

Abstract
Aims
Statins, inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase, possess pleiotropic effects that have been extended to modulation of various cellular behaviors. This study aimed to examine whether statins modulate vascular endothelial growth factor A (VEGF-A) expression in human retinal pigment epithelium (RPE) cells.
Main methods
Human RPE cells (h1RPE7), damaged by hydroquinone (HQ) + advanced glycation endproducts (AGE) in an in vitro AMD model, were treated with atorvastatin or lovastatin for 24 h. The expression of VEGF-A and receptor for AGE (RAGE) was evaluated by real-time RT-PCR. VEGF-A secretion was measured by ELISA. To investigate the impact of RAGE on VEGF-A expression, small interfering RNA (siRNA) for RAGE (siRAGE) was introduced into h1RPE7 cells and VEGF-A expression was measured by real-time RT-PCR. Deletions of VEGF-A and RAGE promoters were performed and transcriptional activities were measured after the addition of statins to HQ + AGE-damaged RPE cells.
Key findings
The mRNA levels of VEGF-A and RAGE and the levels of VEGF-A in the culture medium were increased by HQ + AGE. Both atorvastatin and lovastatin attenuated HQ + AGE-induced VEGF-A and RAGE expression. These statins also decreased VEGF-A levels in the culture medium. RNA interference of RAGE attenuated the up-regulation of VEGF-A in the HQ + AGE treated cells. The deletion analysis demonstrated that these statins attenuated RAGE promoter activation in HQ + AGE-damaged RPE cells.
Significance
Statins attenuated HQ + AGE-induced VEGF expression by decreasing RAGE expression. As VEGF is an important factor in developing wet AMD, statins could decrease the risk of wet-type AMD and be used as preventive medicines.
Keywords: Biological sciences, Ophthalmology, Biochemistry, Cell biology
1. Introduction
Age-related macular degeneration (AMD) is an important cause of irreversible visual disorders in elderly patients; the number of AMD patients in 2020 is estimated to be 196 million worldwide, increasing to 288 million by 2040 [1, 2, 3].
The pathogenesis of AMD has traditionally been classified into early and late stages with dry and wet forms. Wet AMD is characterized by choroidal neovascularization and may cause severe visual loss. Several studies have found that vascular endothelial growth factor (VEGF-A) expression in retinal pigment epithelial (RPE) cells is directly related to the pathogenesis/progression of wet AMD, and anti-VEGF drugs are therefore effective for AMD treatment [4, 5, 6]. Anti-VEGF drugs can decrease the progress of this disease, but they exert limited effects on visual acuity. Therefore, development of novel therapeutic agents is required. Statins, commonly used as cholesterol-lowering medications, are inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase and are reported to be useful for decreasing the risk of wet-type AMD [7]. However, the molecular mechanism by which statins reduce the risk of AMD remains unclear. Recently, we reported that the combination of hydroquinone (HQ; benzene-1,4-diol) and advanced glycation endproducts (AGE) increased the expression of VEGF-A in RPE cells and that VEGF induced RPE cell proliferation as an autocrine growth factor [8]. In this study, we examined the effects of statins on the expression of VEGF-A and on receptor for AGE (RAGE) using the HQ + AGE-induced in vitro AMD model.
2. Materials and methods
2.1. Cell culture
Human RPE cells (h1RPE7 cells) [9], were purchased from the European Collection of Cell Cultures (Salisbury, UK) and were grown in Ham's F10 medium (Gibco®, Life Technologies, Carlsbad, CA) containing 20% (v/v) fetal calf serum, 2 mM glutamine (Nacalai tesque, Kyoto, Japan) and 1 μg/mL puromycin (Gibco®). For the in vitro AMD model, h1RPE7 cells were treated with 300 μg/mL of AGE-bovine serum albumin (BSA) (Calbiochem®, Merck KGaA, Darmstadt, Germany) and 20 μM HQ (Wako Pure Chemical Industries, Ltd., Osaka, Japan) as described [8]. ARPE-19 cells were grown in 1:1 mixture of Dulbecco's modified Eagles medium (Gibco®) and Ham's F12 medium (Gibco®) containing 10% (v/v) fetal calf serum, 100 units/mL penicillin G (Wako), and 100 μg/mL streptomycin (Wako) as described [8].
2.2. Induction of VEGF-A and RAGE messenger RNA
The h1RPE7 cells (4.0 × 105 cells/mL in 24-well plates), damaged by 20 μM HQ + 300 μg/mL AGE, were treated with atorvastatin (PubChem CID: 60823) (100 nM, Sigma-Aldrich, St. Louis, MO) or lovastatin (PubChem CID: 53232) (2 μM, Sigma-Aldrich) in Ham's F10 supplemented with 20% lipid-depleted serum (biowest, Nuaillé, France) for 24 h. After a 24-h incubation, the h1RPE-7 cells were harvested, and total RNA was prepared as described [8, 10, 11, 12]. The PCR primers corresponding to nucleotides 1131–1151 and 1186–1206 for human VEGF-A mRNA (NM_001025370), 780–799 and 969–988 for human RAGE mRNA (NM_001136), 2708–2727 and 3085–3104 for human zonula occludens-1 (ZO-1) mRNA (NM_175610), 1051–1069 and 1120–1130 for human α-smooth muscle actin (αSMA) (NM_001613), 415–436 and 502–520 for human Gremlin (NM_013372), 1584–1603 and 1628–1647 for human transforming growth factor-β1 (TGFβ1) (NM_000660), 1152–1174 and 1565–1586 for human tyrosinase (TYR) (NM_000372), 2524–2543 and 2700–2719 for human MER proto-oncogene tyrosine kinase (MERTK) (NM_006343), and 420–437 and 492–509 for human β-actin mRNA (NM_001101) were synthesized by Nihon Gene Research Laboratories (NGRL; Sendai, Japan) as described [8, 10, 11, 12, 13, 14]. Real-time reverse transcription polymerase chain reaction (RT-PCR) was performed using a SYBR® Fast qPCR kit (KAPA Biosystems, Wilmington, MA) and the Thermal Cycler Dice® Real Time System (Takara Bio Inc., Kusatu, Japan) as described [8, 10, 11, 12, 15]. Target cDNAs were cloned into pBluescript SK(−) plasmid (Stratagene, La Jolla, CA) and sequential 10-fold dilutions from 102–107 copies/μL were prepared. The serial dilutions were run to verify the specificity and to test the sensitivity of the SYBR Green-based real-time RT-PCR. The mRNA expression levels were normalized to the mRNA level of β-actin, which was used to account for differences in the efficiency of reverse transcription between samples.
2.3. Measurement of VEGF-A in the culture medium
The h1RPE7 cells (4.0 × 105 cells/mL in 24-well plates), damaged by 20 μM HQ + 300 μg/mL AGE, were treated with atorvastatin (100 nM) or lovastatin (2 μM). After a 24-h incubation, the cells were given fresh medium and incubated for an additional 3 h at 37 °C. The culture supernatant of the 3-h incubation was collected for measurement of VEGF-A. The concentration of VEGF-A was determined by using a Human VEGF Assay (ELISA; enzyme-linked immunosorbent assay) kit (Immuno-Biological Laboratories Co. Ltd., Fujioka, Japan) according to the manufacturer's instructions.
2.4. RNA interference
Small interfering RNA (siRNA) directed against human RAGE was synthesized by NGRL. The sense sequence of siRNA for human RAGE was 5′-AUCUACAAUUUCUGGCUUCtt-3′ (corresponding to 466–484 of NM_001136). The Silencer® Select human scrambled siRNA was purchased from Ambion® and used as a control. Transfection of siRNAs to h1RPE7 cells was carried out using Lipofectamine® RNAiMAX Reagent (Life Technologies) as described [8, 11, 14, 15]. Cells were transfected with 5 pmol per 24-well culture dish (4.0 × 105 cells/mL in 24-well plates).
2.5. Construction of reporter plasmid and luciferase assay
The reporter constructs were prepared by inserting the 5′-flanking regions of the human VEGF-A gene [16] (–2303 ∼ + 50, –605 ∼ + 50, –188 ∼ + 50, –102 ∼ + 50, –78 ∼ + 50, –64 ∼ + 50, –43 ∼ + 50) and human RAGE gene (–766 ∼ + 29, –253 ∼ + 29, –103 ∼ + 29) upstream of a firefly luciferase reporter gene in the pGL4.17[luc2/Neo] vector (Promega, Madison, WI). Promoter plasmids were transfected into h1RPE7 cells by using Lipofectamine® 3000 (Life Technologies) as described [8]. In brief, h1RPE7 cells were seeded at 1 × 105 cells/well in 24-well plates and promoter plasmids were transfected into the cells. After 24 h, each well received fresh medium containing atorvastatin or lovastatin with HQ + AGE and was then incubated for another 24 h. After the treatment, the cells were washed with 1 ml of phosphate-buffered saline, and cell extracts were prepared in extraction buffer (Life Technologies (Tropix®): 0.1 M potassium phosphate, pH 7.8/0.2% Triton X–100). To monitor transfection efficiency, pCMV-SPORT-βgal plasmid (Life Technologies) was co-transfected in all experiments at a 1:10 dilution. Luciferase activity was measured using a PicaGene® Luciferase assay system (Toyo-ink, Tokyo, Japan) and was normalized by the β-galactosidase activity as described [8, 12, 14, 15, 17, 18].
2.6. Data analysis
Results are expressed as mean ± SE. Statistical significance was determined by Student's t-test using GraphPad Prism software (GraphPad Software, La Jolla, CA).
3. Results
3.1. VEGF-A gene expression was decreased by statins in the HQ + AGE-treated h1RPE7 cells
VEGF-A was demonstrated to be an important factor in choroidal angiogenesis and RPE maintenance [19]. Using real-time RT-PCR, we found that the level of VEGF-A mRNA in h1RPE7 cells was increased by HQ + AGE (P = 0.0026 vs control). The addition of both lovastatin and atorvastatin attenuated the HQ + AGE-induced VEGF expression (P = 0.0002 and P = 0.00153, respectively) (Fig. 1).
Fig. 1.

Induction of VEGF-A expression by the addition of lovastatin or atorvastatin to HQ + AGE-damaged h1RPE7 cells. The h1RPE7 cells were treated with no addition (control), or no addition, lovastatin or atorvastatin in the presence of HQ + AGE for 24 h. VEGF-A mRNA was measured by real-time RT-PCR using β-actin as an endogenous control. Data are expressed as mean ± SE for each group (n = 4).
3.2. Statins decreased secreted VEGF-A from the HQ + AGE-treated h1RPE7 cells
We next measured the concentration of VEGF-A in the h1RPE7 cell culture medium by ELISA and found that it was markedly increased by the treatment with HQ + AGE (P = 0.001). Both the additions of lovastatin and atorvastatin attenuated the HQ + AGE-induced VEGF secretion (P = 0.0208 and P = 0.0231, respectively) (Fig. 2).
Fig. 2.
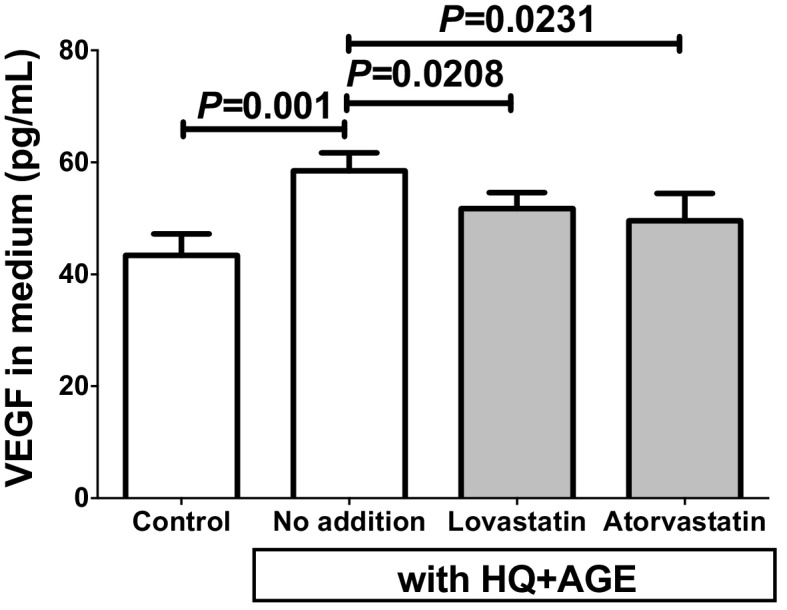
VEGF concentrations in h1RPE7 culture medium. The h1RPE7 human RPE cells were treated with no addition (control), or no addition, lovastatin or atorvastatin in the presence of HQ + AGE for 24 h. Data are expressed as mean ± SE for each group (n = 4).
3.3. RAGE gene expression was decreased by statins in the HQ + AGE-treated h1RPE7 cells
AGE may influence many signaling pathways, initiated by binding a series of cell surface receptors. The most famous AGE receptor is the multi-ligand receptor for AGE, RAGE [20]. Some other AGE receptors were described as AGE-receptor complexes (AGE-R1/OST-48, AGE-R2/80K-H, AGE-R3/galectin-3) [21, 22] and members of the scavenger receptor (SR) family (SR-A [23]; SR-B:CD36 [24, 25]; SR-BI [26]; SR-E:LOX-1 [27]; FEEL-1; FEEL-2 [28]). Using real-time RT-PCR, we found that the level of RAGE mRNA in h1RPE7 cells was increased by HQ + AGE (P = 0.0313). Both the additions of lovastatin and atorvastatin attenuated the HQ + AGE-induced RAGE expression (P = 0.014 and P = 0.0301, respectively) (Fig. 3).
Fig. 3.

Effects of lovastatin or atorvastatin on expression of RAGE in the HQ + AGE-damaged h1RPE7 cells. The h1RPE7 cells were treated with no addition (control), or no addition, lovastatin or atorvastatin in the presence of HQ + AGE for 24 h. RAGE mRNA levels were measured by real-time RT-PCR using β-actin as an endogenous control. Data are expressed as mean ± SE for each group (n = 4).
3.4. Downregulation of RAGE attenuated the VEGF increase in the HQ + AGE-treated cells
To determine the mechanism of increased HQ + AGE-stimulated VEGF expression, the RAGE gene was downregulated by the RNA interference. Knockdown of RAGE inhibited the HQ + AGE-induced VEGF upregulation (P = 0.0444), whereas HQ + AGE treatment induced VEGF expression in scrambled RNA-introduced cells (Fig. 4). These results indicated that HQ + AGE-induced VEGF upregulation was mediated by the AGE-RAGE pathway.
Fig. 4.

Effect of siRNA against RAGE on HQ + AGE-induced VEGF expression. SiRNA of RAGE was transfected into h1RPE7 cells and the cells were incubated with HQ + AGE for 24 h. VEGF-A mRNA levels were measured by real-time RT-PCR using β-actin as an endogenous control. Data are expressed as mean ± SE for each group (n = 4).
3.5. RAGE expression in the statin-treated cells was regulated by the transcriptional level
To identify the type of regulation responsible for RAGE and VEGF-A gene expression in the HQ + AGE-treated h1RPE7 cells, a 795-bp fragment containing a 766-bp promoter region of the human RAGE gene and a 2353-bp fragment containing a 2303-bp promoter region of the human VEGF-A gene were fused to the luciferase gene, respectively. Progressive deletions of the RAGE and VEGF-A promoter genes were performed and the deleted constructs were transfected into h1RPE7 cells. The deletion down to position –253 of the RAGE promoter did not significantly alter the expression of the reporter gene, but an additional deletion to nucleotide -103 caused a remarkable decrease of promoter activity in the RAGE gene (21% activity of the –766 construct). Both the additions of lovastatin and atorvastatin attenuated the HQ + AGE-induced RAGE gene transcription in the –766 and –253 constructs (77% in the –766 and –253 constructs by lovastatin; 77% and 71% in the –766 and –253 constructs by atorvastatin, respectively). However, the inhibitory effects of statins were abolished in the -103 promoter construct (Fig. 5A).
Fig. 5.

Promoter analyses of RAGE (A) and VEGF-A (B). The activities of deleted promoters of the human RAGE and VEGF-A genes are shown. A series of luciferase constructs containing promoter fragments with various 5′-ends were transfected into h1RPE7 cells. After reporter plasmid(s) were introduced into h1RPE7 cells without HQ, AGE, nor statins, cells were incubated for 24 h, and medium was replaced with fresh medium containing atorvastatin or lovastatin with HQ + AGE and was then incubated for another 24 h. The promoter activity was normalized to the activity of co-transfected β-galactosidase plasmid and expressed relative to the activity of –766 without statin in RAGE promoter (A) and -2303 without stain in VEGF promoter (B). Values are mean ± SE for each group (n = 4). Statistical significance was expressed by A and B (vs –766 RAGE promoter without stain), and C and D (vs –253 RAGE promoter without stain).
On the other hand, the deletion down to position -102 of VEGF promoter did not significantly alter the expression of the reporter gene, maintaining 65%-69% promoter activity, but an additional deletion to nucleotide –78 caused a significant decrease of promoter activity in the VEGF gene (36%). The further deletion to –43 abolished the promoter activity (∼2%). The addition of lovastatin and atorvastatin did not decrease but rather stimulated, the HQ + AGE-induced VEGF gene transcription (P < 0.0001 and P = 0.0029 in the -2303 construct, respectively). This stimulatory tendency in the VEGF promoter by statin stimulation was observed in all the constructs except for the –43 construct, which lost VEGF promoter activity (Fig. 5B)
These results indicate that RAGE expression in statin-treated cells was regulated at the transcriptional level, and that the VEGF gene in these cells is not regulated by transcription but by a post-transcriptional mechanism.
4. Discussion
AMD is a progressive disease and one of the most common causes of severe visual disorders in elderly patients. AMD is multifactorial; cigarette smoking [29, 30], diabetes mellitus [31], obesity [32], and hypertension [33, 34] have all been reported as risk factors for its pathogenesis and progression. However, the molecular effects of these risk factors remain elusive. Cigarette smoke contains several pro-oxidant compounds among which HQ is the most abundant and important. HQ causes oxidative damage to RPE cells in vitro and in vivo, and it might play a key role in the pathogenesis of AMD [35, 36, 37, 38]. AGE, which are generated by non-enzymatic reactions between glucose and protein, called the Maillard reaction, are linked to several age-related conditions, including Alzheimer's disease, atherosclerosis, diabetic complications, and AMD [39, 40, 41, 42, 43].
We recently studied the molecular pathogenesis of AMD, and reported that the combined addition of HQ + AGE stimulated VEGF expression and cell proliferation in RPE cells (ARPE-19 and h1RPE7) [8]. Judged by gene expression of TYR, encoding a key enzyme for melanin synthesis, and MERTK, implicated in phagocytosis of outer segments (Fig. 6), h1RPE7 cells seemed to be more functional than ARPE-19 cells. In addition, h1RPR7 cells grew as contact-inhibited monolayers and exhibited as epithelial morphology (Fig. 7). Thus, we used h1RPE7 cells as RPE cells in this study. The exposure RPE cells (h1RPE7 cells) to HQ + AGE did not change expression of ZO-1, αSMA, Gremlin, and TGFβ1 (Fig. 8), indicating that the experimental system seemed to be essentially free from epithelial-mesenchymal transition of retinal pigment epithelial cells. In the present study, we used this model (HQ + AGE in h1RPE7 cells) as an in vitro AMD model to seek a new therapy for the wet AMD.
Fig. 6.

Expression of TYR and MERTK mRNAs in h1RPE7 and ARPE-19 cells. TYR and MERTK mRNAs were measured by real-time RT-PCR using β-actin as an endogenous control. Data are expressed as mean ± SE for each group (n = 4).
Fig. 7.

Phase-contrast photomicrograph of h1RPE7 cells. h1RPE7 cells were grown in Ham's F10 medium supplemented with 20% (v/v) fetal calf serum, 2 mM glutamine, and 1 μg/mL puromycin.
Fig. 8.

Expression of ZO-1, αSMA, Gremlin, and TGFβ1 in h1RPE7 human RPE cells treated with no addition or HQ + AGE. Human RPE cells (h1RPE7) were treated with no addition or HQ + AGE for 24 h. ZO-1, αSMA, Gremlin, and TGFβ1 mRNAs were measured by real-time RT-PCR using β-actin as an endogenous control. Data are expressed as mean ± SE for each group (n = 4). P = 0.7313 (ZO-1), P = 0.1263 (αSMA), P = 0.4494 (Gremlin), and P = 0.8480 (TGFβ1).
Statins (e.g., atorvastatin and lovastatin), common cholesterol-lowering medications, are inhibitors for 3-hydroxy-3-methylglutaryl coenzyme A reductase, and have recently been reported to be useful for decreasing the risk of wet-type AMD [44]. Statin-induced decrease in serum low-density lipoprotein (LDL) cholesterol might reduce LDL cholesterol deposits in the drusen, which plays a key role in the pathogenesis of AMD [45]. In addition to lipid-lowering effects, statins also exert anti-inflammatory effects through inhibition of the inflammatory pathway [46, 47]. Statins decrease the plasma concentrations of C-reactive protein through attenuation of the amount and function of inflammatory cells [48, 49]. Robinson et al. demonstrated that flavastain downregulated VEGF expression in animal model [50]. Moreover, statins were shown to reduce the intravitreal VEGF levels in patients with idiopathic epiretinal membrane [51], and to reduce drusenoid pigment epithelial detachments and improvement in visual activity [6]. However, the underlying molecular mechanisms by which statins reduce the VEGF expression and secretion remains unclear, especially in RPE cells.
In this study, we examined the effects of statins on the expression of VEGF-A and RAGE using an in vitro AMD model, and showed that both atorvastatin and lovastatin attenuated the HQ + AGE-induced expression of RAGE and VEGF. Based on RNA interference experiments with RAGE mRNA, the HQ + AGE-induced expression of VEGF-A mRNA is mediated through the AGE-RAGE pathway. Deletion analyses of the RAGE promoter indicated that the promoter activity of RAGE without statins was maintained to the deletion to –253 and the inhibition of RAGE promoter activity by statins remained complete (Fig. 5A). An additional deletion to nucleotide -103 caused a significant decrease in promoter activity without statins and the statin-induced promoter inhibition also disappeared. As region –253 ∼ -103 was reported to contain Sp1-binding sites important for RAGE gene transcription [52, 53], the Sp1-binding sequences may be important for the HQ + AGE-stimulated RAGE transcription as well as the suppression of the transcription by statins. Motoyama et al. reported that the VEGF promoter was inhibited by atorvastatin via the induction of sterol regulatory element-binding protein (SREBP) in human smooth muscle cells [54]. In our study, VEGF expression seemed to be regulated at the post-transcriptional level not at the transcriptional level (Fig. 5B). Recently, microRNA (miRNA)-mediated regulation has been recognized as a post-transcriptional regulation mechanism; miRNAs appear to regulate the gene expression of more than 60% of the protein-coding genes of the human genome [55]. Hao et al. recently showed that miR-146a is highly related to the expression of VEGF-A in RPE cells [56]. Although the mechanism used by statins to reduce the HQ + AGE-induced expression of VEGF-A mRNA is elusive, statins could reduce the risk of wet AMD by decreasing the expression of RAGE and VEGF-A and therefore may be potential new drugs for preventing and treating wet AMD.
5. Conclusions
Statins attenuated HQ + AGE-induced VEGF expression by decreasing RAGE expression, which was attenuated at the transcriptional level. By decreasing the levels of VEGF-A, a key factor in the pathogenesis of wet AMD, statins may potentially be used for preventing and treating this disease.
Declarations
Author contribution statement
Hiroki Tsujinaka, Asako Itaya-Hironaka, Shin Takasawa: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Akiyo Yamauchi, Sumiyo Sakuramoto-Tsuchida, Ryogo Shobatake, Mai Makino, Naonori Masuda, Hiromasa Hirai, Nahoko Ogata: Performed the experiments; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Funding statement
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (Grant-in-Aid for Encouragement of Scientists; KAKENHI Grant Number #26931058).
Competing interest statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
References
- 1.Wong W.L., Su X., Li X., Cheung C.M., Klein R., Cheng C.Y., Wong T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob. Health. 2014;2:e106–e116. doi: 10.1016/S2214-109X(13)70145-1. [DOI] [PubMed] [Google Scholar]
- 2.Pascolini D., Mariotti S.P. Global estimates of visual impairment: 2010. Br. J. Ophthalmol. 2012;96:614–618. doi: 10.1136/bjophthalmol-2011-300539. [DOI] [PubMed] [Google Scholar]
- 3.Congdon N., O'Colmain B., Klaver C.C., Klein R., Muñoz B., Friedman D.S., Kempen J., Taylor H.R., Mitchell P. Eye Diseases Prevalence Research Group, Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. 2004;122:477–485. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- 4.Kvanta A., Algvere P.V., Berglin L., Seregard S. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Invest. Ophthalmol. Vis. Sci. 1996;37:1929–1934. [PubMed] [Google Scholar]
- 5.Tong Y., Zhao K.K., Feng D., Biswal M., Zhao P.Q., Wang Z.Y., Zhang Y. Comparison of the efficacy of anti-VEGF monotherapy versus PDT and intravitreal anti-VEGF combination treatment in AMD: a Meta-analysis and systematic review. Int. J. Ophthalmol. 2016;9:1028–1037. doi: 10.18240/ijo.2016.07.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vavvas D.G., Daniels A.B., Kapsala Z.K., Goldfarb J.W., Ganotakis E., Loewenstein J.I., Young L.H., Gragoudas E.S., Eliott D., Kim I.K., Tsilimbaris M.K., Miller J.W. Regression of some high-risk features of age-related macular degeneration (AMD) in patients receiving intensive statin treatment. EBioMedicine. 2016;5:198–203. doi: 10.1016/j.ebiom.2016.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbosa D.T.Q., Mendes T.S., Cíntron-Colon H.R., Wang S.Y., Bhisitkul R.B., Singh K., Lin S.C. Age-related macular degeneration and protective effect of HMG Co-A reductase inhibitors (statins): results from the National Health and Nutrition Examination Survey 2005-2008. Eye (Lond). 2014;28:472–480. doi: 10.1038/eye.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsujinaka H., Itaya-Hironaka A., Yamauchi A., Sakuramoto-Tsuchida S., Ota H., Takeda M., Fujimura T., Takasawa S., Ogata N. Human retinal pigment epithelial cell proliferation by the combined stimulation of hydroquinone and advanced glycation end-products via up-regulation of VEGF gene. Biochem. Biophys. Rep. 2015;2:123–131. doi: 10.1016/j.bbrep.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lund R.D., Adamson P., Sauvé Y., Keegan D.J., Girman S.V., Wang S., Winton H., Kanuga N., Kwan A.S.L., Beauchène L., Zerbib A., Hetherington L., Couraud P.-O., Coffey P., Greenwood J. Subretinal transplantation of genetically modified human cell lines attenuates loss of visual function in dystrophic rats. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9942–9947. doi: 10.1073/pnas.171266298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kyotani Y., Ota H., Itaya-Hironaka A., Yamauchi A., Sakuramoto-Tsuchida S., Zhao J., Ozawa K., Nagayama K., Ito S., Takasawa S., Kimura H., Uno M., Yoshizumi M. Intermittent hypoxia induces the proliferation of rat vascular smooth muscle cell with the increases in epidermal growth factor family and erbB2 receptor. Exp. Cell Res. 2013;319:3042–3050. doi: 10.1016/j.yexcr.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 11.Ota H., Itaya-Hironaka A., Yamauchi A., Sakuramoto-Tsuchida S., Miyaoka T., Fujimura T., Tsujinaka H., Yoshimoto K., Nakagawara K., Tamaki S., Takasawa S., Kimura H. Pancreatic β cell proliferation by intermittent hypoxia via up-regulation of Reg family genes and HGF gene. Life Sci. 2013;93:664–672. doi: 10.1016/j.lfs.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Ota H., Tamaki S., Itaya-Hironaka A., Yamauchi A., Sakuramoto-Tsuchida S., Morioka T., Takasawa S., Kimura H. Attenuation of glucose-induced insulin secretion by intermittent hypoxia via down-regulation of CD38. Life Sci. 2012;90:206–211. doi: 10.1016/j.lfs.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Yoshimoto K., Fujimoto T., Itaya-Hironaka A., Miyaoka T., Sakuramoto-Tsuchida S., Yamauchi A., Takeda M., Kasai T., Nakagawara K., Nonomura A., Takasawa S. Involvement of autoimmunity to REG, a regeneration factor, in patients with primary Sjögren's syndrome. Clin. Exp. Immunol. 2013;174:1–9. doi: 10.1111/cei.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamauchi A., Itaya-Hironaka A., Sakuramoto-Tsuchida S., Takeda M., Yoshimoto K., Miyaoka T., Fujimura T., Tsujinaka H., Tsuchida C., Ota H., Takasawa S. Synergistic activations of REG Iα and REG Iβ promoters by IL-6 and glucocorticoids through JAK/STAT pathway in human pancreatic β cells. J. Diabetes Res. 2015;2015:173058. doi: 10.1155/2015/173058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujimura T., Fujimoto T., Itaya-Hironaka A., Miyaoka T., Yoshimoto K., Yamauchi A., Sakuramoto-Tsuchida S., Kondo S., Takeda M., Tsujinaka H., Azuma M., Tanaka Y., Takasawa S. Interleukin-6/STAT pathway is responsible for the induction of gene expression of REG Iα, a new auto-antigen in Sjögren's syndrome patients, in salivary duct epithelial cells. Biochem. Biophys. Rep. 2015;2:69–74. doi: 10.1016/j.bbrep.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tischer E., Mitchell R., Hartman T., Silva M., Gospodarowicz D., Fiddes J.C., Abraham J.A. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem. 1991;266:11947–11954. [PubMed] [Google Scholar]
- 17.Nakagawa K., Takasawa S., Nata K., Yamauchi A., Itaya-Hironaka A., Ota H., Yoshimoto K., Sakuramoto-Tsuchida S., Miyaoka T., Takeda M., Unno M., Okamoto H. Prevention of Reg I-induced β-cell apoptosis by IL-6/dexamethasone through activation of HGF gene regulation. Biochim. Biophys. Acta. 2013;1833:2988–2995. doi: 10.1016/j.bbamcr.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 18.Takasawa S., Ikeda T., Akiyama T., Nata K., Nakagawa K., Shervani N.J., Noguchi N., Murakami-Kawaguchi S., Yamauchi A., Takahashi I., Tomioka-Kumagai T., Okamoto H. Cyclin D1 activation through ATF-2 in Reg-induced pancreatic β-cell regeneration. FEBS Lett. 2006;580:585–591. doi: 10.1016/j.febslet.2005.12.070. [DOI] [PubMed] [Google Scholar]
- 19.Witmer A.N., Vrensen G.F., Van Noorden C.J., Schlingemann R.O. Vascular endothelial growth factors and angiogenesis in eye disease. Prog. Retin. Eye Res. 2003;22:1–29. doi: 10.1016/s1350-9462(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto Y., Kato I., Doi T., Yonekura H., Ohashi S., Takeuchi M., Watanabe T., Yamagishi S., Sakurai S., Takasawa S., Okamoto H., Yamamoto H. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J. Clin. Invest. 2001;108:261–268. doi: 10.1172/JCI11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y.M., Mitsuhashi T., Wojciechowicz D., Shimizu N., Li J., Stitt A., He C., Banerjee D., Vlassara H. Molecular identity and cellular distribution of advanced glycation endproduct receptors: Relationship of p60 to OST-48 and p90 to 80K-H membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 1996;93:11047–11052. doi: 10.1073/pnas.93.20.11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vlassara H., Li Y.M., Imani F., Wojciechowicz D., Yang Z., Liu F.T., Cerami A. Identification of galectin-3 as a high-affinity binding protein for advanced glycation endproducts (AGE): a new member of the AGE-receptor complex. Mol. Med. 1995;1:634–646. [PMC free article] [PubMed] [Google Scholar]
- 23.Araki N., Higashi T., Mori T., Shibayama R., Kawabe Y., Kodama T., Takahashi K., Shichiri M., Horiuchi S. Macrophage scavenger receptor mediates the endocytic uptake and degradation of advanced glycation end products of the Maillard reaction. Eur. J. Biochem. 1995;230:408–415. doi: 10.1111/j.1432-1033.1995.0408h.x. [DOI] [PubMed] [Google Scholar]
- 24.Ohgami N., Nagai R., Ikemoto M., Arai H., Kuniyasu A., Horiuchi S., Nakayama H. CD36, a member of class B scavenger receptor family, is a receptor for advanced glycation end products. Ann. N. Y. Acad. Sci. 2001;947:350–355. doi: 10.1111/j.1749-6632.2001.tb03961.x. [DOI] [PubMed] [Google Scholar]
- 25.Ohgami N., Nagai R., Ikemoto M., Arai H., Miyazaki A., Hakamata H., Horiuchi S., Nakayama H. CD36 serves as a receptor for advanced glycation endproducts (AGE) J. Diabetes Complications. 2002;16:56–59. doi: 10.1016/s1056-8727(01)00208-2. [DOI] [PubMed] [Google Scholar]
- 26.Ohgami N., Nagai R., Ikemoto M., Arai H., Kuniyasu A., Horiuchi S., Nakayama H. CD36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. J. Biol. Chem. 2001;276:3195–3202. doi: 10.1074/jbc.M006545200. [DOI] [PubMed] [Google Scholar]
- 27.Jono T., Miyazaki A., Nagai R., Sawamura T., Kitamura T., Horiuchi S. Lectin-like oxidized low density lipoprotein receptor-1 (LOX-1) serves as an endothelial receptor for advanced glycation end products (AGE) FEBS Lett. 2002;511:170–174. doi: 10.1016/s0014-5793(01)03325-7. [DOI] [PubMed] [Google Scholar]
- 28.Tamura Y., Adachi H., Osuga J., Ohashi K., Yahagi N., Sekiya M., Okazaki H., Tomita S., Iizuka Y., Shimano H., Nagai R., Kimura S., Tsujimoto M., Ishibashi S. FEEL-1 and FEEL-2 are endocytic receptors for advanced glycation end products. J. Biol. Chem. 2003;278:12613–12617. doi: 10.1074/jbc.M210211200. [DOI] [PubMed] [Google Scholar]
- 29.Cong R., Zhou B., Sun Q., Gu H., Tang N., Wang B. Smoking and the risk of age-related macular degeneration: a meta-analysis. Ann. Epidemiol. 2008;18:647–656. doi: 10.1016/j.annepidem.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Khan J.C., Thurlby D.A., Shahid H., Clayton D.G., Yates J.R.W., Bradley M., Moore A.T., Bird A.C. Genetic Factors in AMD Study. Smoking and age related macular degeneration: the number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br. J. Ophthalmol. 2006;90:75–80. doi: 10.1136/bjo.2005.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi J.K., Lym Y.L., Moon J.W., Shin H.J., Cho B. Diabetes mellitus and early age-related macular degeneration. Arch. Ophthalmol. 2011;129:196–199. doi: 10.1001/archophthalmol.2010.355. [DOI] [PubMed] [Google Scholar]
- 32.Seddon J.M., Cote J., Davis N., Rosner B. Progression of age-related macular degeneration: association with body mass index, waist circumference, and waist-hip ratio. Arch. Ophthalmol. 2003;121:785–792. doi: 10.1001/archopht.121.6.785. [DOI] [PubMed] [Google Scholar]
- 33.Hyman L., Schachat A.P., He Q., Leske M.C. Age-Related Macular Degeneration Risk Factors Study Group, Hypertension, cardiovascular disease, and age-related macular degeneration. Arch. Ophthalmol. 2000;118:351–358. doi: 10.1001/archopht.118.3.351. [DOI] [PubMed] [Google Scholar]
- 34.van Leeuwen R., Ikram M.K., Vingerling J.R., Witteman J.C.M., Hofman A., de Jong P.T.V.M. Blood pressure, atherosclerosis, and the incidence of age-related maculopathy; the Rotterdam Study. Invest. Ophthalmol. Vis. Sci. 2003;44:3771–3777. doi: 10.1167/iovs.03-0121. [DOI] [PubMed] [Google Scholar]
- 35.Cao S., Walker G.B., Wang X., Cui J.Z., Matsubara J.A. Altered cytokine profiles of human retinal pigment epithelium: oxidant injury and replicative senescence. Mol. Vis. 2013;19:718–728. [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma A., Patil J.A., Gramajo A.L., Seigel G.M., Kuppermann B.D., Kenney C.M. Effects of hydroquinone on retinal and vascular cells in vitro. Indian J. Ophthalmol. 2012;60:189–193. doi: 10.4103/0301-4738.95869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertram K.M., Baglole C.J., Phipps R.P., Libby R.T. Molecular regulation of cigarette smoke induced-oxidative stress in human retinal pigment epithelial cells: implications for age-related macular degeneration. Am. J. Physiol. Cell Physiol. 2009;297:C1200–C1210. doi: 10.1152/ajpcell.00126.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strunnikova N., Zhang C., Teichberg D., Cousins S.W., Baffi J., Becker K.G., Csaky K.G. Survival of retinal pigment epithelium after exposure to prolonged oxidative injury: a detailed gene expression and cellular analysis. Invest. Ophthalmol. Vis. Sci. 2004;45:3767–3777. doi: 10.1167/iovs.04-0311. [DOI] [PubMed] [Google Scholar]
- 39.Son S.M., Jung E.S., Shin H.J., Byun J., Mook-Jung I. Aβ-induced formation of autophagosomes is mediated by RAGE-CaMKKβ-AMPK signaling. Neurobiol. Aging. 2012;33(1006):e11–e23. doi: 10.1016/j.neurobiolaging.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 40.Barlovic D.P., Soro-Paavonen A., Jandeleit-Dahm K.A. RAGE biology, atherosclerosis and diabetes. Clin. Sci. (Lond) 2011;121:43–55. doi: 10.1042/CS20100501. [DOI] [PubMed] [Google Scholar]
- 41.Myint K.M., Yamamoto Y., Doi T., Kato I., Harashima A., Yonekura H., Watanabe T., Shinohara H., Takeuchi M., Tsuneyama K., Hashimoto N., Asano M., Takasawa S., Okamoto H., Yamamoto H. RAGE control of diabetic nephropathy in a mouse model: effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes. 2006;55:2510–2522. doi: 10.2337/db06-0221. [DOI] [PubMed] [Google Scholar]
- 42.Win M.T.T., Yamamoto Y., Munesue S., Saito H., Han D., Motoyoshi S., Kamal T., Ohara T., Watanabe T., Yamamoto H. Regulation of RAGE for attenuating progression of diabetic vascular complications. Exp. Diabetes Res. 2012;2012:894605. doi: 10.1155/2012/894605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Howes K.A., Liu Y., Dunaief J.L., Milam A., Frederick J.M., Marks A., Baehr W. Receptor for advanced glycation end products and age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2004;45:3713–3720. doi: 10.1167/iovs.04-0404. [DOI] [PubMed] [Google Scholar]
- 44.Ma L., Wang Y., Du J., Wang M., Zhang R., Fu Y. The association between statin use and risk of age-related macular degeneration. Sci. Rep. 2015;5:18280. doi: 10.1038/srep18280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y., Wang M., Zhang X., Zhang Q., Nie J., Zhang M., Liu X., Ma L. The association between the lipids levels in blood and risk of age-related macular degeneration. Nutrients. 2016;8:663. doi: 10.3390/nu8100663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim K.J., Kim K.S., Kim N.R., Chin H.S. Effects of simvastatin on the expression of heme oxygenase-1 in human RPE cells. Invest. Ophthalmol. Vis. Sci. 2012;53:6456–6464. doi: 10.1167/iovs.12-9658. [DOI] [PubMed] [Google Scholar]
- 47.Qian J., Keyes K.T., Long B., Chen G., Ye Y. Impact of HMG-CoA reductase inhibition on oxidant-induced injury in human retinal pigment epithelium cells. J. Cell. Biochem. 2011;112:2480–2489. doi: 10.1002/jcb.23173. [DOI] [PubMed] [Google Scholar]
- 48.Ridker P.M., Rifai N., Pfeffer M.A., Sacks F., Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation. 1999;100:230–235. doi: 10.1161/01.cir.100.3.230. [DOI] [PubMed] [Google Scholar]
- 49.Vine A.K., Stader J., Branham K., Musch D.C., Swaroop A. Biomarkers of cardiovascular disease as risk factors for age-related macular degeneration. Ophthalmology. 2005;112:2076–2080. doi: 10.1016/j.ophtha.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 50.Robinson R., Ho C.E., Tan Q.S., Luu C.D., Moe K.T., Cheung C.Y., Wong T.Y., Barathi V.A. Fluvastatin downregulates VEGF-A expression in TNF-α-induced retinal vessel tortuosity. Invest. Ophthalmol. Vis. Sci. 2011;52:7423–7431. doi: 10.1167/iovs.11-7912. [DOI] [PubMed] [Google Scholar]
- 51.Tuuminen R., Loukovaara S. Statin medication in patients with epiretinal membrane is associated with low intravitreal EPO, TGF-beta-1, and VEGF levels. Clin. Ophthalmol. 2016;10:921–928. doi: 10.2147/OPTH.S105686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J., Schmidt A.M. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J. Biol. Chem. 1997;272:16498–16506. doi: 10.1074/jbc.272.26.16498. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka N., Yonekura H., Yamagishi S., Fujimori H., Yamamoto Y., Yamamoto H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-α through nuclear factor-kappa B, and by 17β-estradiol through Sp-1 in human vascular endothelial cells. J. Biol. Chem. 2000;275:25781–25790. doi: 10.1074/jbc.M001235200. [DOI] [PubMed] [Google Scholar]
- 54.Motoyama K., Fukumoto S., Koyama H., Emoto M., Shimano H., Maemura K., Nishizawa Y. SREBP inhibits VEGF expression in human smooth muscle cells. Biochem. Biophys. Res. Commun. 2006;342:354–360. doi: 10.1016/j.bbrc.2006.01.139. [DOI] [PubMed] [Google Scholar]
- 55.Friedman R.C., Farh K.K., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hao Y., Zhou Q., Ma J., Zhao Y., Wang S. miR-146a is upregulated during retinal pigment epithelium (RPE)/choroid aging in mice and represses IL-6 and VEGF-A expression in RPE cells. J. Clin. Exp. Ophthalmol. 2016;7(562) doi: 10.4172/2155-9570.1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]