

Abstract
The host macrophage response is now well recognized as a predictor of the success or failure of biomaterial implants following placement. More specifically, shifts from an “M1” pro-inflammatory towards a more “M2-like” anti-inflammatory macrophage polarization profile have been shown to result in enhanced material integration and/or tissue regeneration downstream. As a result, a number of biomaterials-based approaches to controlling macrophage polarization have been developed. However, the ability to promote such activity is predicated upon an in-depth, context-dependent understanding of the host response to biomaterials. Recent work has shown the impacts of both tissue location and tissue status (i.e. underlying pathology) upon the host innate immune response to implants, representing a departure from a focus upon implant material composition and form. Thus, the ideas of “biocompatibility,” the host macrophage reaction, and ideal material requirements and modification strategies may need to be revisited on a patient, tissue, and disease basis. Immunosenescence, dysregulation of macrophage function, and delayed resolution of immune responses in aged individuals have all been demonstrated, suggesting that the host response to biomaterials in aged individuals should differ from that in younger individuals. However, despite the increasing usage of implantable medical devices in aged patients, few studies examining the effects of aging upon the host response to biomaterials and the implications of this response for long-term integration and function have been performed. The objective of the present manuscript is to review the putative effects of aging upon the host response to implanted materials and to advance the hypothesis that age-related changes in the local microenvrionement, with emphasis on the extracellular matrix, play a previously unrecognized role in determining the host response to implants.
Keywords: Macrophage, Polarization, Host response, Aging, Implant
1. Introduction
The host immune response to implantable materials has been well studied over the last five decades. The inflammatory response and foreign body reaction are now well recognized predictors of the downstream success or failure of implants. However, regardless of the downstream outcome of tissue remodeling following the placement of an implant, the early stages of the host immune response are largely the same. That is, following placement all materials – regardless of design or composition – are subject to some degree of protein adsorption, an early neutrophil response, and subsequent invasion of the site of implantation by mononuclear phagocytes. While this represents an oversimplification of a complex process (which has been reviewed at length elsewhere [1–3]), the view that these events – eventually leading to an inflammatory response at the implant surface – represent a negative occurrence has led to a number of approaches to evade the early host immune response including, tuning of surface topography [4–8], porosity [9–14], and chemistry [15–18] of the material, as well as the use of non-fouling surfaces and coatings [19–21] and decorating surfaces with matricellular proteins to prevent non-self recognition [22–24]. While these techniques have undoubtedly shown promise, prevention of long-term interaction of protein or cells with implants has yet to be demonstrated.
Recently, many studies have begun to suggest that the interaction of host immune cells with implanted materials may be an integral and beneficial component of the response which leads to a positive remodeling outcome (see [25,26] for review). Such results have led to renewed interest in the interaction of host inflammatory cells with implants. This is particularly true with regard to macrophages as they have long been considered to be master regulators of the host immune response to implants. Just as macrophages have now been shown to be capable of affecting both beneficial and detrimental outcomes in the processes of wound healing following injury, the response to pathogen, and cancer (all of which are reviewed in [27,28]), it is now well recognized that macrophage populations which interact with implants also have heterogeneous phenotypes and functions which are predictive of downstream outcomes.
In the biomaterials literature, macrophages are most commonly segregated into “M1” pro-inflammatory and “M2” anti-inflammatory/ regulatory subsets as shown in Fig. 1. In reality, macrophage phenotype can hardly be segregated into two distinct phenotypes [29]. Rather, macrophages display a wide variety of context and tissue-dependent phenotypic characteristics, which may include aspects of both M1 and M2 phenotypes. Therefore, we will discuss the host response to implants using these terms as they represent a simplified framework for discussion of the host macrophage response and as they are used ubiquitously throughout the biomaterials literature.
Fig. 1.
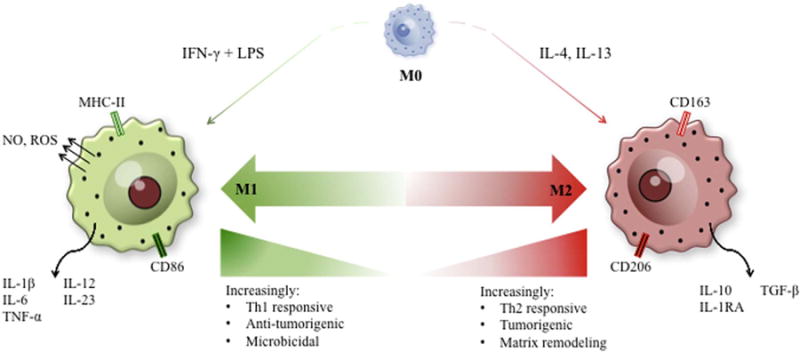
Macrophage polarization exists as a spectrum of phenotypic and functional states. Macrophage polarization states can best be described as a spectrum ranging from antimicrobial, pro-inflammatory M1 macrophages to immunotolerant, anti-inflammatory M2 macrophages. While this review simplifies macrophage polarization states to a basic M1/M2 paradigm for ease of discussion, it is important to note that these extreme polarization states are rare in vivo and macrophage polarization relies on the integration of cytokine signaling in the local microenvironment to polarize macrophages to some intermediate state along the polarization spectrum.
Briefly, studies have now demonstrated that an early shift from an “M1” pro-inflammatory macrophage phenotype to a more “M2-like” anti-inflammatory phenotype is associated with improved implant integration and/or tissue regeneration, depending upon the type of implant and it’s intended purpose (i.e. permanent implants versus regenerative medicine-based approaches to tissue reconstruction) [13,30–36]. As a result, multiple strategies have now been devised to promote shifts in macrophage phenotype at the host-tissue interface.
These include novel surface topographies, coatings, and drug delivery strategies among an increasing number of other strategies (which have been recently reviewed elsewhere [37–39]). In general, however, all of these strategies are targeted at shifting the acute phases of the host macrophage response, demonstrating the critical nature of the earliest events in the innate immune response and suggesting that strategies which seek to exploit rather than avoid the innate immune response will meet with increased success.
With the renewed interest in macrophage polarization within sites of implantation has come an increased understanding of the importance of the tissue and disease specific context into which the implant is placed. Briefly, each tissue may possess a distinct subset of resident macrophages and respond to injury or inflammatory insult differently [40,41]. For example, though macrophages are described as having a key role in outcomes in each case, the progression and make-up of the post-injury macrophage response in cutaneous, muscular, and central nervous tissues is distinct (see [26] for a discussion). Similarly, differences in the progression and intensity of the host response can be observed when the same material is placed in two different locations. For example, polyether-polyurethane implants elicited different chemokine profiles when implanted in intraperitoneal versus subcutaneous spaces in mice [42]; and polypropylene implants were observed to elicit a stronger inflammatory reaction when implanted into the vagina compared to the abdomen in rabbits [43,44]. Similarly, intraperitoneal implants in diabetic rats were associated with increases in inflammatory factors as compared to non-diabetic animals [45]; and increased fibrosis was observed following implantation of materials in lupus-prone mouse models [46]. Recent studies have also shown changes in the host response and downstream remodeling of implants based upon NSAID usage, demonstrating the potential for effects of post-surgical care regimes upon the host response [47]. These studies and others have begun to suggest that both tissue location and disease context is, at least in part, responsible for determining the course and outcome of the inflammatory reaction following the implantation of materials. While it is entirely logical to assume that tissue location and disease context would have important impacts upon the host response to implants, evaluations of the host response to are most commonly carried out in a small number of tissue locations in young, healthy animals.
In order to design “next-generation” implants or modification strategies to improve the performance of currently available materials in a wide array of applications, it is necessary to develop a context-dependent understanding of the host response which will be encountered upon implantation. One such context is aging. Advances in modern medical practice and increased access to medical care have significantly extended lifespan; however, increases in lifespan have not been accompanied by similar increases in healthspan. By 2050, the number of individuals over the age of 65 is expected to increase by 71% to nearly 2 billion individuals worldwide, with the number of US citizens over the age of 65 surpassing 20% by 2030 [48]. Over 75% of the elderly population has at least one chronic condition, often presenting with systemic inflammation [49]. As a result there is an increasing demand for implantable medical devices intended to treat age-related disorders. Examples of such devices include pacemakers, glucose sensors, and total joint implants, among others. Despite the increasing usage of implantable medical devices in aged patients, very few studies have been performed to assess the impacts of aging upon the host response and subsequent remodeling of implanted materials.
The objective of the present manuscript is to review the putative effects of aging upon the host response to implanted materials and to advance the hypothesis that age-related changes in the local micro-envrionement, with emphasis on the extracellular matrix, play a previously unrecognized role in determining the host response to implants. These effects will be examined from a cellular as well as microenvronmental perspective, and the limited evidence demonstrating differences in the host response to materials with age will be presented.
2. Cellular aging of the host immune system
The design of improved biomaterials for context specific applications requires an understanding of the host immune system in various states and pathologies, including aging. Aging has been shown to influence the functions of both innate and adaptive immune cells in scenarios such as wound healing, pathogen response, and vaccination. In this section of the review, we discuss changes in both the adaptive and innate immune response that may have implications for the host response to implanted biomaterials.
2.1. Immunosenescence
Immunosenescence is defined as the state of dysregulated immune function occurring with advanced age, which has been associated with an increased susceptibility of elderly populations to infection, autoimmune disease, and cancer related mortalities [50,51]. While the root word ‘senescence’ is used to classify cells undergoing irreversible cell cycle arrest, immunosenescence does not equate to the replicative senescence observed in fibroblasts, also known as the Hayflick limit [52]. Immunosenescence manifests in both the innate and adaptive immune system as altered circulating cell numbers, delayed migration to sites of infection or injury, and disrupted effector responses to stimulating cytokines [51,53]. Furthermore, immunosenescence is accompanied by elevated systemic levels of inflammatory cytokines and reactive oxygen species that can wreak havoc on tissue homeostasis, and has thus been termed “inflammaging” [54,55]. Inflammatory cytokine production may arise from dysregulated immune cells or other epithelial cells that have undergone senescence and acquire a Senescence-Associated Secretory Phenotype (SASP) [56]. Whether inflammaging is a direct cause, effect, or cyclical process surrounding immunosenescence is unclear; however, it is well accepted that chronic inflammatory signals have been linked to numerous age-related disorders including cardiovascular disease, cancer, obesity, metabolic syndrome, osteoporosis, and dementia [57]. Therefore, it follows that immunosenescence and inflammaging in the elderly population will lead to dramatic shifts in the host repose to implanted biomaterials and tissue regeneration, although published studies testing these hypotheses are limited, especially regarding the effect of immune aging upon this response.
2.2. Innate and adaptive immune aging
2.2.1. Adaptive immune aging
The adaptive immune response provides acquired antigen-specific immunity, leading to highly effective cell destruction, immune regulation and memory. The effector cells of the adaptive immune system, T-cells and B-cells, are derived from lymphoid progenitor cells in the bone marrow and mature in secondary lymphoid tissues. Generation of the adaptive immune system is robust in young individuals, as is necessary to establish a diverse immune repertoire. However, lymphopoeisis is significantly diminished in elderly individuals, in part due to involution of the thymus and a skewing of hematopoietic progenitor cells towards the myeloid lineage [58–60]. As a result, the number of T-cell and B-cell progenitors in the bone marrow and thymus are markedly reduced with age [61–64].
Advanced age is associated with low numbers of naïve T-cells, persistent memory T-cells, and reduced diversity of the T-cell receptor (TCR) repertoire, with a large proportion of TCRs directed towards persistent cytomegalovirus (CMV) infection [65–68]. A reduction in naïve T-cells has been attributed to involution of the thymus, which begins as early as puberty in mammals [58,62]. Activation of T-cells is also impaired with aging. T-cell activation occurs following interaction with antigen-presenting cells (APCs) forming an immune “synapse.” Several defects in the synapse have been reported, including glycosylation of costimulatory molecules, such as CD28, or changes in lipid membrane properties [69,70]. Perhaps the single most important consequence of synapse dysfunction is reduced production of interleukin-2 (IL-2), as exogenous IL-2 can rescue many of the age-related deficits of T-cell activation [71,72]. Age-related changes of other T-cell produced cytokines, such as interleukin-1(IL-1), interleukin-10 (IL-10) and interferons, vary across literature and are likely context specific.
Similarly, B-cells undergo important changes with aging. B-cell precursors do not proliferate as extensively in elderly mice as they do in young mice, and they undergo higher rates of apoptosis[73]. Changes in the bone marrow, including decreased secretion of interleukin-7 (IL-7) by stromal cells, have been implicated in B-cell development in aged individuals [74,75]. Both human and murine B-cells exhibit impaired class switching, limiting the specific phenotypes of antibody producing B-cells crucial for responding to invading pathogens [76].
The deficits in lymphocyte cell number and function play a large role in the deficits of the elderly population to respond to vaccination and to clear both bacterial and viral infections, especially following other stressors such as injury, surgery, and biomaterial implantation including artificial joints, stents, and pace-makers. The co-morbidity of surgical procedures and biomaterial implantation with infection warrants further investigation into the adaptive immune system of aging population. Furthermore, it has been recently shown that lack of adaptive cells, specifically T helper 2 (Th2) subsets removes the regenerative response seen during normal biomaterial remodeling [77]. Reduction in numbers of lymphocytes, including Th2 cells, could lead to reduced regulation of the regenerative response to biomaterials by these cells.
2.2.2. Innate immune aging
The innate immune system provides a rapid response to infection or injury initiated by pathogen associated molecular signals (PAMPs) and damage associated molecular signals (DAMPs). Innate immune cells include granulocytes (neutrophils, eosinophils, basophils, mast cells), innate lymphoid cells (natural killer cells), and antigen presenting cells (dendritic cells, macrophages) [78,79]. Each of these innate immune cells provides a tightly regulated, specific function that orchestrates tissue wound repair and response to foreign bodies with the ultimate goal of restoring tissue homeostasis. In the elderly population, immunosenescence of innate immune cells and inflammatory tissue environments effects innate immune processes.
Neutrophils are the first immune cell responders in the foreign body reaction. While the number and phagocytic capacity of neutrophils is reported to be maintained in the elderly, delays in migration and reduction of respiratory burst have been reported [80]. A renewed interest in natural killer (NK) cells and other classes of innate lymphoid cells has emerged in recent years. While innate lymphoid cells are not heavily implicated in the foreign body response, much remains to be characterized. With aging, NK cells have reported diminished target binding due to changes in cell signaling and impaired activation, leading to changes in effector function, such as release of granzyme, perforin, and other cytokines [81]. Dendritic cells are potent antigen presenting cells, known to be particularly important for T-cell activation. DCs become activated after contacting biomaterials, increase expression of co-stimulatory molecules (CD80/CD86), and promote T-cell proliferation and Th1 cytokine production [82,83]. Growing evidence suggests that DC activation is impaired in aging, along with impaired capacity to cross tissue barriers, and to induce cytokine production from T-cells, likely changing the response of DCs to implanted biomaterials [84,85].
Monocytes and macrophages are vitally important cells in the foreign body response, where macrophage polarization can be used as a determinant of the host response outcome (ref). Although the output of immune cells from the bone marrow is weighted towards myeloid lineage with aging, the absolute monocyte number is unchanged between young and aged individuals [86,87]. However, an increased percentage of non-classical monocytes (CD14+ CD16++), and a reduction in classical monocytes (CD14+ CD16−) has been reported in elderly individuals [86,87]. Like other innate immune cells, macrophage recruitment is delayed in aging, possibly due to a reduction in expression of the integrin very late antigen-4 (VLA-4) on monocytes, and vascular cell adhesion molecule-1 (VCAM-1) on endothelial cells [88]. Furthermore, a reduction in major histocompatibility complex (MHC-II) expression has been observed in both human and murine macrophages, leading to a reduction in antigen-presentation capacity and cross-talk with the adaptive immune system [89,90]. An increase in prostaglandin E2 production by macrophages has been postulated as a possible explanation for the reduction in MHC-II expression [91]. Toll-like receptors (TLRs) including TLR-1, TLR-2, and TLR-4, important mediators for PAMPs and DAMPs, are also reported to decrease on aged peritoneal and splenic macrophages, perhaps contributing to the susceptibility of the elderly to bacterial, mycotic, and viral infections [92,93].
Classically activated “M1” macrophages are very efficient at producing pro-inflammatory cytokines, nitric oxide, and performing phagocytosis. With aging, phagocytosis has been reported to decrease, perhaps associated with a reduction in the cellular receptors or responsiveness to cytokines [86,94,95]. Furthermore, macrophage senescence and quiescence induced in vitro has been reported to lead to a reduction in phagocytosis in response to biomaterials [96]. In addition, decreases in nitric oxide and superoxide production by aged macrophages have been reported in rats[97]. Alternatively activated “M2” macrophages are crucial for clearing helminth infections, and have important roles in the resolution of inflammatory reactions, including new matrix deposition and anti-inflammatory cytokine production. Changes in “M2” macrophage function are less well documented in the literature. However, “M2” macrophages have been found to accumulate in aged skeletal muscle and result in the detrimental replacement of muscle fibers with fibrotic tissue over time [98]. Advanced age may also impair the ability of macrophages to polarize to either M1 or M2 phenotypes. The gene signature of macrophages isolated from bone marrow and polarized to M1 or M2 phenotypes was mostly unchanged between young and aged mice, with the exception of increased Fizz-1 (M2 marker) expression on aged cells [99]. However, macrophages isolated from mouse spleen had a reduction in both “M1” and “M2” gene expression patterns when stimulated with polarizing cytokines, suggesting deficits in polarization capacity [99].
While less well documented than the adaptive immune system, changes in innate immune function have a significant impact on the host response to infection, biomaterials and the normal wound healing process. Further studies that systemically examine the effect of aging on the innate immune response to biomaterials are necessary to further the development of biocompatible and bioactive materials.
2.3. Bone marrow elicited versus tissue-resident macrophages in aging and the host response
In recent years, there has been a conceptual revolution in the understanding of macrophage development and contribution to disease pathology. A dichotomy in the origin and function of macrophages has been realized by lineage tracing studies demonstrating that tissue-resident macrophages arise from the yolk sac and fetal liver during embryogenesis, and can be maintained into adulthood independently of the bone marrow, contrary to the previously accepted view of the mononuclear phagocyte system [100–102]. Tissue-resident macrophages are present in virtually every tissue with unique gene signature profiles, including in the liver (Kupffer cells), brain (microglia), lungs (alveolar macrophages), bone (osteoclasts), skin (Langerhans's cells), Peyer's patches in the gut, red and white pulp in the spleen, the peritoneal cavity, and the interstitium of organs such as kidney, heart, and pancreas. Macrophages residing in certain organs, such as the colon, spleen, peritoneum, and heart, are replaced regularly by bone marrow derived monocytes, whereas the brain receives virtually no input from monocytes under homeostasis [102,103]. Tissue imprinting has been linked to changes in the enhancer regions of the genome of these macrophages, imparting tissue specific functions [104]. Despite the prenatal origin of these tissue-resident macrophages and the effects of tissue imprinting, monocyte derived macrophages have the capacity to take on a seemingly identical phenotype to Kupffer cells, including self-renewal, when the niche is made available in the liver [103].
The macrophage lineage tracing experiments reveal a large gap of knowledge in the field of regenerative medicine and biomaterials, as it is currently unknown whether tissue-derived or bone marrow elicited macrophages are the primary responders in the host response. Given the tissue-specific functions and “M2” polarization of tissue-resident macrophages versus the “M1” profile of elicited macrophages, it is of interest to determine which populations are contributing to the host response to improve immunomodulation in site and age-specific contexts.
Indeed, several studies have identified that tissue-resident macrophages, but not bone marrow derived macrophages, become impaired with aging [95,99]. Recent work from our group also demonstrates minimal differences between young and aged bone marrow derived macrophage functions, including nitric oxide production and phagocytosis [105,106]. Interestingly, when bone marrow derived macrophages are pre-exposed to degradation products from young and aged extracellular matrix or “tissue cues”, differences in function arise following polarization with M1-inducing cytokines [107]. Taken together, these studies suggest that tissue microenvironments dictate a large portion of the observed immunosenescence of macrophages.
3. Tissue microenvironmental changes with aging
In addition to cellular changes with age that effect the host response to natural biomaterials, changes in the local tissue microenvironment can also play a large role in the resultant response upon implantation. Apart from age-related dysfunction and changes in the immune response, there are also observed changes in cells accessory to the immune response, extracellular matrix, and the concentration of many different molecules that effect wound healing. In this section of the review, we highlight how changes in the local implantation micro-environment could affect the innate immune response to implanted biomaterials.
3.1. Extracellular matrix composition and mechanics change with age
The extracellular matrix (ECM) comprises the protein, glycosaminoglycans, proteoglycans and other molecules secreted by tissue resident cells that provides structure and signaling cues to the cells of that tissue in a reciprocally dynamic relationship [108]. The ECM acts as a local microenvironment which influences tissue homeostasis as well as response to injury [109]. The ECM is specific to a given tissue and comprises a unique composition of structural and associated molecules which support the function of the resident cells [110]. The extracellular matrix can influence cellular phenotypes and function through the interaction of an assortment of binding proteins including integrins, discoidin domain receptors, syndecans and many others [110]. Therefore the use of decellularized tissues, also known as ECM scaffolds, have been studied as natural biomaterials for tissue repair [108]. ECM scaffolds initiate the foreign body response and have been shown to promote “M2” macrophage polarization, in part dependent on Th2 cytokine response, leading to constructive tissue remodeling (see [111] for an in-depth review of the host response to ECM scaffolds). However, it is unclear how aging of both the local microenvironment and the source age of natural biomaterials influences the outcome of the foreign body response.
Few studies tying compositional changes in aging tissue to age related changes in the host response have been performed. However, Tottey, et al. demonstrated that source animal age was linked to changes in both the compositional make up and physical properties of decellularized tissue scaffolds composed of small intestinal submucosa (SIS) [112]. Briefly, small intestinal submucosa was harvested from 3, 12, 26, and 52 week old pigs and decellularized using a peracetic acid washing procedure. The resultant tissue was then tested mechanically and subjected to compositional testing for growth factors, collagen, and glycosaminoglycan content. The results showed a trend toward increasing tissue thickness, reduced tensile modulus, and resistance to collagenase degradation with aging. Compositional analysis showed changes in bFGF and VEGF content with age including increases in both growth factors between 3 and 26 weeks with decreased content thereafter. Analysis of glycosaminoglycan content showed a steady drop in content with aging from the 3–52 week old source animals. These changes were associated with changes in the mitogenesis, metabolism and migration of perivascular progenitor cells exposed to the materials.
A follow-up study utilized the same decellularized small intestinal extracellular matrix as an implant in a well described model of partial thickness abdominal wall defect repair [113]. The results of this study demonstrated that scaffolds derived from younger animals (3 and 12 weeks) were associated with significantly improved remodeling (i.e. formation of innervated skeletal muscle bundles) of the abdominal skeletal muscle as compared to 26 or 52-week old source tissues and untreated and autograft treated controls. Investigation of the early host response revealed that the materials derived from younger animals (3 and 12 weeks) were also associated with a decreased ratio of M1:M2 macrophages within the site of implantation as compared to 26 or 52-week old source tissues and untreated and autograft treated controls.
3.2. Extracellular matrix modifications
It is unclear whether the compositional changes in the implanted scaffolds described above were responsible for the observed changes in the host response. While it is hardly possible to evaluate the role of the hundreds of individual components within each tissue, a number of modifications of extracellular matrix molecules have been observed to occur with aging. Here we describe advanced glycation end products (AGEs) as one potential modification of ECM molecules which occurs with age; however, numerous other modifications resulting from changes in SPARC, LOX, and the effects of reactive oxygen species have been described with aging, but are not described here due to space limitations.
Advanced glycation end products, a heterogeneous group of over 20 different molecules, are formed through the non-enzymatic glycation of long-lived proteins, lipids, or nucleic acids [114,115]. AGE formation in vivo tends to be a progressive process that occurs with increasing age or following certain lifestyle choices, such as diets high in glucose or exposure to environmental toxins[114,116]. AGEs are capable of protein cross-linking, resulting in the reduced ability of cellular machinery to degrade damaged proteins, in turn leading to tissue stiffening and dysfunction [117]. AGEs can also promote formation and accumulation of reactive oxidative species (ROS) through the blockade of the 20S subunit of the intracellular proteasome[116,118]. Since the 20S subunit is typically responsible for the degradation of oxidized intracellular proteins, blockade of this intracellular machinery by AGEs, coupled with reduced antioxidant to oxidative species ratios in aged individuals, allows for a feed-forward loop of oxidative species formation, subsequently resulting in enhanced AGE formation in vivo [114,118,119].
Additionally, AGEs have been shown to promote inflammatory processes through several distinct signaling pathways, with the receptor for advanced glycation end products (RAGE) pathway being one the best characterized [114]. RAGE is a pattern recognition receptor of the immunoglobulin receptor family and is widely expressed across a variety of cell types, including macrophages [120], T-lymphocytes [121], B-lymphocytes [122], dendritic cells [123], endothelial cells [124], fibroblasts [125], smooth muscle cells [120], and neuronal cell [114,126,127]. Additionally, RAGE is able to bind a variety of ligands in addition to AGEs, such as lipopolysaccharide, calgranulin/S100 family ligands, or high mobility group box protein 1 [114,122]. Since RAGE signaling is dependent on both cell and ligand type, RAGE signaling tends to be complex and studied in a context dependent manner [122]. Due to space limitations, this review will focus only on monocyte/macrophage RAGE signaling. However, it is important to note that this is a limited view of the microenvironmental response to AGE accumulation and RAGE signaling in other cell types in important for the tissue response to AGE accumulation.
In macrophage populations, ligand binding to RAGE has been shown to induce M1, pro-inflammatory macrophage states through the upregulation of MHC-II expression on the cell surface and secretion of pro-inflammatory cytokines [128,129]. While it has not been determined definitively how ligand binding to RAGE impacts M2, pro-remodeling states in macrophages, some data exists that suggests RAGE-ligand binding promotes the down-regulation of M2 genes [130]. However, a recent study has shown that AGE-mediated RAGE activation has no impact on M2 marker expression[131]. Thus, further studies will need to be performed to fully elucidate how RAGE signaling can impact M2 gene and surface marker expression. Additionally, while the effects of RAGE-ligand binding have been fairly well elucidated in macrophage populations, the specific signaling pathways utilized in these cell populations have not. RAGE signaling pathways in monocyte and macrophage populations has been shown to be dependent on both the RAGE-bound ligand as well as the extracellular microenvironment [122]. For example, monocyte RAGE stimulation with AGEs resulted in signaling through PKC, ERK1/2, p38–but not JNK – to activate master inflammatory regulator gene NF-κB [132]. However, under hypoxic conditions Chang et al. demonstrated that monocyte RAGE stimulation did not signal through this pathway but rather led to the translocation of PKCβII and subsequent JNK activation, resulting in downstream Egr-1 activation [133]. Additionally, macrophages have been shown to signal through p38, p44/42, JNK, and MAPK pathways [128]. Thus, one can see the importance of ligand and extracellular environment in determining the RAGE signaling propagation pathway.
3.3. Tissue stiffness
Traditionally, modulation of cellular phenotype and functionality were thought to be predominantly mediated by secreted biochemical signals that can be sequestered by the extracellular matrix. However, recent studies have identified that the mechanical properties of extracellular matrix can also dictate cellular behaviors during development, homeostasis, and during disease pathogenesis. While mechanosensitive cell migration and proliferation serve important roles during development and tissue homeostasis, these processes can have deleterious consequences following the dysregulation of extracellular matrix stiffness during aging or tissue injury[134–136]. While extracellular matrix stiffening has been fairly well characterized in a number of pathologies, a cohesive mechanism of age-related matrix stiffening in the absence of any co-morbidity has yet to be fully elucidated. As previously discussed, accumulation of cross-linking advanced glycation end products in aging tissues is the best characterized mechanism of age-related extracellular matrix stiffening. However, several other mechanisms of ECM stiffening have been characterized within distinct tissue sites. One such mechanism is the calcification of extracellular matrix proteins with relatively low turnover rates [134]. This process has best been elucidated in the context of age-related arterial wall stiffening. It has been shown that calcium levels in the arterial wall rise with increasing age, which in turn allows for the calcification of low turnover proteins such as elastin [137–139]. Elastin calcification can subsequently cause fragmentation of elastin fibers, which in turn results in arterial stiffening as elastic fibers are lost and arterial wall stiffness relies increasingly on collagen content within the matrix, which has an elastic modulus substantially greater than that of elastin [134,140,141]. Another modulator of ECM stiffness is age-related alterations in matrix metalloprotease (MMP) and tissue inhibitor of metalloproteases (TIMP) expression [135,142]. In a study conducted by Bonnema et. al., the authors found elevated plasma levels of MMP-2, MMP-7, TIMP-1, TIMP-2, and TIMP-4 in aged individuals with no evidence of cardiovascular disease [135]. Upon further examination of the MMP/TIMP ratios in the subjects, the authors found the MMP/TIMP ratios favored extracellular matrix deposition in aged individuals [135]. The authors also found a statistically significant negative correlation between MMP-7 and TIMP-1,-4 expression levels and left ventricular volume to mass ratio and mitral E/A ratios, both of which are predictors of cardiovascular dysfunction [135]. This observed age-related alteration in plasma MMP and TIMP levels in the absence of other co-morbidity is interesting as MMP expression levels in pathological microenvironments have been shown to play roles in the progression of many diseases − including cardiovascular disease [134,142], cancer [143–145], neural disorders [146], and arthritis [147]. Future studies should seek to better elucidate the relationship between this age-related change in MMP and TIMP expression and disease pathogenesis. It must be noted that while each mechanism of extracellular matrix stiffness modulation was considered as an isolated phenomenon, these processes are not isolated in aging individuals and instead function in concert, often in the presence of additional co-morbidities, to promote tissue specific alterations in ECM elastic modulus that can in turn promote cellular dysfunction and disease progression.
As is noted above, the effects of substrate stiffness upon mechanotransduction in many cell types across multiple biologic processes is well described, mechanotransduction has only recently been investigated in macrophage populations. The evidence that exists suggest that macrophages are responsive to substrate, stress and strain, and cell shape; however, a general consensus as to what the specific effects of each of these factors upon the inflammatory response has yet to be defined (see [148,149] for recent reviews on this topic). Thus, while it is likely that soluble factors within the local microenvironment are the predominant cues which drive polarization, crosslinking and structural modification of ECM leading to dysregulation of the mechanical environment may play a role in macrophage mediated pathogenesis. Similarly, such changes in the local environment would also be expected to have an effect upon the tissue-implant interface and the host inflammatory response.
4. Assessing the host response to implants in aged animal models
There are few, if any reports in the literature which have investigated the host response to implants in aged animals. However, a recent study from our group sought to elucidate the impacts of aging upon the host response to polypropylene mesh implanted into 8-week (young) and 18-month-old mice (aged) [105]. The findings of the study suggested that the host macrophage response to polypropylene mesh implants was delayed, dysregulated, and unresolved in the aged animals as compared to the young. Briefly, there was decreased recruitment of macrophages to the site of implantation at 3 and 7 days post-implantation. Further, the population which was present in the aged animals was found to be expressing significantly more iNOS and less Arginase-1 than was observed in young animals, indicating a more M1 skewed response. Analysis of tissue remodeling outcomes at 90 days demonstrated similar composition and thickness of the tissue capsule surrounding the implants; however, 18 month old animals were observed to have a significantly higher number of macrophages within the tissue capsule, suggesting an unresolved inflammatory response. These results, as displayed in Fig. 2, correlate well with the findings of dysregulated, and unresolved host responses in studies of the response pathogen and in wound healing in aged individuals.
Fig. 2.
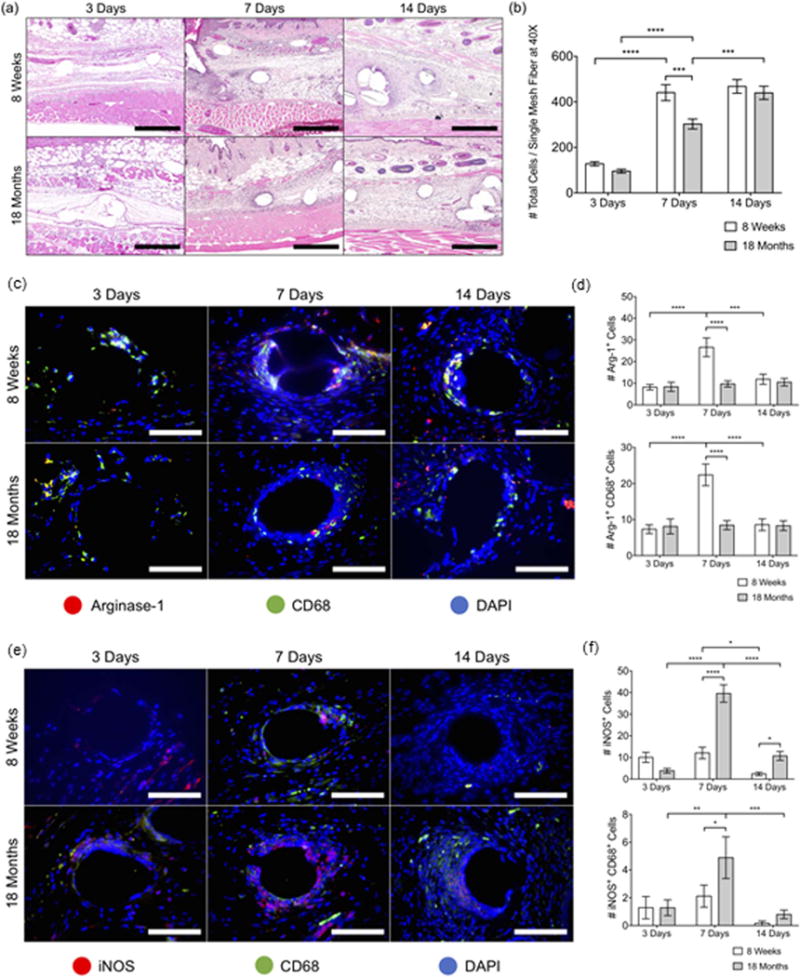
The early host response to implanted materials in young and aged C57BL/6 mice. (a) Images of H & E-stained tissue cross-sections at 10× and (b) total cell counts (DAPI) surrounding single mesh fibers in 40× images at 3, 7, and 14 days. Scale bars represent 200 mm. Fluorescence microscopy images of (c) Arginase-1 (red) CD68 (green) co-immunolabeling and (e) iNOS (red) CD68 (green) coimmunolabeling at a single mesh fiber at 3, 7, and 14 days. DAPI was used to stain cell nuclei. Scale bars represent 50 mm. Cell count image analysis of (d) Arg- 1+, Arg-1+ CD68+ cells and (f) iNOS+, iNOS+ CD68+ cells at 3, 7, and 14 days. Bars represent the mean ± SEM. Statistical significance as (*) p < 0.05, (**) p < 0.01, (***) p < 0.001, (****) p < 0.0001. All other differences are nonsignificant. N = 7.
This study also examined the M1 and M2 polarization profile of bone marrow derived macrophages harvested from the young and aged mice. Few differences between M1 and M2 polarized macrophages were observed, suggesting that the ability to effectively polarize is maintained in aged rodents. Thus, the results of the study showed that though there were significant differences in the host response between young and aged animals, these differences were likely not accounted for by age-related cell-intrinsic defects, and that the local tissue environment likely played a role. While the role of changes in the local tissue microenvironment were not investigated in depth in the study, a reduction in the production of glycosaminoglycan constituents, including hyaluronan, at early time points in the host response was observed and could have led to a reduction in the recruitment of early responding cells and/or changes in their polarization profile.
5. Conclusions
While few studies to-date have investigated the host response to biomaterials in aged animals, those which have been performed demonstrate that the response is delayed, dysregulated, and chronically activated. The exact mechanisms which underlie these findings are not yet clear; however, herein, we present evidence that these observations may be a result of dysregulation of both the innate immune system and the local tissue microenvironment into which the implant is placed. Additional studies will be necessary to design effective materials, implants, and tissue reconstruction strategies to meet the needs of an increasingly aged population.
Acknowledgments
This work was supported by National Institutes of Health awardsK12HD043441 (BNB), R03AG043606 (BNB), R01GM121558 (BNB) and T32EB001026 (STL, MJH, and ECS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- AGE
advanced glycation end-products
- APCs
anatigen presenting cells
- CMV
cytomegalovirus
- DAMPs
damage associated molecular signals
- ECM
extracellular matrix
- ERK
extracellular-regulated signal kinase
- EGR
early growth response protein
- IL-1
interleukin-1
- IL-2
interleukin-2
- IL-7
interleukin-7
- IL-10
interleukin-10
- JNK
c-Jun N-terminal kinase
- LOX
lysyl oxidase
- MHC
major histocompatibility complex
- MAPK
mitogen-activated protein kinase
- MMP
matrix metalloprotease
- NK
natural killer
- PAMPs
pathogen associated molecular signals
- PKC
protein kinase C
- RAGE
receptor for advanced glycation end products
- ROS
reactive oxidative species
- SPARC
secreted protein acidic and cysteine rich
- SASP
senescence associated secretory phenotype
- SIS
small intestinal submucosa
- TCR
T-cell receptor
- TIMP
tissue inhibitor of metalloproteases
- TLRs
toll-like receptors
- VCAM-1
vascular cell adhesion molecule-1
- VLA-4
very late antigen-4
References
- 1.Anderson JM. Inflammatory response to implants. ASAIO Trans. 1988;34(2):101–107. doi: 10.1097/00002480-198804000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Anderson JM, Rodriguez A, Chang DT. Foreign body reaction to biomaterials. Semin Immunol. 2008;20(2):86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klopfleisch R, Jung F. The pathology of the foreign body reaction against biomaterials. J Biomed Mater Res A. 2016 doi: 10.1002/jbm.a.35958. [DOI] [PubMed] [Google Scholar]
- 4.Rice JM, et al. Quantitative assessment of the response of primary derived human osteoblasts and macrophages to a range of nanotopography surfaces in a single culture model in vitro. Biomaterials. 2003;24(26):4799–4818. doi: 10.1016/s0142-9612(03)00381-8. [DOI] [PubMed] [Google Scholar]
- 5.Paul NE, et al. Topographical control of human macrophages by a regularly microstructured polyvinylidene fluoride surface. Biomaterials. 2008;29(30):4056–4064. doi: 10.1016/j.biomaterials.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Bota PC, et al. Biomaterial topography alters healing in vivo and monocyte/ macrophage activation in vitro. J Biomed Mater Res A. 2010;95(2):649–657. doi: 10.1002/jbm.a.32893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collie AM, et al. Differential monocyte/macrophage interleukin-1beta production due to biomaterial topography requires the beta2 integrin signaling pathway. J Biomed Mater Res A. 2011;96(1):162–169. doi: 10.1002/jbm.a.32963. [DOI] [PubMed] [Google Scholar]
- 8.Hotchkiss KM, et al. Titanium surface characteristics, including topography and wettability, alter macrophage activation. Acta Biomater. 2016;31:425–434. doi: 10.1016/j.actbio.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brauker JH, et al. Neovascularization of synthetic membranes directed by membrane microarchitecture. J Biomed Mater Res. 1995;29(12):1517–1524. doi: 10.1002/jbm.820291208. [DOI] [PubMed] [Google Scholar]
- 10.Fleckman P, et al. Cutaneous and inflammatory response to long-term percutaneous implants of sphere-templated porous/solid poly(HEMA) and silicone in mice. J Biomed Mater Res A. 2012;100(5):1256–1268. doi: 10.1002/jbm.a.34012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukano Y, et al. Epidermal and dermal integration into sphere-templated porous poly(2-hydroxyethyl methacrylate) implants in mice. J Biomed Mater Res A. 2010;94(4):1172–1186. doi: 10.1002/jbm.a.32798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karp RD, et al. Tumorigenesis by Millipore filters in mice: histology and ultrastructure of tissue reactions as related to pore size. J Natl Cancer Inst. 1973;51(4):1275–1285. doi: 10.1093/jnci/51.4.1275. [DOI] [PubMed] [Google Scholar]
- 13.Sussman EM, et al. Porous implants modulate healing and induce shifts in local macrophage polarization in the foreign body reaction. Ann Biomed Eng. 2014;42(7):1508–1516. doi: 10.1007/s10439-013-0933-0. [DOI] [PubMed] [Google Scholar]
- 14.Underwood RA, et al. Quantifying the effect of pore size and surface treatment on epidermal incorporation into percutaneously implanted sphere-templated porous biomaterials in mice. J Biomed Mater Res A. 2011;98(4):499–508. doi: 10.1002/jbm.a.33125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blaszykowski C, Sheikh S, Thompson M. Surface chemistry to minimize fouling from blood-based fluids. Chem Soc Rev. 2012;41(17):5599–5612. doi: 10.1039/c2cs35170f. [DOI] [PubMed] [Google Scholar]
- 16.Brodbeck WG, et al. Influence of biomaterial surface chemistry on the apoptosis of adherent cells. J Biomed Mater Res. 2001;55(4):661–668. doi: 10.1002/1097-4636(20010615)55:4<661::aid-jbm1061>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 17.MacEwan MR, et al. Student Research Award in the Undergraduate Degree Candidate category, 30th Annual Meeting of the Society for Biomaterials, Memphis, Tennessee, April 27–30, 2005. Monocyte/lymphocyte interactions and the foreign body response: in vitro effects of biomaterial surface chemistry. J Biomed Mater Res A. 2005;74(3):285–93. doi: 10.1002/jbm.a.30316. [DOI] [PubMed] [Google Scholar]
- 18.Wilson CJ, et al. Mediation of biomaterial-cell interactions by adsorbed proteins: a review. Tissue Eng. 2005;11(1–2):1–18. doi: 10.1089/ten.2005.11.1. [DOI] [PubMed] [Google Scholar]
- 19.Kingshott P, Griesser HJ. Surfaces that resist bioadhesion. Curr Opin Solid State Mater Sci. 1999;4(4):403–412. [Google Scholar]
- 20.Zhang L, et al. Zwitterionic hydrogels implanted in mice resist the foreign-body reaction. Nat Biotechnol. 2013;31(6):553–556. doi: 10.1038/nbt.2580. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Z, et al. Blood compatibility of surfaces with superlow protein adsorption. Biomaterials. 2008;29(32):4285–4291. doi: 10.1016/j.biomaterials.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 22.Finley MJ, et al. Intracellular signaling mechanisms associated with CD47 modified surfaces. Biomaterials. 2013;34(34):8640–8649. doi: 10.1016/j.biomaterials.2013.07.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finley MJ, et al. Diminished adhesion and activation of platelets and neutrophils with CD47 functionalized blood contacting surfaces. Biomaterials. 2012;33(24):5803–5811. doi: 10.1016/j.biomaterials.2012.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tengood JE, Levy RJ, Stachelek SJ. The use of CD47-modified biomaterials to mitigate the immune response. Exp Biol Med (Maywood) 2016;241(10):1033–1041. doi: 10.1177/1535370216647130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown BN, et al. Macrophage polarization: an opportunity for improved outcomes in biomaterials and regenerative medicine. Biomaterials. 2012;33(15):3792–3802. doi: 10.1016/j.biomaterials.2012.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown BN, Sicari BM, Badylak SF. Rethinking regenerative medicine: a macrophage-centered approach. Front Immunol. 2014;5:510. doi: 10.3389/fimmu.2014.00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mills CD. M1 and M2 macrophages oracles of health and disease. Crit Rev Immunol. 2012;32(6):463–488. doi: 10.1615/critrevimmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- 28.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murray PJ, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown BN, et al. Macrophage phenotype as a predictor of constructive remodeling following the implantation of biologically derived surgical mesh materials. Acta Biomater. 2012;8(3):978–987. doi: 10.1016/j.actbio.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo R, et al. Substrate modulus of 3D-printed scaffolds regulates the regenerative response in subcutaneous implants through the macrophage phenotype and Wnt signaling. Biomaterials. 2015;73:85–95. doi: 10.1016/j.biomaterials.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keeney M, et al. Mutant MCP-1 protein delivery from layer-by-layer coatings on orthopedic implants to modulate inflammatory response. Biomaterials. 2013;34(38):10287–10295. doi: 10.1016/j.biomaterials.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mokarram N, et al. Effect of modulating macrophage phenotype on peripheral nerve repair. Biomaterials. 2012;33(34):8793–8801. doi: 10.1016/j.biomaterials.2012.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spiller KL, et al. Sequential delivery of immunomodulatory cytokines to facilitate the M1-to-M2 transition of macrophages and enhance vascularization of bone scaffolds. Biomaterials. 2015;37:194–207. doi: 10.1016/j.biomaterials.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown BN, et al. Macrophage phenotype and remodeling outcomes in response to biologic scaffolds with and without a cellular component. Biomaterials. 2009;30(8):1482–1491. doi: 10.1016/j.biomaterials.2008.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hachim D, et al. Shifts in macrophage phenotype at the biomaterial interface via IL-4 eluting coatings are associated with improved implant integration. Biomaterials. 2017;112:95–107. doi: 10.1016/j.biomaterials.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alvarez MM, et al. Delivery strategies to control inflammatory response: modulating M1-M2 polarization in tissue engineering applications. J Control Release. 2016 doi: 10.1016/j.jconrel.2016.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garash R, et al. Drug delivery strategies to control macrophages for tissue repair and regeneration. Exp Biol Med (Maywood) 2016;241(10):1054–1063. doi: 10.1177/1535370216649444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vishwakarma A, et al. Engineering immunomodulatory biomaterials to tune the inflammatory response. Trends Biotechnol. 2016;34(6):470–482. doi: 10.1016/j.tibtech.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 40.Davies LC, et al. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okabe Y, Medzhitov R. Tissue biology perspective on macrophages. Nat Immunol. 2016;17(1):9–17. doi: 10.1038/ni.3320. [DOI] [PubMed] [Google Scholar]
- 42.Mendes JB, et al. Host response to sponge implants differs between subcutaneous and intraperitoneal sites in mice. J Biomed Mater Res B Appl Biomater. 2007;83(2):408–415. doi: 10.1002/jbm.b.30810. [DOI] [PubMed] [Google Scholar]
- 43.Pierce LM, et al. Long-term histologic response to synthetic and biologic graft materials implanted in the vagina and abdomen of a rabbit model. Am J Obstet Gynecol. 2009;200(5):546 e1–8. doi: 10.1016/j.ajog.2008.12.040. [DOI] [PubMed] [Google Scholar]
- 44.Pierce LM, et al. Biomechanical properties of synthetic and biologic graft materials following long-term implantation in the rabbit abdomen and vagina. Am J Obstet Gynecol. 2009;200(5):549 e1–8. doi: 10.1016/j.ajog.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 45.Oviedo-Socarras T, et al. Diabetes alters inflammation, angiogenesis, and fibrogenesis in intraperitoneal implants in rats. Microvasc Res. 2014;93:23–29. doi: 10.1016/j.mvr.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 46.Campos PP, et al. Alterations in the dynamics of inflammation: proliferation and apoptosis in subcutaneous implants of lupus-prone mice. Histol Histopathol. 2011;26(4):433–442. doi: 10.14670/HH-26.433. [DOI] [PubMed] [Google Scholar]
- 47.Dearth CL, et al. Inhibition of COX1/2 alters the host response and reduces ECM scaffold mediated constructive tissue remodeling in a rodent model of skeletal muscle injury. Acta Biomater. 2016;31:50–60. doi: 10.1016/j.actbio.2015.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ortman Jennifer M, Howard Hogan VAV. An Aging Nation: The Older Population in the United States. US Census Bureau. 2014 [Google Scholar]
- 49.(NCOA), N.C.o.A. Chronic Disease Management. 2015 [Google Scholar]
- 50.Sebastián C, Lloberas J, Celada A, et al. Molecular and cellular aspects of macrophage aging. In: Fulop T, editor. Handbook on Immunosenescence. Springer; Netherlands: 2009. pp. 919–945. [Google Scholar]
- 51.Stahl EC, Brown BN. Cell therapy strategies to combat immunosenescence. Organogenesis. 2015;11(4):159–172. doi: 10.1080/15476278.2015.1120046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 53.Aw D, Silva AB, Palmer DB. Immunosenescence: emerging challenges for an ageing population. Immunology. 2007;120(4):435–446. doi: 10.1111/j.1365-2567.2007.02555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 55.Franceschi C, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128(1):92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 56.Coppe JP, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu BP, Chung HY. Adaptive mechanisms to oxidative stress during aging. Mech Ageing Dev. 2006;127(5):436–443. doi: 10.1016/j.mad.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 58.Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol. 2004;5(2):133–139. doi: 10.1038/ni1033. [DOI] [PubMed] [Google Scholar]
- 59.Lynch HE, et al. Thymic involution and immune reconstitution. Trends Immunol. 2009;30(7):366–373. doi: 10.1016/j.it.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rossi DJ, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005;102(26):9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson KM, Owen K, Witte PL. Aging and developmental transitions in the B cell lineage. Int Immunol. 2002;14(11):1313–1323. doi: 10.1093/intimm/dxf092. [DOI] [PubMed] [Google Scholar]
- 62.Min H, Montecino-Rodriguez E, Dorshkind K. Reduction in the developmental potential of intrathymic T cell progenitors with age. J Immunol. 2004;173(1):245–250. doi: 10.4049/jimmunol.173.1.245. [DOI] [PubMed] [Google Scholar]
- 63.Montecino-Rodriguez E, Berent-Maoz B, Dorshkind K. Causes: consequences, and reversal of immune system aging. J Clin Invest. 2013;123(3):958–965. doi: 10.1172/JCI64096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller JP, Allman D. The decline in B lymphopoiesis in aged mice reflects loss of very early B-lineage precursors. J Immunol. 2003;171(5):2326–2330. doi: 10.4049/jimmunol.171.5.2326. [DOI] [PubMed] [Google Scholar]
- 65.Blackman MA, Woodland DL. The narrowing of the CD8 T cell repertoire in old age. Curr Opin Immunol. 2011;23(4):537–542. doi: 10.1016/j.coi.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cicin-Sain L, et al. Cytomegalovirus infection impairs immune responses and accentuates T-cell pool changes observed in mice with aging. PLoS Pathog. 2012;8(8):e1002849. doi: 10.1371/journal.ppat.1002849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olsson J, et al. Age-related change in peripheral blood T-lymphocyte subpopulations and cytomegalovirus infection in the very old: the Swedish longitudinal OCTO immune study. Mech Ageing Dev. 2000;121(1–3):187–201. doi: 10.1016/s0047-6374(00)00210-4. [DOI] [PubMed] [Google Scholar]
- 68.Wikby A, et al. Expansions of peripheral blood CD8 T-lymphocyte subpopulations and an association with cytomegalovirus seropositivity in the elderly: the Swedish NONA immune study. Exp Gerontol. 2002;37(2–3):445–453. doi: 10.1016/s0531-5565(01)00212-1. [DOI] [PubMed] [Google Scholar]
- 69.Weng NP, Akbar AN, Goronzy J. CD28(−) T cells: their role in the age-associated decline of immune function. Trends Immunol. 2009;30(7):306–312. doi: 10.1016/j.it.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nguyen DH, Espinoza JC, Taub DD. Cellular cholesterol enrichment impairs T cell activation and chemotaxis. Mech Ageing Dev. 2004;125(9):641–650. doi: 10.1016/j.mad.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 71.Haynes L, et al. Interleukin 2: but not other common gamma chain-binding cytokines, can reverse the defect in generation of CD4 effector T cells from naive T cells of aged mice. J Exp Med. 1999;190(7):1013–1024. doi: 10.1084/jem.190.7.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haynes L, Lefebvre JS. Age-related deficiencies in antigen-specific CD4 t cell responses: lessons from mouse models. Aging Dis. 2011;2(5):374–381. [PMC free article] [PubMed] [Google Scholar]
- 73.Min H, Montecino-Rodriguez E, Dorshkind K. Effects of aging on the common lymphoid progenitor to pro-B cell transition. J Immunol. 2006;176(2):1007–1012. doi: 10.4049/jimmunol.176.2.1007. [DOI] [PubMed] [Google Scholar]
- 74.Labrie JE, 3rd, et al. Bone marrow microenvironmental changes underlie reduced RAG-mediated recombination and B cell generation in aged mice. J Exp Med. 2004;200(4):411–423. doi: 10.1084/jem.20040845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stephan RP, Reilly CR, Witte PL. Impaired ability of bone marrow stromal cells to support B-lymphopoiesis with age. Blood. 1998;91(1):75–88. [PubMed] [Google Scholar]
- 76.Frasca D, et al. Aging down-regulates the transcription factor E2A: activation-induced cytidine deaminase, and Ig class switch in human B cells. J Immunol. 2008;180(8):5283–5290. doi: 10.4049/jimmunol.180.8.5283. [DOI] [PubMed] [Google Scholar]
- 77.Sadtler K, et al. Developing a pro-regenerative biomaterial scaffold microenvironment requires T helper 2 cells. Science. 2016;352(6283):366–370. doi: 10.1126/science.aad9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91(3):295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 79.Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. 2016;17(7):765–774. doi: 10.1038/ni.3489. [DOI] [PubMed] [Google Scholar]
- 80.Fulop T, et al. Signal transduction and functional changes in neutrophils with aging. Aging Cell. 2004;3(4):217–226. doi: 10.1111/j.1474-9728.2004.00110.x. [DOI] [PubMed] [Google Scholar]
- 81.Solana R, et al. Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol. 2012;24(5):331–341. doi: 10.1016/j.smim.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 82.Babensee JE. Interaction of dendritic cells with biomaterials. Semin Immunol. 2008;20(2):101–108. doi: 10.1016/j.smim.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 83.Frick JS, Grunebach F, Autenrieth IB. Immunomodulation by semi-mature dendritic cells: a novel role of Toll-like receptors and interleukin-6. Int J Med Microbiol. 2010;300(1):19–24. doi: 10.1016/j.ijmm.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 84.Rhee I, et al. Control of dendritic cell migration: T cell-dependent immunity, and autoimmunity by protein tyrosine phosphatase PTPN12 expressed in dendritic cells. Mol Cell Biol. 2014;34(5):888–899. doi: 10.1128/MCB.01369-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Steinmann GG, Klaus B, Muller-Hermelink HK. The involution of the ageing human thymic epithelium is independent of puberty. A morphometric study. Scand J Immunol. 1985;22(5):563–575. doi: 10.1111/j.1365-3083.1985.tb01916.x. [DOI] [PubMed] [Google Scholar]
- 86.Hearps AC, et al. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell. 2012;11(5):867–875. doi: 10.1111/j.1474-9726.2012.00851.x. [DOI] [PubMed] [Google Scholar]
- 87.Seidler S, et al. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. 2010;11:30. doi: 10.1186/1471-2172-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ashcroft GS, Horan MA, Ferguson MW. Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Lab Invest. 1998;78(1):47–58. [PubMed] [Google Scholar]
- 89.Herrero C, et al. Immunosenescence of macrophages: reduced MHC class II gene expression. Exp Gerontol. 2002;37(2–3):389–394. doi: 10.1016/s0531-5565(01)00205-4. [DOI] [PubMed] [Google Scholar]
- 90.Zissel G, Schlaak M, Muller-Quernheim J. Age-related decrease in accessory cell function of human alveolar macrophages. J Investig Med. 1999;47(1):51–56. [PubMed] [Google Scholar]
- 91.Wu D, Meydani SN. Mechanism of age-associated up-regulation in macrophage PGE2 synthesis. Brain Behav Immun. 2004;18(6):487–494. doi: 10.1016/j.bbi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 92.van Duin D, et al. Age-associated defect in human TLR-1/2 function. J Immunol. 2007;178(2):970–975. doi: 10.4049/jimmunol.178.2.970. [DOI] [PubMed] [Google Scholar]
- 93.Boehmer ED, et al. Aging negatively skews macrophage TLR2- and TLR4-mediated pro-inflammatory responses without affecting the IL-2-stimulated pathway. Mech Ageing Dev. 2005;126(12):1305–1313. doi: 10.1016/j.mad.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 94.De La Fuente M. Changes in the macrophage function with aging. Comp Biochem Physiol A Comp Physiol. 1985;81(4):935–938. doi: 10.1016/0300-9629(85)90933-8. [DOI] [PubMed] [Google Scholar]
- 95.Linehan E, et al. Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging Cell. 2014;13(4):699–708. doi: 10.1111/acel.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Holt DJ, Grainger DW. Senescence and quiescence induced compromised function in cultured macrophages. Biomaterials. 2012;33(30):7497–7507. doi: 10.1016/j.biomaterials.2012.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Alvarez E, et al. Nitric oxide and superoxide anion production decrease with age in resident and activated rat peritoneal macrophages. Cell Immunol. 1996;169(1):152–155. doi: 10.1006/cimm.1996.0103. [DOI] [PubMed] [Google Scholar]
- 98.Wang Y, et al. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell. 2015;14(4):678–688. doi: 10.1111/acel.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mahbub S, Deburghgraeve CR, Kovacs EJ. Advanced age impairs macrophage polarization. J Interferon Cytokine Res. 2012;32(1):18–26. doi: 10.1089/jir.2011.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336(6077):86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 101.Gomez Perdiguero E, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518 (7540) 2015:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yona S, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Scott CL, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. 2016;7:10321. doi: 10.1038/ncomms10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Amit I, Winter DR, Jung S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat Immunol. 2016;17(1):18–25. doi: 10.1038/ni.3325. [DOI] [PubMed] [Google Scholar]
- 105.Hachim D, Wang N, Lopresti ST, Stahl EC, Umeda YU, Rege RD, Carey ST, Mani D, Brown BN. Effects of aging upon the host immune response to implants. J Biomed Mater Res Part A. 2017;105A:1281–1292. doi: 10.1002/jbm.a.36013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hachim D, et al. Effects of aging upon the host response to polypropylene MESH. Female Pelvic Med Reconstr Surg. 2015;21(5):S10. [Google Scholar]
- 107.LoPresti ST, Dash S, Brown BN. Effects of age-related changes in ECM upon host macropahge responses. Tissue Eng Part A. 2015;21(S1):S–344. [Google Scholar]
- 108.Brown BN, Badylak SF. Extracellular matrix as an inductive scaffold for functional tissue reconstruction. Transl Res. 2014;163(4):268–285. doi: 10.1016/j.trsl.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840(8):2506–2519. doi: 10.1016/j.bbagen.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(24):4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Keane TJ, Badylak SF. The host response to allogeneic and xenogeneic biological scaffold materials. J Tissue Eng Regen Med. 2015;9(5):504–511. doi: 10.1002/term.1874. [DOI] [PubMed] [Google Scholar]
- 112.Tottey S, et al. The effect of source animal age upon extracellular matrix scaffold properties. Biomaterials. 2011;32(1):128–136. doi: 10.1016/j.biomaterials.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sicari BM, et al. The effect of source animal age upon the in vivo remodeling characteristics of an extracellular matrix scaffold. Biomaterials. 2012;33(22):5524–5533. doi: 10.1016/j.biomaterials.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ott C, et al. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014;2:411–429. doi: 10.1016/j.redox.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kasper M, Funk RH. Age-related changes in cells and tissues due to advanced glycation end products (AGEs) Arch Gerontol Geriatr. 2001;32(3):233–243. doi: 10.1016/s0167-4943(01)00103-0. [DOI] [PubMed] [Google Scholar]
- 116.John WG, Lamb EJ. The Maillard or browning reaction in diabetes. Eye (Lond) 1993;7(Pt 2):230–237. doi: 10.1038/eye.1993.55. [DOI] [PubMed] [Google Scholar]
- 117.Badenhorst D, et al. Cross-linking influences the impact of quantitative changes in myocardial collagen on cardiac stiffness and remodelling in hypertension in rats. Cardiovasc Res. 2003;57(3):632–641. doi: 10.1016/s0008-6363(02)00733-2. [DOI] [PubMed] [Google Scholar]
- 118.Valencia JV, et al. Advanced glycation end product ligands for the receptor for advanced glycation end products: biochemical characterization and formation kinetics. Anal Biochem. 2004;324(1):68–78. doi: 10.1016/j.ab.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 119.Vlassara H, Uribarri J. Advanced glycation end products (AGE) and diabetes: cause, effect, or both? Curr Diab Rep. 2014;14(1):453. doi: 10.1007/s11892-013-0453-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang F, et al. Anti-receptor for advanced glycation end products therapies as novel treatment for abdominal aortic aneurysm. Ann Surg. 2009;250(3):416–423. doi: 10.1097/SLA.0b013e3181b41a18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen Y, et al. RAGE ligation affects T cell activation and controls T cell differentiation. J Immunol. 2008;181(6):4272–4278. doi: 10.4049/jimmunol.181.6.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kierdorf K, Fritz G. RAGE regulation and signaling in inflammation and beyond. J Leukoc Biol. 2013;94(1):55–68. doi: 10.1189/jlb.1012519. [DOI] [PubMed] [Google Scholar]
- 123.Dumitriu IE, et al. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174(12):7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 124.Pollreisz A, et al. Receptor for advanced glycation endproducts mediates pro-atherogenic responses to periodontal infection in vascular endothelial cells. Atherosclerosis. 2010;212(2):451–456. doi: 10.1016/j.atherosclerosis.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu Y, et al. AGEs increased migration and inflammatory responses of adventitial fibroblasts via RAGE: MAPK and NF-kappaB pathways. Atherosclerosis. 2010;208(1):34–42. doi: 10.1016/j.atherosclerosis.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 126.Neeper M, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267(21):14998–15004. [PubMed] [Google Scholar]
- 127.Chavakis T, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med. 2003;198(10):1507–1515. doi: 10.1084/jem.20030800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kokkola R, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61(1):1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 129.Webster L, et al. Induction of TNF alpha and IL-1 beta mRNA in monocytes by methylglyoxal- and advanced glycated endproduct-modified human serum albumin. Biochem Soc Trans. 1997;25(2):250S. doi: 10.1042/bst025250s. [DOI] [PubMed] [Google Scholar]
- 130.Juranek JK, et al. RAGE deficiency improves postinjury sciatic nerve regeneration in type 1 diabetic mice. Diabetes. 2013;62(3):931–943. doi: 10.2337/db12-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jin X, et al. Advanced glycation end products enhance macrophages polarization into M1 phenotype through activating RAGE/NF-kappaB pathway. BioMed Res Int. 2015;2015:732450. doi: 10.1155/2015/732450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shanmugam N, et al. Regulation of cyclooxygenase-2 expression in monocytes by ligation of the receptor for advanced glycation end products. J Biol Chem. 2003;278(37):34834–34844. doi: 10.1074/jbc.M302828200. [DOI] [PubMed] [Google Scholar]
- 133.Chang JS, et al. Oxygen deprivation triggers upregulation of early growth response-1 by the receptor for advanced glycation end products. Circ Res. 2008;102(8):905–913. doi: 10.1161/CIRCRESAHA.107.165308. [DOI] [PubMed] [Google Scholar]
- 134.Kohn JC, Lampi MC, Reinhart-King CA. Age-related vascular stiffening: causes and consequences. Front Genet. 2015;6:112. doi: 10.3389/fgene.2015.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bonnema DD, et al. Effects of age on plasma matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs) J Card Fail. 2007;13(7):530–540. doi: 10.1016/j.cardfail.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Handorf AM, et al. Tissue stiffness dictates development: homeostasis, and disease progression. Organogenesis. 2015;11(1):1–15. doi: 10.1080/15476278.2015.1019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Urry DW. Neutral sites for calcium ion binding to elastin and collagen: a charge neutralization theory for calcification and its relationship to atherosclerosis. Proc Natl Acad Sci U S A. 1971;68(4):810–814. doi: 10.1073/pnas.68.4.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Urry DW, Onishi T. Calcium ion binding to carbonyls of elastin hexapeptide. Bioinorg Chem. 1974;3(4):305–313. doi: 10.1016/s0006-3061(00)82010-4. [DOI] [PubMed] [Google Scholar]
- 139.Otto CM, et al. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. 1999;341(3):142–147. doi: 10.1056/NEJM199907153410302. [DOI] [PubMed] [Google Scholar]
- 140.Elliott RJ, McGrath LT. Calcification of the human thoracic aorta during aging. Calcif Tissue Int. 1994;54(4):268–273. doi: 10.1007/BF00295949. [DOI] [PubMed] [Google Scholar]
- 141.Gaillard V, et al. Pioglitazone improves aortic wall elasticity in a rat model of elastocalcinotic arteriosclerosis. Hypertension. 2005;46(2):372–379. doi: 10.1161/01.HYP.0000171472.24422.33. [DOI] [PubMed] [Google Scholar]
- 142.Lindsey ML, Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther. 2012;30(1):31–41. doi: 10.1111/j.1755-5922.2010.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ren F, et al. Overexpression of MMP family members functions as prognostic biomarker for breast cancer patients: a systematic review and meta-analysis. PLoS One. 2015;10(8):e0135544. doi: 10.1371/journal.pone.0135544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gong L, et al. Prognostic impact of serum and tissue MMP-9 in non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget. 2016;7(14):18458–18468. doi: 10.18632/oncotarget.7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sun DW, et al. Prognostic significance of MMP-7 expression in colorectal cancer: a meta-analysis. Cancer Epidemiol. 2015;39(2):135–142. doi: 10.1016/j.canep.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 146.Vafadari B, Salamian A, Kaczmarek L. MMP-9 in translation: from molecule to brain physiology, pathology, and therapy. J Neurochem. 2016;139(Suppl. 2):91–114. doi: 10.1111/jnc.13415. [DOI] [PubMed] [Google Scholar]
- 147.Sun HB. Mechanical loading, cartilage degradation, and arthritis. Ann N Y Acad Sci. 2010;1211:37–50. doi: 10.1111/j.1749-6632.2010.05808.x. [DOI] [PubMed] [Google Scholar]
- 148.McWhorter FY, Davis CT, Liu WF. Physical and mechanical regulation of macrophage phenotype and function. Cell Mol Life Sci. 2015;72(7):1303–1316. doi: 10.1007/s00018-014-1796-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Springer NL, Fischbach C. Biomaterials approaches to modeling macrophage-extracellular matrix interactions in the tumor microenvironment. Curr Opin Biotechnol. 2016;40:16–23. doi: 10.1016/j.copbio.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]