Microbiology 160: 2681–2693. doi: 10.1099/mic.0.082503-0 [DOI] [PubMed] [Google Scholar]
- 5.Yousef-Coronado F, Travieso ML, Espinosa-Urgel M (2008) Different, overlapping mechanisms for colonization of abiotic and plant surfaces by Pseudomonas putida. FEMS Microbiol Lett 288: 118–124. doi: 10.1111/j.1574-6968.2008.01339.x [DOI] [PubMed] [Google Scholar]
- 6.Yousef-Coronado F, Soriano MI, Yang L, Molin S, Espinosa-Urgel M (2011) Selection of hyperadherent mutants in Pseudomonas putida biofilms. Microbiology-Sgm 157: 2257–2265. [DOI] [PubMed] [Google Scholar]
- 7.Gjermansen M, Nilsson M, Yang L, Tolker-Nielsen T (2010) Characterization of starvation-induced dispersion in Pseudomonas putida biofilms: genetic elements and molecular mechanisms. Mol Microbiol 75: 815–826. doi: 10.1111/j.1365-2958.2009.06793.x [DOI] [PubMed] [Google Scholar]
- 8.Duque E, de la Torre J, Bernal P, Molina-Henares MA, Alaminos M, Espinosa-Urgel M, et al. (2013) Identification of reciprocal adhesion genes in pathogenic and non-pathogenic Pseudomonas. Environ Microbiol 15: 36–48. doi: 10.1111/j.1462-2920.2012.02732.x [DOI] [PubMed] [Google Scholar]
- 9.Martinez-Gil M, Yousef-Coronado F, Espinosa-Urgel M (2010) LapF, the second largest Pseudomonas putida protein, contributes to plant root colonization and determines biofilm architecture. Mol Microbiol 77: 549–561. doi: 10.1111/j.1365-2958.2010.07249.x [DOI] [PubMed] [Google Scholar]
- 10.El-Kirat-Chatel S, Beaussart A, Boyd CD, O'Toole GA, Dufrene YF (2014) Single-cell and single-molecule analysis deciphers the localization, adhesion, and mechanics of the biofilm adhesin LapA. ACS Chem Biol 9: 485–494. doi: 10.1021/cb400794e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Espinosa-Urgel M, Salido A, Ramos JL (2000) Genetic analysis of functions involved in adhesion of Pseudomonas putida to seeds. J Bacteriol 182: 2363–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinsa SM, Espinosa-Urgel M, Ramos JL, O'Toole GA (2003) Transition from reversible to irreversible attachment during biofilm formation by Pseudomonas fluorescens WCS365 requires an ABC transporter and a large secreted protein. Mol Microbiol 49: 905–918. [DOI] [PubMed] [Google Scholar]
- 13.Lahesaare A, Ainelo H, Teppo A, Kivisaar M, Heipieper HJ, Teras R (2016) LapF and its regulation by Fis affect the cell surface hydrophobicity of Pseudomonas putida. PLoS One 11: e0166078 doi: 10.1371/journal.pone.0166078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navarro MVAS, Newell PD, Krasteva PV, Chatterjee D, Madden DR, O'Toole GA, et al. (2011) Structural basis for c-di-GMP-mediated inside-out signaling controlling periplasmic proteolysis. Plos Biology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newell PD, Boyd CD, Sondermann H, O'Toole GA (2011) A c-di-GMP effector system controls cell adhesion by inside-out signaling and surface protein cleavage. Plos Biology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez-Gil M, Ramos-Gonzalez MI, Espinosa-Urgel M (2014) Roles of cyclic Di-GMP and the Gac system in transcriptional control of the genes coding for the Pseudomonas putida adhesins LapA and LapF. J Bacteriol 196: 1484–1495. doi: 10.1128/JB.01287-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao Y, Nie H, Liu H, Luo X, Chen W, Huang Q (2016) C‐di‐GMP regulates the expression of lapA and bcs operons via FleQ in Pseudomonas putida KT2440. Environ Microbiol Reports 8: 659–666. [DOI] [PubMed] [Google Scholar]
- 18.Jimenez-Fernandez A, Lopez-Sanchez A, Jimenez-Diaz L, Navarrete B, Calero P, Platero AI, et al. (2016) Complex interplay between FleQ, cyclic diguanylate and multiple sigma factors coordinately regulates flagellar motility and biofilm development in Pseudomonas putida. PLoS One 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuste L, Hervas AB, Canosa I, Tobes R, Jimenez JI, Nogales J, et al. (2006) Growth phase-dependent expression of the Pseudomonas putida KT2440 transcriptional machinery analysed with a genome-wide DNA microarray. Environ Microbiol 8: 165–177. doi: 10.1111/j.1462-2920.2005.00890.x [DOI] [PubMed] [Google Scholar]
- 20.Jakovleva J, Teppo A, Velts A, Saumaa S, Moor H, Kivisaar M, et al. (2012) Fis regulates the competitiveness of Pseudomonas putida on barley roots by inducing biofilm formation. Microbiology 158: 708–720. doi: 10.1099/mic.0.053355-0 [DOI] [PubMed] [Google Scholar]
- 21.Saldana Z, Xicohtencatl-Cortes J, Avelino F, Phillips AD, Kaper JB, Puente JL, et al. (2009) Synergistic role of curli and cellulose in cell adherence and biofilm formation of attaching and effacing Escherichia coli and identification of Fis as a negative regulator of curli. Environ Microbiol 11: 992–1006. doi: 10.1111/j.1462-2920.2008.01824.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lahesaare A, Moor H, Kivisaar M, Teras R (2014) Pseudomonas putida Fis binds to the lapF promoter in vitro and represses the expression of LapF. PLoS One 9: e115901 doi: 10.1371/journal.pone.0115901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prigent-Combaret C, Zghidi-Abouzid O, Effantin G, Lejeune P, Reverchon S, Nasser W (2012) The nucleoid-associated protein Fis directly modulates the synthesis of cellulose, an essential component of pellicle-biofilms in the phytopathogenic bacterium Dickeya dadantii. Mol Microbiol 86: 172–186. doi: 10.1111/j.1365-2958.2012.08182.x [DOI] [PubMed] [Google Scholar]
- 24.Travers A, Muskhelishvili G (2005) DNA supercoiling—a global transcriptional regulator for enterobacterial growth? Nat Rev Microbiol 3: 157–169. doi: 10.1038/nrmicro1088 [DOI] [PubMed] [Google Scholar]
- 25.Cameron AD, Stoebel DM, Dorman CJ (2011) DNA supercoiling is differentially regulated by environmental factors and FIS in Escherichia coli and Salmonella enterica. Mol Microbiol 80: 85–101. doi: 10.1111/j.1365-2958.2011.07560.x [DOI] [PubMed] [Google Scholar]
- 26.Amzallag GN (2004) Adaptive changes in bacteria: a consequence of nonlinear transitions in chromosome topology? J Theor Biol 229: 361–369. doi: 10.1016/j.jtbi.2004.04.001 [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez-Gil G, Bringmann P, Kahmann R (1996) FIS is a regulator of metabolism in Escherichia coli. Mol Microbiol 22: 21–29. [DOI] [PubMed] [Google Scholar]
- 28.Bradley MD, Beach MB, de Koning AP, Pratt TS, Osuna R (2007) Effects of Fis on Escherichia coli gene expression during different growth stages. Microbiology 153: 2922–2940. doi: 10.1099/mic.0.2007/008565-0 [DOI] [PubMed] [Google Scholar]
- 29.Gutierrez-Rios RM, Freyre-Gonzalez JA, Resendis O, Collado-Vides J, Saier M, Gosset G (2007) Identification of regulatory network topological units coordinating the genome-wide transcriptional response to glucose in Escherichia coli. BMC Microbiol 7: 53 doi: 10.1186/1471-2180-7-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lenz DH, Bassler BL (2007) The small nucleoid protein Fis is involved in Vibrio cholerae quorum sensing. Mol Microbiol 63: 859–871. doi: 10.1111/j.1365-2958.2006.05545.x [DOI] [PubMed] [Google Scholar]
- 31.Teras R, Jakovleva J, Kivisaar M (2009) Fis negatively affects binding of Tn4652 transposase by out-competing IHF from the left end of Tn4652. Microbiology 155: 1203–1214. doi: 10.1099/mic.0.022830-0 [DOI] [PubMed] [Google Scholar]
- 32.Martinez-Garcia E, de Lorenzo V (2011) Engineering multiple genomic deletions in Gram-negative bacteria: analysis of the multi-resistant antibiotic profile of Pseudomonas putida KT2440. Environ Microbiol 13: 2702–2716. doi: 10.1111/j.1462-2920.2011.02538.x [DOI] [PubMed] [Google Scholar]
- 33.Kivistik PA, Putrins M, Puvi K, Ilves H, Kivisaar M, Hõrak R (2006) The ColRS two-component system regulates membrane functions and protects Pseudomonas putida against phenol. J Bacteriol 188: 8109–8117. doi: 10.1128/JB.01262-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller JH (1992) A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 35.Sharma RC, Schimke RT (1996) Preparation of electrocompetent E. coli using salt-free growth medium. Biotechniques 20: 42–44. [DOI] [PubMed] [Google Scholar]
- 36.Antoine R, Locht C (1992) Isolation and molecular characterization of a novel broad-host-range plasmid from Bordetella bronchiseptica with sequence similarities to plasmids from gram-positive organisms. Mol Microbiol 6: 1785–1799. [DOI] [PubMed] [Google Scholar]
- 37.Kues U, Stahl U (1989) Replication of plasmids in gram-negative bacteria. Microbiol Rev 53: 491–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77: 61–68. [DOI] [PubMed] [Google Scholar]
- 39.Choi KH, Kumar A, Schweizer HP (2006) A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods 64: 391–397. doi: 10.1016/j.mimet.2005.06.001 [DOI] [PubMed] [Google Scholar]
- 40.Wong SM, Mekalanos JJ (2000) Genetic footprinting with mariner-based transposition in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 97: 10191–10196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sambrook J, Russel DW (2001) Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 42.Gama-Castro S, Salgado H, Peralta-Gil M, Santos-Zavaleta A, Muniz-Rascado L, Solano-Lira H, et al. (2011) RegulonDB version 7.0: transcriptional regulation of Escherichia coli K-12 integrated within genetic sensory response units (Gensor Units). Nucleic Acids Res 39: D98–105. doi: 10.1093/nar/gkq1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Medina-Rivera A, Defrance M, Sand O, Herrmann C, Castro-Mondragon JA, Delerce J, et al. (2015) RSAT 2015: Regulatory Sequence Analysis Tools. Nucleic Acids Res 43: W50–W56. doi: 10.1093/nar/gkv362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teras R, Hõrak R, Kivisaar M (2000) Transcription from fusion promoters generated during transposition of transposon Tn4652 is positively affected by integration host factor in Pseudomonas putida. J Bacteriol 182: 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hawley DK, McClure WR (1983) Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res 11: 2237–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finkel SE, Johnson RC (1992) The Fis protein: it's not just for DNA inversion anymore. Mol Microbiol 6: 3257–3265. [DOI] [PubMed] [Google Scholar]
- 47.Shao Y, Feldman-Cohen LS, Osuna R (2008) Functional Characterization of the Escherichia coli Fis-DNA Binding Sequence. J Mol Biol 376: 771–785. doi: 10.1016/j.jmb.2007.11.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D'Arrigo I, Bojanovic K, Yang XC, Rau MH, Long KS (2016) Genome-wide mapping of transcription start sites yields novel insights into the primary transcriptome of Pseudomonas putida. Environ Microbiol 18: 3466–3481. doi: 10.1111/1462-2920.13326 [DOI] [PubMed] [Google Scholar]
- 49.Kojic M, Venturi V (2001) Regulation of rpoS gene expression in Pseudomonas: involvement of a TetR family regulator. J Bacteriol 183: 3712–3720. doi: 10.1128/JB.183.12.3712-3720.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jovcic B, Bertani I, Venturi V, Topisirovic L, Kojic M (2008) 5' Untranslated region of the Pseudomonas putida WCS358 stationary phase sigma factor rpoS mRNA is involved in RpoS translational regulation. J Microbiol 46: 56–61. doi: 10.1007/s12275-007-0127-2 [DOI] [PubMed] [Google Scholar]
- 51.Ramos-Gonzalez MI, Molin S (1998) Cloning, sequencing, and phenotypic characterization of the rpoS gene from Pseudomonas putida KT2440. J Bacteriol 180: 3421–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ilves H, Horak R, Kivisaar M (2001) Involvement of sigma(S) in starvation-induced transposition of Pseudomonas putida transposon Tn4652. J Bacteriol 183: 5445–5448. doi: 10.1128/JB.183.18.5445-5448.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, et al. (2006) An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A 103: 2833–2838. doi: 10.1073/pnas.0511100103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mendoza-Vargas A, Olvera L, Olvera M, Grande R, Vega-Alvarado L, Taboada B, et al. (2009) Genome-wide identification of transcription start sites, promoters and transcription factor binding sites in E. coli. PLoS One 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Conway T, Creecy JP, Maddox SM, Grissom JE, Conkle TL, Shadid TM, et al. (2014) Unprecedented high-resolution view of bacterial operon architecture revealed by RNA sequencing. Mbio 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramos-Gonzalez MI, Campos MJ, Ramos JL (2005) Analysis of Pseudomonas putida KT2440 gene expression in the maize rhizosphere: in vivo [corrected] expression technology capture and identification of root-activated promoters. J Bacteriol 187: 4033–4041. doi: 10.1128/JB.187.12.4033-4041.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mann EE, Wozniak DJ (2012) Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol Rev 36: 893–916. doi: 10.1111/j.1574-6976.2011.00322.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FS (2016) Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res 44: D646–653. doi: 10.1093/nar/gkv1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tanaka K, Takayanagi Y, Fujita N, Ishihama A, Takahashi H (1993) Heterogeneity of the principal sigma factor in Escherichia coli: the rpoS gene product, sigma 38, is a second principal sigma factor of RNA polymerase in stationary-phase Escherichia coli. Proc Natl Acad Sci U S A 90: 8303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaal T, Ross W, Estrem ST, Nguyen LH, Burgess RR, Gourse RL (2001) Promoter recognition and discrimination by E sigma(s) RNA polymerase. Mol Microbiol 42: 939–954. [DOI] [PubMed] [Google Scholar]
- 61.Jyot J, Dasgupta N, Ramphal R (2002) FleQ, the major flagellar gene regulator in Pseudomonas aeruginosa, binds to enhancer sites located either upstream or atypically downstream of the RpoN binding site. Journal of Bacteriology 184: 5251–5260. doi: 10.1128/JB.184.19.5251-5260.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
a restrictases are shown in brackets. b sequences recognized by endonucleases are underlined, the nucleotides mutated in Fis binding sites or -10 boxes are shown in bold and sequences complementary to another oligonucleotide are indicated in bold and italics.
(DOCX)
Data from at least 5 independent measurements is shown. 95% confidence intervals are shown in parentheses. Letters a-c depict different homogeneity groups according to ANOVA post hoc Bonferroni test. Identical letters denote non-significant differences (P>0.05) between averages of β-galactosidase activity.
(DOCX)
Data Availability Statement
All relevant data are included within the paper and its Supporting Information files.