

Abstract
Background
Our previous studies suggested certain β-adrenoceptor blockers (β-blockers) attenuate the asthma phenotype in ovalbumin driven murine models of asthma. However, the ovalbumin model has been criticized for lack of clinical relevance.
Methods
We tested the non-selective β-blockers, carvedilol and nadolol, in house dust mite (HDM) driven murine asthma models where drugs were administered both pre- and post- development of the asthma phenotype. We measured inflammation, mucous metaplasia, and airway hyper-responsiveness (AHR). We also measured the effects of the β-blockers on extracellular-signal regulated kinases (ERK 1/2) phosphorylation in lung homogenates.
Results
We show that nadolol, but not carvedilol, attenuated inflammation and mucous metaplasia, and had a moderate effect attenuating AHR. Following HDM exposure, ERK1/2 phosphorylation was elevated, but the level of phosphorylation was unaffected by β-blockers, suggesting ERK1/2 phosphorylation becomes dissociated from the asthma phenotype.
Conclusion
Our findings in HDM models administering drugs both pre- and post-development of the asthma phenotype are consistent with previous results using ovalbumin models and show differential effects for nadolol and carvedilol on the asthma phenotype. Lastly, our data suggest that ERK1/2 phosphorylation may be involved in development of the asthma phenotype, but may have a limited role in maintaining the phenotype.
Keywords: β-adrenoceptor blockers, house dust mite, murine model, asthma, extracellular-signal regulated kinases 1/2
1. Introduction
Asthma is a chronic disease of the airways, which occurs in people from all age groups. According to the ‘Global Asthma Report’, approximately 334 million people have asthma worldwide (The Global Asthma Report 2014). The numbers continue to increase making asthma a serious health and economic burden. The most often prescribed bronchodilators for asthma therapy are β2 adrenoceptor (β2AR) agonists. For decades, β2AR agonists have been modified to enhance their effectiveness by improving their receptor selectivity and increasing their duration of action. However, studies have found that prolonged use of certain long acting β2AR agonists such as salmeterol is associated with a loss of asthma control, and a small increase in asthma related deaths [1–6].
We have previously shown that mice lacking epinephrine, the endogenous agonist for the β2AR, do not develop the asthma phenotype in ovalbumin allergen-driven models of asthma [7]. These mice lack the enzyme phenylethnolamine N-methyl transferase (PNMT), which is required for the final step in the synthesis of epinephrine, and have no detectable levels of circulating epinephrine [7]. In these PNMT-KO mice, chronic treatment with the long acting β2AR agonists, salmeterol and formoterol restored the asthma phenotype [8]. Also, treatment with the β-blocker, nadolol attenuated the asthma phenotype in wild type (WT) mice and did not restore the phenotype in PNMT-KO mice [9]. These results suggested β2AR agonists, but not antagonists were able to restore the asthma phenotype. However, other non-selective β-blockers such as carvedilol and propranolol did restore development of the asthma phenotype in PNMT-KO mice and had no effect on the phenotype in WT mice [9].
Furthermore, pilot clinical trials have shown that chronic nadolol treatment of mild asthmatics produced a dose-dependent increase in the amount of methacholine needed to elicit a 20% reduction in forced expiratory volume in 1 second (PC20 methacholine) [10, 11], while another study showed no benefits of chronic propranolol treatment in a different set of asthmatics [12]. Thus clinical trials also suggest mechanistic differences between different β-blockers in asthma. Several in vitro studies indicate that despite the ability of β-blockers to inhibit the canonical Gs-cAMP pathway at the β2AR, they differ in their activity at the extracellular signal-regulated kinase 1/2 (ERK1/2) pathway. In these studies, nadolol, timolol, metoprolol and ICI 118,551 inhibited the ERK1/2 pathway, while carvedilol and propranolol activated the ERK1/2 pathway [13–15]. Therefore, our in vivo results, and the limited data from clinical trials, suggested a correlation between the differential effects of β-blockers in the murine asthma model and their in vitro activation profiles at the ERK1/2 pathway [9–12]. Moreover, several studies have implicated ERK1/2 phosphorylation in the pathogenesis of asthma [16–18]. For example, administration of U0126, a mitogen activated protein kinase kinase (MEK1/2) inhibitor that inhibits ERK1/2 activation, also attenuated the asthma phenotype in an ovalbumin-driven murine asthma model [16].
However, as noted, all of our previous studies used ovalbumin as the antigen inducing the asthma phenotype. A shortcoming of ovalbumin-driven murine asthma models is their limited clinical relevance [19]. Also, persistent exposure of mice to ovalbumin could lead to immune tolerance and diminished airway inflammation [20]. To address some of these limitations, the present studies were performed using house dust mite (HDM) extract as the source of antigens. House dust mites are a common aeroallergen causing a range of respiratory symptoms in humans including asthma [21]. The whole body extract from the species, Dermatophagoides pteronyssinus has been used as a clinically more relevant allergen in animal models of asthma [19, 21–23]. For example, a study showed that intranasal delivery of purified HDM extract for 10 consecutive days produced a robust airway inflammatory response in mice accompanied by airway hyper-responsiveness [22]. In this study, the inflammatory response resolved after 4 weeks and could be restored by subsequent re-exposure to the HDM extract [22]. Despite the differences in the allergens involved, ovalbumin and HDM driven models of asthma are qualitatively similar in terms of the phenotypes they produce in mice. To further ensure that the two models are also similar in the cytokine/chemokine profile that drives the asthma phenotypes, we also characterized the HDM model for the presence of TH1- and TH2-associated cytokines and chemokines.
In these studies we tested the β-blockers, nadolol and carvedilol in murine models of asthma utilizing house dust mite (HDM) extract. These β-blockers were chosen because as discussed above, they have opposing effects on the asthma phenotype in the PNMT-KO and WT mice [9], and their activities differ at the ERK1/2 pathway in in vitro studies [13–15]. In addition, our previous studies had shown only a ‘prophylactic’ effect of β-blockers (the drugs were administered prior to the development of asthma phenotype) [9, 24]. To further add clinical relevance, in the present study we investigated both the ‘prophylactic’ and ‘therapeutic’ effects (where drugs were administered after an asthma phenotype had been established) in HDM models.
Lastly, in an attempt to elucidate a potential mechanism for the differential effects of nadolol and carvedilol, we investigated ERK1/2 phophorylation in whole lung homogenates. The rationale for this was the correlation we had noted between the ability of ligands to inhibit ERK1/2, and the ability of ligands to attenuate the asthma phenotype. Additionally, previous studies implicate ERK1/2 as having a role in the development of the asthma phenotype [9, 16–18]. Therefore, in the present studies, we examined ERK1/2 phosphorylation in whole lung in both the ovalbumin and the HDM driven murine models of asthma.
2. Materials and Methods
2.1 Animals
All animal experiments were performed in compliance with the ARRIVE guidelines. All procedures and protocols were approved by the Institutional Animal Care and Use Committee at the University of Houston (Protocol # 13-021, 16-022) which follows all NIH guidelines.
Male Balb/c mice (4–8 weeks old) purchased from Jackson Laboratories (Bar Harbor, ME, USA) were used in this study. Mice were housed in specific pathogen free conditions with ALPHA-dri® bedding. They were maintained at 22–24°C and 45–48% humidity under a 12h light/dark cycle, and were provided food and water ad libitum. Age-matched mice were randomly assigned to the different experimental groups.
2.2 Protocol Timelines and Drug Administration
We employed house dust mite models to develop the asthma phenotypes in mice. In these models, Balb/c mice were challenged with 25 μg of HDM protein (Greer Laboratories, Lenoir, North Carolina, USA) in 10 μl sterile saline by once daily intranasal delivery according to the timelines laid out for the ‘prophylactic’ and ‘therapeutic’ protocols (Fig. 1). Three or 4 animals of the same treatment group were housed in each cage. The calculated amounts of drugs were mixed with powdered chow and filled sufficient quantities of food (~5g per animal) in J-feeders to last a single day, and the feeders were replenished with fresh food every day.
Figure 1. Treatment Protocol.
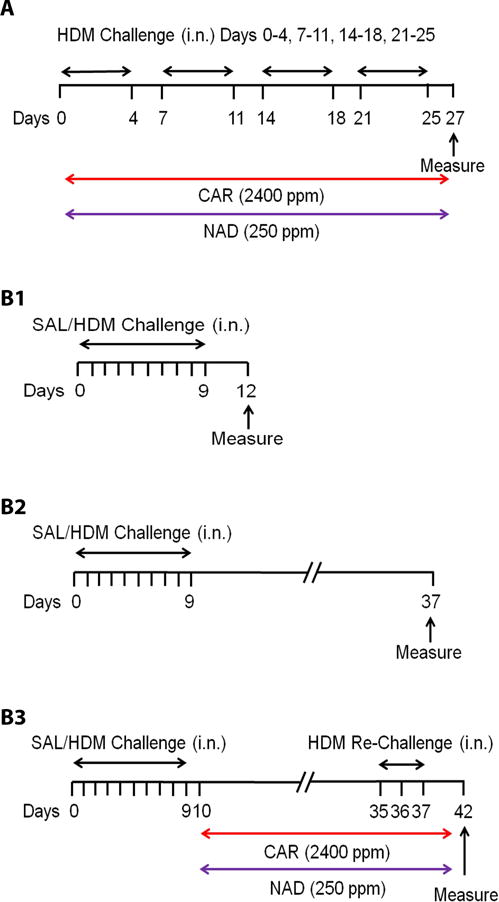
Balb/c mice were challenged with saline or 25 μg HDM protein by intranasal delivery in HDM driven (A) ‘prophylactic’ and (B1–B3) ‘therapeutic’ models of asthma. (A) In the ‘prophylactic’ model, mice were challenged for 5 days a week for 4 weeks. Groups of mice received vehicle, 2400 ppm of carvedilol (red bar) or 250 ppm of nadolol (purple bar) mixed with powdered rodent chow provided ad libitum for 4 weeks. (B1–B3) In the ‘therapeutic’ model, mice were initially challenged for 10 days and a group was evaluated on day 12. The remaining mice received vehicle, 2400 ppm of carvedilol (red bar) or 250 ppm of nadolol (purple bar) mixed with powdered rodent chow provided ad libitum. (B2) A vehicle treated group was evaluated 4 weeks after the last HDM challenge on day 37. (B3) The remaining vehicle and drug groups were re-challenged with HDM on days 35–37 and evaluated 5 days later on day 42. Drug treatment continued till day 42.
2.2.1. ‘Prophylactic’ model of asthma
In the ‘prophylactic’ model, mice were treated with β-blockers prior to development of the asthma phenotype. To establish the phenotype in the ‘prophylactic’ model, mice were challenged with saline or HDM protein for 5 days a week over a period of 4 weeks. During this time, groups of mice received 2400 ppm of carvedilol (Patterson Veterinary, Blythewood, SC, USA) or 250 ppm of nadolol (Sigma Aldrich, St. Louis, MO, USA) orally. As described above, the drugs were mixed with powdered rodent chow and provided to mice ad libitum. On day 28 of the protocol, all groups were evaluated for the various parameters of asthma.
2.2.2. ‘Therapeutic’ model of asthma
In the ‘therapeutic’ model, drug treatment was started after the asthma phenotype had been established. In the ‘therapeutic’ model, the mice were initially challenged with saline or HDM protein for 10 consecutive days. Seventy-two hours after the last challenge (protocol day 12), a control and a vehicle group were evaluated for the asthma phenotype. After the phenotype had been established, the remaining mice were started on drugs, carvedilol (2400 ppm) or nadolol (250 ppm), or vehicle administered orally in rodent chow for 4 weeks. Another vehicle group was evaluated four weeks after the last HDM challenge (protocol day 37), while the remaining vehicle and drug treatment groups were re-challenged with HDM (25 μg) for 3 days (protocol days 35, 36, 37) and evaluated 5 days later (protocol day 42). In the latter groups, vehicle or drug treatments continued till day 42.
2.2.3. Ovalbumin sensitization/challenge model of asthma
In the ovalbumin driven murine asthma model, mice were immunized with 0.4 mg/kg/day ovalbumin adsorbed to 2 mg alum intraperitoneally on days 0, 7 and 14. Following sensitization, mice were challenged with saline or 1 mg/kg/day ovalbumin by intranasal delivery on days 24–28 [7]. Mice were evaluated for the asthma parameters on day 29, 24 hours after the last ovalbumin challenge.
2.3 Broncho-Alveolar Lavage (BAL)
Mice were euthanized with 100 mg/kg i.p. pentobarbital sodium (Patterson Veterinary, Blythewood, SC, USA) and their tracheas were cannulated using a 20 G luer stub adapter. After isolating the left lung lobe with a hemostat, the right lobes were lavaged with 500 μl saline to obtain the broncho-alveolar lavage fluid (BALF). The total and differential cells counts were determined using a Hemocytometer (Hausser Scientific, Horsham, PA) and Wright-Geimsa staining (Sigma Aldrich, St. Louis, MO, USA) as described previously [7]. The numbers of eosinophils were determined in 5 random fields on the slides using 40× magnification on a light microscope and expressed as eosinophils/ml of BALF.
2.4 Mucous Metaplasia
Following BALF collection, the hemostat was released and the lungs were fixed with cold 10 % neutral buffered formalin (Sigma Aldrich, St. Louis, MO, USA) perfused via the tracheal cannula and further fixed for 24 hours at 4°C. Afterwards, the left lobe was sectioned transversely and embedded into paraffin blocks for further sectioning. Five μm thick transverse sections were obtained using a microtome, collected onto positively charged glass slides and stained with periodic acid fluorescent Schiff’s stain (PAFS). Images of bronchial airway sections were captured at 40× magnification as explained previously [7, 25, 26]. For images measured on a μm scale, the mucin volume density was calculated in a blinded manner using ImageJ software (National Institutes of Health) by dividing the area of mucin staining by the product of the length of basement membrane and 4/π, and expressed as nl/mm2.
2.5 Airway Hyper-responsiveness (AHR)
Mice were anesthetized with 240 mg/kg ketamine and 48 mg/kg xylazine for measurement of airway hyper-responsiveness using the Flexivent® (Scireq, Montreal, Canada) with an in-line nebulizer. After establishing the loss of corneal and nociceptive reflexes, the mice were tracheostomized and the tracheas cannulated with a 20 G luer stub adapter. Mice were ventilated at 150 breaths per minute by a small animal ventilator within the Flexivent, and heart rate and EKG were monitored. Total respiratory system resistance (Rrs) was measured using the forced oscillation technique in response to incrementing doses of inhaled methacholine (0–50 mg/ml) administered through the nebulizer. Airway sensitivity (PC100), determined by the concentration of methacholine that causes doubling of the baseline airway resistance and airway reactivity (K), by the slope of the Rrs-methacholine dose-response curve, were calculated using non-linear regression analysis and fitting the dose-response curve to an exponential growth function as described in [9]. If heart rate fell below 40 bpm, the experiment was terminated and the mouse euthanized immediately.
2.6 Immunoblotting
After collecting BALF, the right lung lobes were removed, washed in saline and snap-frozen in liquid nitrogen. The tissues were homogenized in buffer containing protease and phosphatase inhibitors and protein levels were determined by Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Waltham, MA, USA). Twenty-five μg of protein from each sample was subjected to 4–20% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with milk powder (5%) in 50 mM Tris, 0.15 M NaCl and 0.1% Tween 20 (TBS-T) and immunoblotted with mouse monoclonal anti-P-p44/42 MAPK (pERK1/2) and anti-p44/42 MAPK (ERK1/2) antibodies (Cell Signaling, Danvers, MA, USA) using the recommended antibody dilutions.
2.7 Cytokine and Chemokine Analysis in Broncho-Alvelolar Lavage Fluid (BALF)
Cytokines and chemokines were measured in the BALF collected from mice subjected to the HDM-driven ‘prophylactic’ model. Based on a study that had performed a time-course [27], the time-point for collection of BALF samples for cytokine analysis was set at 4 hours post-final challenge with saline/HDM. This time point showed detection of peak cytokine and chemokine levels [27]. The BALF levels of cytokines IL-13 (TH2), IFN-γ (TH1) and IL-17 (TH17), and chemokines EOTAXIN (TH2) and CXCL1/KC (TH1) were measured by sandwich ELISAs according to the manufacturer’s instructions (R&D Systems,, Inc., Minneapolis, MN, USA).
2.8 Statistical Analysis
Data are expressed as mean ± SEM (standard error of means). Normal distribution of the data was assumed and statistical analysis performed using one-way analysis of variance (one-way ANOVA) followed by Tukey’s multiple comparison tests. Differences were considered statistically significant at p<0.05. Airway sensitivity and reactivity were measured using non-linear regression analysis. All analyses were performed using the Graph Pad Prism 7 software, San Diego, CA.
3. Results
3.1 Effect of β-blockers on inflammatory cell infiltration in BALF
We studied the effects of the β-blockers, carvedilol and nadolol, on inflammatory cellular infiltration in BALF in house dust mite (HDM) driven murine models of asthma, when drug administration began before (‘prophylactic’) and after (‘therapeutic’) the development of an asthma phenotype.
3.1.1. ‘Prophylactic’ Model
In the prophylactic model (Fig. 1A), mice developed a robust increase in both the total cells and eosinophils in BALF after 4 weeks of HDM exposure (Figs. 2A and 2B). Chronic treatment with carvedilol had no effect, while chronic treatment with nadolol significantly attenuated the increase in both the total cells and eosinophils in BALF (Figs. 2A and 2B).
Figure 2. Effect of β-blockers on inflammatory cellular infiltration in broncheo-alveolar lavage fluid (BALF) in the ‘prophylactic’ and ‘therapeutic’ HDM models.
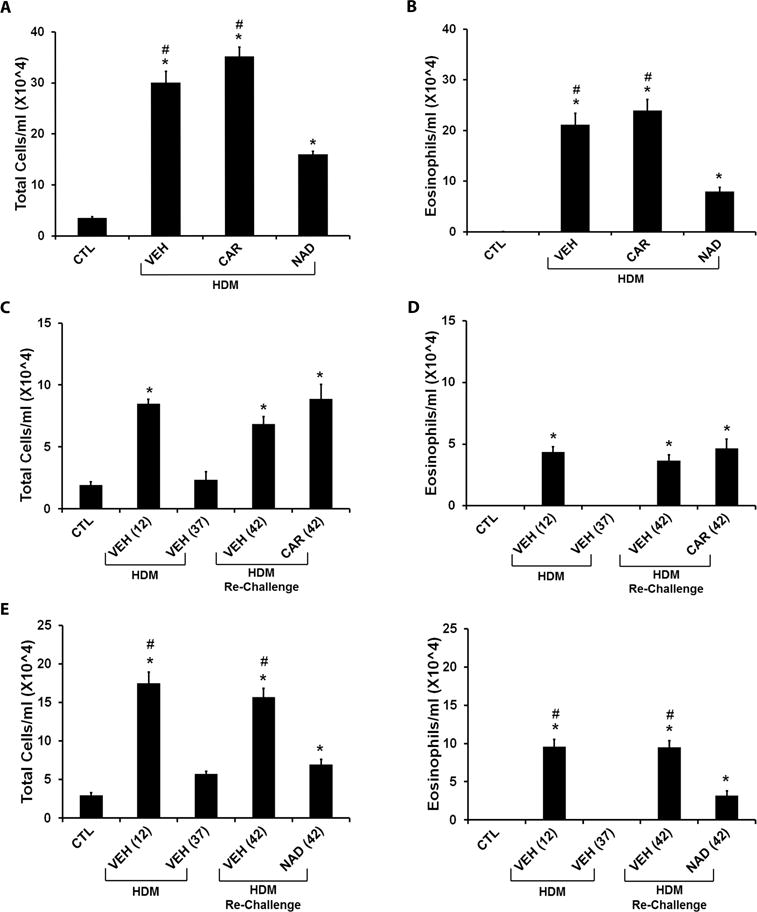
The graphs represent total cells and eosinophil infiltration in BALF collected from Balb/c mice subjected to HDM challenge in the ‘prophylactic’ and ‘therapeutic’ models. (A and B) Effect of β-blockers in the ‘prophylactic’ model. (A) Total cell count and (B) eosinophil count in BALF from HDM challenged mice treated with vehicle, carvedilol or nadolol in comparison to saline control mice. (C and D) Effect of carvedilol in the ‘therapeutic’ model. (C) Total cell count and (D) eosinophil count in BALF of saline control mice compared with mice challenged with HDM and evaluated on days 12 and 37; and mice re-challenged with HDM with or without carvedilol and evaluated on day 42. (E and F) Effect of nadolol in the ‘therapeutic’ model. (E) Comparison of Total cell count and (F) comparison of eosinophil count in BALF of saline control mice compared with mice challenged with HDM and evaluated on days 12 and 37; and mice re-challenged with HDM with or without nadolol and evaluated on day 42. Data are mean ± SEM from 5–8 mice in each group. * represents significance at p<0.05 compared to respective saline control mice. # represents significance at p<0.05 compared to respective nadolol treated mice. The effects of CAR (C and D) and NAD (E and F) are plotted separately because the different time matched control and vehicle groups for each drug treatment resulted in different baselines.
3.1.2. ‘Therapeutic’ Model
In the therapeutic model (Fig. 1B), an initial exposure to HDM extract for 10 days produced a vigorous increase in the total cells (Figs. 2C and 2E) and eosinophils (Figs. 2D and 2F). These increases returned to baseline by day 37 of the protocol (Figs. 2C–2F). Subsequent re-exposure to HDM for just 3 days restored the increase in the total cells (Fig. 2C) and eosinophils (Fig. 2D) after vehicle or carvedilol treatment. However, nadolol treatment caused a significant reduction in the total cells (Fig. 2E) and eosinophils (Fig. 2F) following HDM re-challenge.
3.2 Effect of β-blockers on mucous metaplasia
We measured mucin volume density in histological sections of the airways from mice subjected to the HDM driven ‘prophylactic’ and ‘therapeutic’ models of asthma.
3.2.1. ‘Prophylactic’ Model
In the prophylactic model, mice showed a profound increase in airway mucus production in response to HDM. Chronic carvedilol treatment also showed increased mucus production in the HDM exposed mice compared with the saline control mice (Figs. 3A and 3B). However, chronic nadolol treatment significantly attenuated the mucus production in the HDM exposed mice (Figs. 3A and 3B).
Figure 3. Effect of β-blockers on airway mucin content in the ‘prophylactic’ and ‘therapeutic’ HDM models.
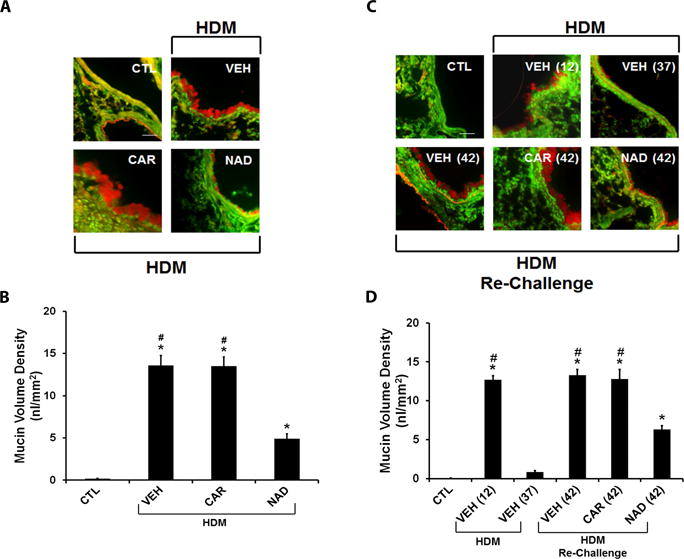
Airway sections from mouse lungs were stained with periodic acid fluorescent Schiff’s stain (PAFS) for mucin (red) content in airway epithelia (green). (A and B) Mucin stained images of airway sections from the (A) ‘prophylactic’ and (B) ‘therapeutic’ models. (C) Morphometric quantification of mucin volume density in the ‘prophylactic’ model from HDM challenged mice treated with vehicle, carvedilol or nadolol in comparison to saline control mice. (D) Morphometric quantification of mucin volume density in the ‘therapeutic’ model from saline control mice compared with mice challenged with HDM and evaluated on days 12 and 37; and vehicle, carvedilol or nadolol treated mice re-challenged with HDM and evaluated on day 42. Data are mean ± SEM from 5–8 mice in each group. * represents significance at p<0.05 compared to respective saline control mice. # represents significance at p<0.05 compared to respective nadolol treated mice.
3.2.2. ‘Therapeutic’ Model
In the therapeutic model, an initial 10 days of HDM exposure increased the airway mucus production, which resolved after 4 weeks (Figs. 3C and 3D). Re-exposure to HDM restored the airway mucus production, which was significantly reduced by chronic nadolol treatment, but not carvedilol treatment in this model (Figs. 3C and 3D).
3.3 Effect of β-blockers on Airway Hyper-responsiveness (AHR)
We examined the prophylactic and therapeutic effects of the β2AR blockers, carvedilol and nadolol, on airway hyper-responsiveness (AHR) in HDM models of asthma. Total respiratory airway resistance (Rrs) was measured in response to incrementing doses of inhaled methacholine (0–50 mg/ml). Airway sensitivity (PC100) and reactivity were calculated using non-linear regression analysis.
3.3.1. ‘Prophylactic’ Model
Consistent with the effects on airway inflammation and mucus hypersecretion, HDM exposure significantly increased Rrs in the prophylactic model, which was not affected by carvedilol treatment, and only moderately decreased by nadolol treatment (Figs. 4A and 4B). HDM exposure significantly lowered the dose of methacholine that caused doubling of the baseline airway resistance (PC100), suggesting an increase in airway sensitivity. Carvedilol treatment did not affect, whereas nadolol treatment prevented, the increase in airway sensitivity (Fig. 4C). Similarly, nadolol, but not carvedilol treatment during HDM exposure decreased airway reactivity in the prophylactic model (Fig. 4D).
Figure 4. Effect of β-blockers on airway hyper-responsiveness (AHR) in the ‘prophylactic’ HDM model.
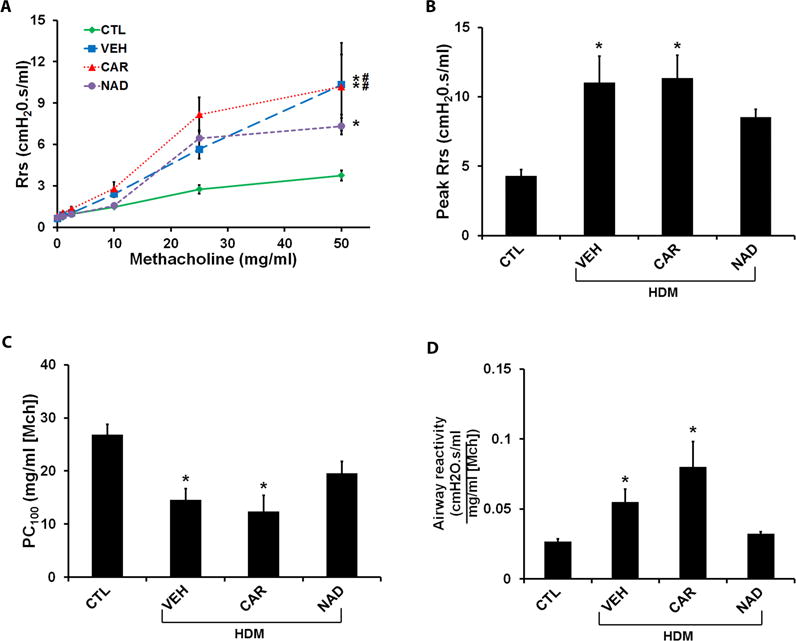
Total respiratory system resistance (Rrs) in response to incrementing doses of nebulized methacholine (0–50 mg/ml) was measured by the forced oscillation technique. Rrs was calculated as the average of three peak resistance responses at each dose of methacholine. The peak Rrs to methacholine, airway sensitivity (measured as the provocative dose of methacholine that causes doubling of the baseline airway resistance, PC100) and airway reactivity (the slope, K of the Rrs-methacholine dose response curve) were plotted as measures of AHR. (A–D) Rrs in response to methacholine (0–50 mg/ml), Peak Rrs, PC100 and K measured in the ‘prophylactic model’ in HDM challenged mice with or without carvedilol or nadolol treatment in comparison to saline control mice. Data are mean ± SEM from 7–8 mice in each group. * represents significance at p<0.05 compared to respective saline control mice. # represents significance at p<0.05 compared to respective nadolol treated mice.
3.3.2. ‘Therapeutic’ Model
In the therapeutic model, HDM exposure significantly increased the Rrs compared with control mice, which remained elevated even after 4 weeks (Fig. 5A). Subsequent re-exposure to HDM maintained the increased Rrs, which was moderately reduced by nadolol treatment, but was unaffected by carvedilol treatment (Fig. 5A). HDM exposure also showed an increase in the peak Rrs over saline controls, which was lowered by both carvedilol and nadolol treatments (Fig. 5B). With respect to airway sensitivity (Fig. 5C) and airway reactivity (Fig. 5D), neither carvedilol nor nadolol produced an effect on these parameters following HDM exposure.
Figure 5. Effect of β-blockers on airway hyper-responsiveness (AHR) in the ‘therapeutic’ HDM model.
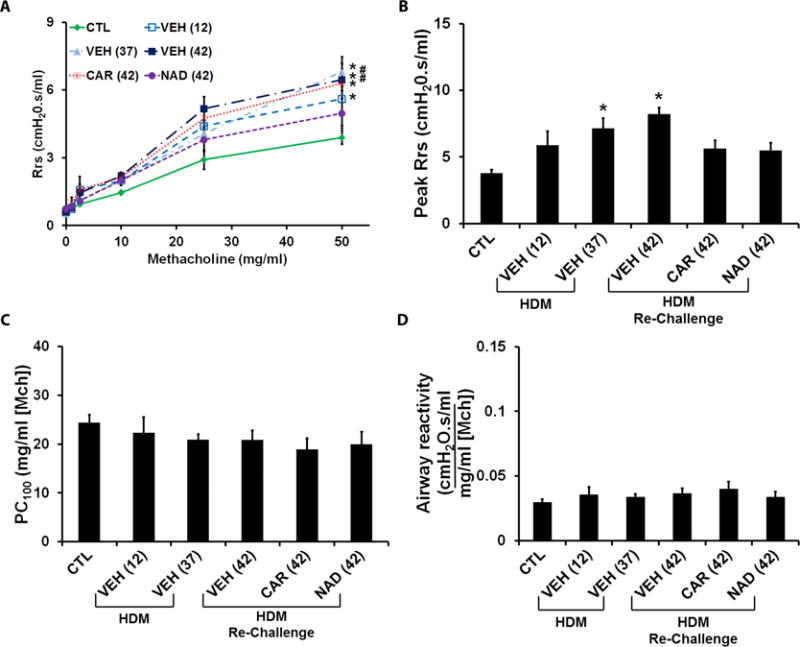
Total respiratory system resistance (Rrs) was measured in response to incrementing doses of nebulized methacholine (0–50 mg/ml) in the ‘therapeutic’ model. (A) Rrs measured in saline control mice compared with mice challenged with HDM and evaluated on days 12 day 37; and vehicle, carvedilol or nadolol treated mice re-challenged with HDM and evaluated on day 42. (B–D) Peak Rrs, PC100 and K measured in saline control mice and the HDM challenged mice with or without carvedilol or nadolol treatment. Data are mean ± SEM from 7–13 mice in each group. * represents significance at p<0.05 compared to respective saline control mice. # represents significance at p<0.05 compared to respective nadolol treated mice.
3.4 ERK1/2 Expression in the lungs
To study ERK1/2 phosphorylation in murine models of asthma, we measured the expression of phosphorylated and total ERK1/2 in lung tissue homogenate by western blotting in the HDM models of asthma. As reported by others [17, 18], HDM exposure significantly elevated the levels of pERK1/2 in total lung homogenate in the prophylactic model (Figs. 6A and 6B). However, drug therapy had no effect on the increased pERK1/2 expression in this model (Figs. 6A and 6B). In the therapeutic model, pERK expression remained upregulated even at 4 weeks after the last HDM exposure, when airway inflammation and mucous metaplasia had been resolved. HDM re-challenge further increased the pERK expression, which again was unaffected by nadolol treatment (Suppl. Fig. S1). These results suggest a dissociation of ERK1/2 phosphorylation from the inflammation and mucous metaplasia associated with the asthma phenotype.
Figure 6. Extracellular-signal regulated kinase (ERK1/2) phosphorylation in lungs homogenates from the murine asthma models.
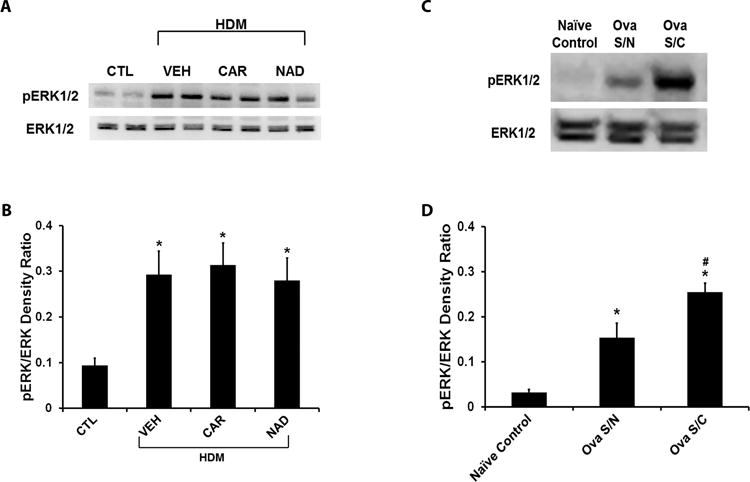
(A and B) ERK1/2 phosphorylation in the ‘prophylactic’ HDM model of asthma. (A) Representative western blot and (B) quantification of the phospho-ERK/ERK density ratio in HDM challenged mice with or without carvedilol or nadolol treatment in comparison to saline control mice. (C and D) ERK1/2 phosphorylation in the ovalbumin-driven murine asthma model. (C) Representative western blot and (D) quantification of the phospho-ERK/ERK density ratio in mice sensitized and challenged with ovalbumin (Ova S/C) in comparison to mice sensitized but not challenged with ovalbumin (Ova S/N) and naïve control mice. (A and B) Data are mean ± SEM from 6 mice in each group. * represents significance at p<0.05 compared to naïve control mice. # represents significance at p<0.05 compared to Ova S/N mice. (C and D) Data are mean ± SEM from 4–5 mice in each group. * represents significance at p<0.05 compared to saline control mice.
We also measured ERK1/2 phosphorylation in an ovalbumin driven murine model of asthma. In this model, ovalbumin sensitization, in the absence of ovalbumin challenge (Ova S/N), significantly increased ERK1/2 phosphorylation over naïve controls, and ovalbumin challenge (Ova S/C) further increased the ERK1/2 phosphorylation (Figs. 6C and 6D), suggesting that allergen sensitization is sufficient to significantly upregulate ERK1/2 phosphorylation even in the absence of the asthma phenotype. Airway inflammation and mucous metaplasia were measured after the challenge and confirmed the presence of asthma phenotype (Suppl. Fig. S2 and S3).
3.5 Effect of β-blockers on inflammatory cytokine and chemokine profile in BAL fluid
To characterize the HDM-driven murine model for the immune response driving the asthma phenotype, we measured the levels of TH1 and TH2 cytokines and chemokines in BALF collected from mice subjected to the ‘prophylactic’ model. In this model, HDM exposure increased the level of the TH2 cytokine IL-13 but not of the TH1 and TH17 cytokines IFN-γ and IL-17, respectively (Suppl. Fig. 4). Nadolol, but not carvedilol, attenuated the HDM induced increase in IL-13 level in BAL fluid (Suppl. Fig. 4A). The HDM exposure also increased the BALF concentration of the eosinophilic chemoattractant EOTAXIN, which was significantly attenuated by nadolol, but not carvedilol treatment (Suppl. Fig. 5C). The level of the neutrophilic chemoattractant CXCL1/KC was only moderately increased by HDM exposure in mice. This increase was further enhanced by carvedilol treatment but was unaffected by nadolol treatment (Suppl. Fig. 5D). Overall, the levels of the chemokines EOTAXIN and KC correlated well with the levels of eosinophils and neutrophils, respectively in BALF. The Pearson correlation coefficient, r for EOTAXIN/eosinophils was 0.6952 and for KC/neutrophils was 0.8654; the p values for the correlations were 0.0003 and <0.0001, respectively (data not shown).
4. Discussion
The objective of the present study was to evaluate the effects of β-blockers on HDM-induced asthma models when treatment was initiated prior to development of the asthma phenotype (prophylactic) or after development of the phenotype (therapeutic). We also evaluated the role of inhibition of ERK 1/2 phosphorylation in the beneficial effects of the β-blockers on the asthma phenotype. Our results indicate that nadolol treatment, whether prophylactic or therapeutic, prevented or reversed the HDM-induced asthma phenotype, respectively. Results reported here also indicate that the β-blocker nadolol did not reduce ERK 1/2 phosphorylation observed in the lung after HDM sensitization, in spite of the fact that nadolol prevented the development of the asthma phenotype. Therefore, we conclude that β-blockers such as nadolol, which inhibit ERK 1/2 activation can prevent the initiation and reverse the development of HDM-induced asthma, but that this effect is not associated with long-term reduction of ERK1/2 activation. We further conclude that ERK 1/2 activation by HDM treatment persists even after prevention or elimination of the HDM-induced asthma phenotype.
Our current results regarding β-blocker reduction of the asthma phenotype characteristics are similar to those we had obtained in ovalbumin models. Some investigators are critical of the ovalbumin model because of its low clinical relevance [22]. On the other hand, house dust mites are a natural source of aeroallergens and often implicated in human asthma [21, 22]. The whole body extract from dust mites is frequently employed in acute and chronic murine models of asthma without significant development of immune tolerance [22, 28]. Therefore, we used house dust mite (HDM) extract as the source of allergens in the present study. Like ovalbumin, exposure to HDM increased the number of total cells and eosinophils in BALF. Other studies have shown HDM exposure increases both eosinophil and neutrophil counts in BALF, with the response depending on the protocol for allergen challenge and the time point of evaluating the parameters after the final HDM exposure [29]. Here, we measured the asthma parameters 48 hours after the final HDM challenge in the prophylactic model, as this has been shown to be the time point to detect peak eosinophil numbers [29]. For similar reasons BALF was characterized 72 hours after the initial challenge and 5 days after re-challenge in the ‘therapeutic’ model [22]. Neutrophils, being relatively short-lived, peak at 6–12 hours after the final HDM challenge [29], and were not detected in BALF in our HDM models. Consistent with our previous results in the ovalbumin model, chronic nadolol treatment, but not carvedilol treatment, attenuated the eosinophilic infiltration in BALF and the airway mucous metaplasia in both the ‘prophylactic’ and ‘therapeutic’ HDM models. As previously reported, we also observed that inflammation and mucin hypersecretion caused by the initial HDM challenge in the therapeutic model were completely resolved after 4 weeks, and returned after the mice were re-exposed to HDM.
With respect to AHR in the prophylactic model, chronic carvedilol treatment had no effect on the increase in peak airway resistance, airway sensitivity (decrease in PC100 concentration) or airway reactivity, while nadolol treatment attenuated these parameters. The effect of nadolol, however, was less than previously reported in the ovalbumin model. Airway resistance in the nadolol-treated mice was still higher than in the saline controls. In previous studies using the ovalbumin models, we had found that chronic carvedilol treatment decreased the peak airway resistance but increased the airway sensitivity (lowered PC100) [30]. In the HDM ‘therapeutic’ model, both carvedilol and nadolol lowered the peak airway resistance, but did not affect the airway sensitivity and reactivity. In this ‘therapeutic’ model, HDM exposure did not show a significant increase in airway sensitivity and reactivity over saline controls. Therefore it is difficult to make any definitive conclusions about the effects of β-blockers on AHR in the ‘therapeutic’ model. Also, the increase in airway resistance caused by the initial HDM exposure did not resolve after 4 weeks as was observed with the inflammatory and mucous responses. This perhaps suggests a very robust AHR response in the HDM ‘therapeutic’ model. This observation is consistent with a previous report showing that AHR only partially resolves at 9 weeks after the last HDM exposure, whereas airway inflammation resolves completely [28]. Another study showed that while the standard combination therapy with glucocorticoids and β2- agonist attenuated the inflammatory response, it was not effective at attenuating AHR when given concomitantly with HDM challenges [31]. Our results also suggest that the moderate effect of nadolol in the HDM model may be attributable to a very robust and persistent AHR response in this model. Nevertheless, while nadolol did not completely normalize the HDM-induced asthma phenotype as it did in the ovalbumin model, it significantly reduced the characteristics of the asthma phenotype.
Our results show the ovalbumin and HDM-driven murine models of asthma are similar in the asthma phenotypes they produce, and in the effects of β-blockers on these phenotypes. We then also characterized the HDM model for the presence of TH1 and TH2 type inflammatory cytokines and chemokines in BALF. The BALF samples were collected from the subjects in the HDM induced ‘prophylactic’ model at a time-point of 4 hours following the last saline/HDM challenge. The 4 hour time-point was selected based on a previous study in the HDM model showing this time-point to be optimal for detecting peak inflammatory cytokines in BALF [27]. Our results suggest that HDM exposure in mice primarily developed a TH2 mediated immune response as shown by an increase in TH2 cytokine, IL-13 and the eosinophilic chemoattractant, EOTAXIN in BALF. Both TH1 and TH17 cytokines were nearly undetected. These results are similar to previous studies using the ovalbumin models [24]. Furthermore, as in the ovalbumin model, nadolol but not carvedilol attenuated the increase in the TH2 immune response to HDM, suggesting that nadolol’s beneficial effect on the asthma phenotype may be mediated by a reduction of the TH2 immune response [24].
A significant difference between our present study and our previous work is that the previous studies had used models where drug administration started prior to the animals developing the asthma phenotype (testing in a ‘prophylactic model). To further increase the potential clinical relevance of our present studies, we used both a ‘prophylactic’, and a ‘therapeutic’ model where the drug was administered after the mice had developed an asthma phenotype. Using more clinically relevant murine models and conditions has become important because there are clinical studies that support the potential therapeutic benefit of some but not all β-blockers in asthma.
Two pilot studies using nadolol in mild asthmatics have shown a dose-dependent increase in the PC20 methacholine following chronic treatment with nadolol [10, 11], demonstrating reduced airway hyper-responsiveness. On the other hand, other trials using propranolol resulted in no improvement in a different subset of asthmatics [12, 32–35]. Our results provide two important new pieces of information. First, they demonstrate that nadolol is effective in attenuating the asthma phenotype in either the ovalbumin-induced or HDM-induced models. Second, our results demonstrate that nadolol reduces the asthma phenotype regardless of whether treatment is initiated prior to or after development of the asthma phenotype. This second point supports findings of clinical studies where asthma symptoms were reduced by nadolol treatment.
An important question is raised by our results with nadolol and carvedilol in the present study and those with nadolol and propranolol in the clinical studies reported to date. Why do some β-blockers reduce the characteristics of asthma while others do not? Specifically, in our murine models, nadolol and ICI-118, 551 attenuated the asthma phenotype. However, carvedilol and propranolol were ineffective at attenuating the asthma phenotype [9, 24, 36]. In trying to understand the differential effects of β-blockers on the asthma phenotype in the various murine models of asthma, we examined their signaling profiles at the Gs-cAMP and ERK1/2 pathways in in vitro studies [9, 14, 15]. Nadolol and ICI-118,551 are antagonists at the ERK1/2 pathway, while carvedilol and propranolol, which did not attenuate the asthma phenotype, are agonists at the ERK1/2 pathway. The in vitro studies suggest a correlation between the effects of the β-blockers on the asthma phenotype and their ERK1/2 activation profiles [9]. Previous studies also have reported a potential role of the ERK1/2 pathway in mediating the asthma phenotype in murine models [16–18]. In the current study, we found that HDM exposure increased ERK1/2 phosphorylation in whole lung, but the increase was unaffected by the β-blockers treatment in both the ‘prophylactic’ and ‘therapeutic’ models. Furthermore, ERK1/2 phosphorylation remained upregulated even when airway eosinophilia and mucous metaplasia had resolved after 4 weeks in the ‘therapeutic’ model. To further check if ERK1/2 phosphorylation becomes dissociated from the asthma phenotype, we returned to using the ovalbumin model where the asthma phenotype is not present until the mice are challenged. In the ovalbumin model, allergen sensitization was sufficient to increase the phosphorylation of ERK1/2 in whole lung even in the absence of the asthma phenotype. These results suggest that the initial activation of the ERK1/2 pathway produced by allergen exposure, becomes dissociated from the asthma phenotype, and does not appear to be the key factor mediating the opposing effects of the β-blockers blockers on the asthma phenotype. Previous studies have reported increased phosphorylation of ERK1/2 in airway epithelial and smooth muscle cells from asthmatic patients and also in murine models of asthma [17, 18]. However, our present studies only measure ERK1/2 phosphorylation in the whole lung. Therefore, further in vivo studies on cell specific ERK1/2 activation are required to definitively show that the ERK1/2 pathway is not sufficient for perpetuation of the asthma phenotype.
The results of the present study highlight a recently recognized characteristic of some β2 adrenoceptor ligands, that they can preferentially activate one of the several signaling pathways downstream of the receptor [37]. This phenomenon has become widely known as biased signaling [38, 39]. The two major pathways associated with the β2AR are the canonical Gs-cAMP pathway and the ERK1/2 pathway [13, 14, 40]. We therefore reasoned that the differential effects of the β-blockers on the asthma phenotype in the various murine models of asthma might be explained by their signaling profiles at the Gs-cAMP and ERK1/2 pathways in in vitro studies [9, 14, 15]. Despite ICI-118,551, nadolol, carvedilol and propranolol inhibiting the Gs-cAMP pathway, these β-blockers differed in their actions at the ERK1/2 pathway [14]. However, the observations that nadolol did not normalize ERK 1/2 phosphorylation yet attenuated the HDM-induced asthma phenotype suggest that a potential role of ERK 1/2 in this action of nadolol will require additional investigation to resolve.
5. Conclusions
Overall, our data are consistent with our previous findings in the ovalbumin murine models of asthma, where we had observed differential effects of β-blockers on the asthma phenotype. Additionally, we found that nadolol attenuated airway eosinophilia and mucous metaplasia even when administered after the development of the asthma phenotype. Lastly, our data show that drug treatment dissociates ERK1/2 activity from the asthma phenotypes, suggesting a limited role for ERK1/2 in maintaining the asthma phenotype.
Supplementary Material
(A) Representative western blot and (B) quantification of the phospho-ERK/ERK density ratio in HDM challenged mice evaluated on day 12 and 4 weeks later on day 37; and HDM re-challenged mice evaluated on day 42 with or without nadolol treatment in comparison to saline control mice. Data are mean ± SEM from 5 mice in each group. * represents significance at p<0.05 compared to saline control mice.
(A and B) The graphs represent total cells and eosinophil infiltration in BALF collected from Balb/c mice subjected to the ovalbumin model of asthma. (A) Total cell count and (B) eosinophil count in BALF of mice sensitized and challenged with ovalbumin (Ova S/C) in comparison to mice sensitized but not challenged with ovalbumin (Ova S/N) and naïve control mice. Data are mean ± SEM from 4–5 mice in each group. * represents significance at p<0.05 compared to naïve control mice. # represents significance at p<0.05 compared to Ova S/N mice.
Airway sections from mice were stained with periodic acid fluorescent schiff’s stain (PAFS) for mucin (red) content in airway epithelia (green). (A) Mucin stained images from Balb/c mice subjected to the ovalbumin model of asthma. (B) Morphometric quantification of mucin volume density in mice sensitized and challenged with ovalbumin (Ova S/C) in comparison to mice sensitized but not challenged with ovalbumin (Ova S/N) and naïve control mice. Data are mean ± SEM from 4–5 mice in each group. * represents significance at p<0.05 compared to naïve control mice. # represents significance at p<0.05 compared to Ova S/N mice.
The graphs represent the levels of inflammatory cytokines in BALF collected from Balb/c mice subjected to HDM challenge in the ‘prophylactic’ model. Concentration of (A) IL-13, (B) INF-γ and (C) IL-17 in BALF from HDM challenged mice treated with vehicle, carvedilol or nadolol in comparison to saline control mice. Data are mean ± SEM from 5–6 mice in each group.
The graphs represent the levels of inflammatory cells (A and B) and chemokines (C and D) in BALF collected from Balb/c mice subjected to HDM challenge in the ‘prophylactic’ model. Cellular counts of (A) eosinophils and (B) neutrophils, and the concentrations of (C) EOTAXIN and (D) CXCL1/KC in BALF from HDM challenged mice treated with vehicle, carvedilol or nadolol in comparison to saline control mice. Data are mean ± SEM from 5–6 mice in each group. * represents significance at p<0.05 compared to respective saline control mice. # represents significance at p<0.05 compared to respective nadolol treated mice.
Acknowledgments
The authors would like to thank Jason Eriksen at the University of Houston for permitting use of histology equipment in his laboratory. The authors would also like to thank Vaidehi Thanawala at Vapogenix Inc., Houston, Texas for providing technical guidance, and our collaborator Julia Walker at Duke University for providing training using the forced oscillation technique (Flexivent®, Montreal, Canada) to measure airway function.
Funding Source
This work was supported by National Institutes of Health [grant number R01AI110007].
Abbreviations
- AHR
airway hyperresponsiveness
- β2AR
β2-adrenoceptor
- BALF
bronchoalveolar lavage fluid
- cAMP
cyclic adenosine monophosphate
- ERK1/2
extracellular signal-regulated kinase 1/2
- Ig
immunoglobulin
- i.n
intra-nasal
- i.p
intra-peritoneal
- MAPK
mitogen activated protein kinase
- PNMT
phenylethnolamine N-methyl transferase
- Ova S/C
ovalbumin-sensitized and –challenged
- Ova S/N
ovalbumin-sensitized and not challenged
- PAFS
periodic acid fluorescent Schiff
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: none.
References
- 1.Abramson MJ, Walters J, Walters EH. Adverse effects of beta-agonists: are they clinically relevant? American journal of respiratory medicine: drugs, devices, and other interventions. 2003;2(4):287–97. doi: 10.1007/BF03256657. [DOI] [PubMed] [Google Scholar]
- 2.Johnston SL, Edwards MR. Mechanisms of adverse effects of {beta}-agonists in asthma. Thorax. 2009;64(9):739–41. doi: 10.1136/thx.2009.119230. [DOI] [PubMed] [Google Scholar]
- 3.Shore SA, Drazen JM. Beta-agonists and asthma: too much of a good thing? The Journal of clinical investigation. 2003;112(4):495–7. doi: 10.1172/JCI19642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM, S.S. Group The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest. 2006;129(1):15–26. doi: 10.1378/chest.129.1.15. [DOI] [PubMed] [Google Scholar]
- 5.Salpeter SR, Buckley NS, Ormiston TM, Salpeter EE. Meta-analysis: effect of long-acting beta-agonists on severe asthma exacerbations and asthma-related deaths. Annals of internal medicine. 2006;144(12):904–12. doi: 10.7326/0003-4819-144-12-200606200-00126. [DOI] [PubMed] [Google Scholar]
- 6.Xia Y, Kelton CM, Xue L, Guo JJ, Bian B, Wigle PR. Safety of long-acting beta agonists and inhaled corticosteroids in children and adolescents with asthma. Therapeutic advances in drug safety. 2013;4(6):254–63. doi: 10.1177/2042098613504124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thanawala VJ, Forkuo GS, Al-Sawalha N, Azzegagh Z, Nguyen LP, Eriksen JL, Tuvim MJ, Lowder TW, Dickey BF, Knoll BJ, Walker JK, Bond RA. beta2-Adrenoceptor agonists are required for development of the asthma phenotype in a murine model. American journal of respiratory cell and molecular biology. 2013;48(2):220–9. doi: 10.1165/rcmb.2012-0364OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forkuo GS, Kim H, Thanawala VJ, Al-Sawalha N, Valdez D, Joshi R, Parra S, Pera T, Gonnella PA, Knoll BJ, Walker JK, Penn RB, Bond RA. Phosphodiesterase 4 Inhibitors Attenuate the Asthma Phenotype Produced by beta2-Adrenoceptor Agonists in Phenylethanolamine N-Methyltransferase-Knockout Mice. American journal of respiratory cell and molecular biology. 2016;55(2):234–42. doi: 10.1165/rcmb.2015-0373OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thanawala VJ, Valdez DJ, Joshi R, Forkuo GS, Parra S, Knoll BJ, Bouvier M, Leff P, Bond RA. beta-Blockers have differential effects on the murine asthma phenotype. British journal of pharmacology. 2015;172(20):4833–46. doi: 10.1111/bph.13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanania NA, Singh S, El-Wali R, Flashner M, Franklin AE, Garner WJ, Dickey BF, Parra S, Ruoss S, Shardonofsky F, O’Connor BJ, Page C, Bond RA. The safety and effects of the beta-blocker, nadolol, in mild asthma: an open-label pilot study. Pulmonary pharmacology & therapeutics. 2008;21(1):134–41. doi: 10.1016/j.pupt.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanania NA, Mannava B, Franklin AE, Lipworth BJ, Williamson PA, Garner WJ, Dickey BF, Bond RA. Response to salbutamol in patients with mild asthma treated with nadolol. The European respiratory journal. 2010;36(4):963–5. doi: 10.1183/09031936.00003210. [DOI] [PubMed] [Google Scholar]
- 12.Short PM, Williamson PA, Anderson WJ, Lipworth BJ. Randomized placebo-controlled trial to evaluate chronic dosing effects of propranolol in asthma. American journal of respiratory and critical care medicine. 2013;187(12):1308–14. doi: 10.1164/rccm.201212-2206OC. [DOI] [PubMed] [Google Scholar]
- 13.Galandrin S, Bouvier M. Distinct signaling profiles of beta1 and beta2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Molecular pharmacology. 2006;70(5):1575–84. doi: 10.1124/mol.106.026716. [DOI] [PubMed] [Google Scholar]
- 14.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(42):16657–62. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Westhuizen ET, Breton B, Christopoulos A, Bouvier M. Quantification of ligand bias for clinically relevant beta2-adrenergic receptor ligands: implications for drug taxonomy. Molecular pharmacology. 2014;85(3):492–509. doi: 10.1124/mol.113.088880. [DOI] [PubMed] [Google Scholar]
- 16.Duan W, Chan JH, Wong CH, Leung BP, Wong WS. Anti-inflammatory effects of mitogen-activated protein kinase kinase inhibitor U0126 in an asthma mouse model. Journal of immunology. 2004;172(11):7053–9. doi: 10.4049/jimmunol.172.11.7053. [DOI] [PubMed] [Google Scholar]
- 17.Liu W, Liang Q, Balzar S, Wenzel S, Gorska M, Alam R. Cell-specific activation profile of extracellular signal-regulated kinase 1/2, Jun N-terminal kinase, and p38 mitogen-activated protein kinases in asthmatic airways. The Journal of allergy and clinical immunology. 2008;121(4):893–902 e2. doi: 10.1016/j.jaci.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Liu W, Tundwal K, Liang Q, Goplen N, Rozario S, Quayum N, Gorska M, Wenzel S, Balzar S, Alam R. Establishment of extracellular signal-regulated kinase 1/2 bistability and sustained activation through Sprouty 2 and its relevance for epithelial function. Molecular and cellular biology. 2010;30(7):1783–99. doi: 10.1128/MCB.01003-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevenson CS, Birrell MA. Moving towards a new generation of animal models for asthma and COPD with improved clinical relevance. Pharmacology & therapeutics. 2011;130(2):93–105. doi: 10.1016/j.pharmthera.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Swirski FK, Sajic D, Robbins CS, Gajewska BU, Jordana M, Stampfli MR. Chronic exposure to innocuous antigen in sensitized mice leads to suppressed airway eosinophilia that is reversed by granulocyte macrophage colony-stimulating factor. Journal of immunology. 2002;169(7):3499–506. doi: 10.4049/jimmunol.169.7.3499. [DOI] [PubMed] [Google Scholar]
- 21.Willart MA, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, Hammad H. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. The Journal of experimental medicine. 2012;209(8):1505–17. doi: 10.1084/jem.20112691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cates EC, Fattouh R, Wattie J, Inman MD, Goncharova S, Coyle AJ, Gutierrez-Ramos JC, Jordana M. Intranasal exposure of mice to house dust mite elicits allergic airway inflammation via a GM-CSF-mediated mechanism. Journal of immunology. 2004;173(10):6384–92. doi: 10.4049/jimmunol.173.10.6384. [DOI] [PubMed] [Google Scholar]
- 23.Post S, Nawijn MC, Hackett TL, Baranowska M, Gras R, van Oosterhout AJ, Heijink IH. The composition of house dust mite is critical for mucosal barrier dysfunction and allergic sensitisation. Thorax. 2012;67(6):488–95. doi: 10.1136/thoraxjnl-2011-200606. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen LP, Omoluabi O, Parra S, Frieske JM, Clement C, Ammar-Aouchiche Z, Ho SB, Ehre C, Kesimer M, Knoll BJ, Tuvim MJ, Dickey BF, Bond RA. Chronic exposure to beta-blockers attenuates inflammation and mucin content in a murine asthma model. American journal of respiratory cell and molecular biology. 2008;38(3):256–62. doi: 10.1165/rcmb.2007-0279RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim V, Kelemen SE, Abuel-Haija M, Gaughan JP, Sharafkaneh A, Evans CM, Dickey BF, Solomides CC, Rogers TJ, Criner GJ. Small airway mucous metaplasia and inflammation in chronic obstructive pulmonary disease. Copd. 2008;5(6):329–38. doi: 10.1080/15412550802522445. [DOI] [PubMed] [Google Scholar]
- 26.Piccotti L, Dickey BF, Evans CM. Assessment of intracellular mucin content in vivo. Methods in molecular biology. 2012;842:279–95. doi: 10.1007/978-1-61779-513-8_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gregory LG, Causton B, Murdoch JR, Mathie SA, O’Donnell V, Thomas CP, Priest FM, Quint DJ, Lloyd CM. Inhaled house dust mite induces pulmonary T helper 2 cytokine production. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 2009;39(10):1597–610. doi: 10.1111/j.1365-2222.2009.03302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson JR, Wiley RE, Fattouh R, Swirski FK, Gajewska BU, Coyle AJ, Gutierrez-Ramos JC, Ellis R, Inman MD, Jordana M. Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. American journal of respiratory and critical care medicine. 2004;169(3):378–85. doi: 10.1164/rccm.200308-1094OC. [DOI] [PubMed] [Google Scholar]
- 29.Piyadasa H, Altieri A, Basu S, Schwartz J, Halayko AJ, Mookherjee N. Biosignature for airway inflammation in a house dust mite-challenged murine model of allergic asthma. Biology open. 2016;5(2):112–21. doi: 10.1242/bio.014464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Callaerts-Vegh Z, Evans KL, Dudekula N, Cuba D, Knoll BJ, Callaerts PF, Giles H, Shardonofsky FR, Bond RA. Effects of acute and chronic administration of beta-adrenoceptor ligands on airway function in a murine model of asthma. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(14):4948–53. doi: 10.1073/pnas.0400452101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson JR, Pacitto SR, Wong J, Archer EW, Eirefelt S, Miller-Larsson A, Jordana M. Combined budesonide/formoterol therapy in conjunction with allergen avoidance ameliorates house dust mite-induced airway remodeling and dysfunction. American journal of physiology Lung cellular and molecular physiology. 2008;295(5):L780–8. doi: 10.1152/ajplung.90229.2008. [DOI] [PubMed] [Google Scholar]
- 32.Short PM, Anderson WJ, Williamson PA, Lipworth BJ. Effects of intravenous and oral beta-blockade in persistent asthmatics controlled on inhaled corticosteroids. Heart. 2014;100(3):219–23. doi: 10.1136/heartjnl-2013-304769. [DOI] [PubMed] [Google Scholar]
- 33.Anderson WJ, Short PM, Williamson PA, Manoharan A, Lipworth BJ. The inverse agonist propranolol confers no corticosteroid-sparing activity in mild-to-moderate persistent asthma. Clinical science. 2014;127(11):635–43. doi: 10.1042/CS20140249. [DOI] [PubMed] [Google Scholar]
- 34.Kazani S, Israel E. What doesn’t kill may not make you stronger. beta-blockers for asthma. American journal of respiratory and critical care medicine. 2013;187(12):1281. doi: 10.1164/rccm.201305-0815ED. [DOI] [PubMed] [Google Scholar]
- 35.Bond RA. The intrinsic bias of generalizations. American journal of respiratory and critical care medicine. 2014;189(3):359. doi: 10.1164/rccm.201306-1109LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen LP, Lin R, Parra S, Omoluabi O, Hanania NA, Tuvim MJ, Knoll BJ, Dickey BF, Bond RA. Beta2-adrenoceptor signaling is required for the development of an asthma phenotype in a murine model. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(7):2435–40. doi: 10.1073/pnas.0810902106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clarke WP, Bond RA. The elusive nature of intrinsic efficacy. Trends in pharmacological sciences. 1998;19(7):270–6. doi: 10.1016/s0165-6147(97)01138-3. [DOI] [PubMed] [Google Scholar]
- 38.Kenakin T. Biased agonism. F1000 biology reports. 2009;1:87. doi: 10.3410/B1-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kenakin T. Functional selectivity and biased receptor signaling. The Journal of pharmacology and experimental therapeutics. 2011;336(2):296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- 40.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. The Journal of biological chemistry. 2006;281(2):1261–73. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Representative western blot and (B) quantification of the phospho-ERK/ERK density ratio in HDM challenged mice evaluated on day 12 and 4 weeks later on day 37; and HDM re-challenged mice evaluated on day 42 with or without nadolol treatment in comparison to saline control mice. Data are mean ± SEM from 5 mice in each group. * represents significance at p<0.05 compared to saline control mice.
(A and B) The graphs represent total cells and eosinophil infiltration in BALF collected from Balb/c mice subjected to the ovalbumin model of asthma. (A) Total cell count and (B) eosinophil count in BALF of mice sensitized and challenged with ovalbumin (Ova S/C) in comparison to mice sensitized but not challenged with ovalbumin (Ova S/N) and naïve control mice. Data are mean ± SEM from 4–5 mice in each group. * represents significance at p<0.05 compared to naïve control mice. # represents significance at p<0.05 compared to Ova S/N mice.
Airway sections from mice were stained with periodic acid fluorescent schiff’s stain (PAFS) for mucin (red) content in airway epithelia (green). (A) Mucin stained images from Balb/c mice subjected to the ovalbumin model of asthma. (B) Morphometric quantification of mucin volume density in mice sensitized and challenged with ovalbumin (Ova S/C) in comparison to mice sensitized but not challenged with ovalbumin (Ova S/N) and naïve control mice. Data are mean ± SEM from 4–5 mice in each group. * represents significance at p<0.05 compared to naïve control mice. # represents significance at p<0.05 compared to Ova S/N mice.
The graphs represent the levels of inflammatory cytokines in BALF collected from Balb/c mice subjected to HDM challenge in the ‘prophylactic’ model. Concentration of (A) IL-13, (B) INF-γ and (C) IL-17 in BALF from HDM challenged mice treated with vehicle, carvedilol or nadolol in comparison to saline control mice. Data are mean ± SEM from 5–6 mice in each group.
The graphs represent the levels of inflammatory cells (A and B) and chemokines (C and D) in BALF collected from Balb/c mice subjected to HDM challenge in the ‘prophylactic’ model. Cellular counts of (A) eosinophils and (B) neutrophils, and the concentrations of (C) EOTAXIN and (D) CXCL1/KC in BALF from HDM challenged mice treated with vehicle, carvedilol or nadolol in comparison to saline control mice. Data are mean ± SEM from 5–6 mice in each group. * represents significance at p<0.05 compared to respective saline control mice. # represents significance at p<0.05 compared to respective nadolol treated mice.