

Abstract
Circular RNAs (circRNAs) have recently emerged as a large class of novel non-coding RNA species. However, the detailed functional significance of the vast majority of them remains to be elucidated. Most functional characterization studies targeting circRNAs have been limited to resting cells, leaving their role in dynamic cellular responses to stimuli largely unexplored. In this study, we focus on the LPS-induced cytoplasmic circRNA, mcircRasGEF1B, and combine targeted mcircRasGEF1B depletion with high-throughput transcriptomic analysis to gain insight into its function during the cellular response to LPS stimulation. We show that knockdown of mcircRasGEF1B results in altered expression of a wide array of genes. Pathway analysis revealed an overall enrichment of genes involved in cell cycle progression, mitotic division, active metabolism, and of particular interest, NF-κB, LPS signaling pathways, and macrophage activation. These findings expand the set of functionally characterized circRNAs and support the regulatory role of mcircRasGEF1B in immune response during macrophage activation and protection against microbial infections.
Introduction
Circular RNAs (circRNAs) are a unique class of endogenous noncoding RNAs formed by the backsplicing of linear transcripts into a covalently closed circular molecule. Although some circRNAs were initially identified decades ago, they were long considered to be mere alternative splicing by-products of little biological importance1,2. However, primarily thanks to the advent of high-throughput sequencing technologies over the last decade, this perception has now been changed by multiple reports showing that a large number of circRNAs are generated by thousands of loci in the human, mouse, and other genomes, in cell-type specific manner3–6, and that some of these circRNAs are in fact functional. Several algorithms have been developed for the identification of circRNAs to elucidate their functions7–9.
The functions of circRNAs appear to be mostly manifested via post-transcriptional regulatory mechanism. CircRNAs can play the role of miRNA sponges3,5,10–15, by sequestering miRNAs through base pair complementarity, thus keeping them away from their mRNA targets. This is the mechanism of action that has received the most attention so far. However, the potential role of circRNAs as miRNA sponges seems to be limited by the fact that most of them have very few binding sites for specific miRNAs, and indeed, additional mechanisms of circRNA action have been discovered more recently. In addition to serving as miRNA sponges, circRNAs can also play the role of RNA-binding protein decoys, they can regulate alternative splicing, and finally, some may have direct effects on transcription16,17. Recent evidence also suggests the translation ability of circRNA via splicing-dependent, cap-independent manner18.
Despite these advances, the number of functionally characterized circRNAs remains very low in relative to the total number of circRNAs that are generated in mammalian cells, and their mechanisms of action are far from being fully elucidated. One neglected aspect of circRNA biology is the role that circRNAs might play in the regulation of dynamic cellular responses to external stimuli, as most previous studies have focused their efforts on studying circRNAs in resting cells.
Lipopolysaccharide (LPS) is a major component of the outer membrane of Gram-negative bacteria. Upon binding of LPS to the toll-like receptor 4 (TLR4), macrophages are activated leading to a myriad of biological responses that results in the induction of innate immune response along with the release of different immunomodulating molecules including tumour necrosis factor-alpha (TNFα), interleukin 1 (IL-1), IL-619,20, macrophage inflammatory proteins (MIP) and IP1021. The release of these cytokines and chemokines is important for pro-apoptotic activity of the activated macrophage and attracting neutrophils, natural killer cells and activated T-cells for host-cell defense21.
Previously, we identified a circRNA, mcircRasGEF1B, the expression of which is induced upon lipopolysaccharide (LPS) stimulation and which appears to regulate ICAM-1 transcript stability and therefore its protein levels in the mouse macrophages22. LPS stimulation, through the TLR4/LPS pathway, triggers the activation of a large orchestrated transcriptional network involving a wide array of NF-κB responsive genes, and is thus an ideal system to address in details the significance of circRNA species such as mcircRasGEF1B in cellular responses to external stimuli.
To this end, we depleted mcircRasGEF1B in mouse macrophages and studied the effects of its knockdown on transcriptome dynamics during LPS response using RNA sequencing (RNA-seq). Analysis of the RNA-seq data revealed that depletion of mcircRasGEF1B results in the misregulation of hundreds of genes, enriched for functional categories involved in cell cycle progression, mitotic division, metabolic activity, NF-κB and LPS signaling pathways, and macrophage activation. These findings confirm that mcircRasGEF1B plays a functional role in the process of cellular response to LPS stimulation.
Results and Discussion
Characterizing the effects of mcircRasGEF1B knockdown on the transcriptome during LPS stimulation
Previous study demonstrated that knocking down mcircRasGEF1B reduces transcript and protein levels of the LPS-induced ICAM-1 gene by destabilizing its mature mRNA products. However, the question of to what extend mcircRasGEF1B is an important regulator of the inflammatory network remains open. To address this question, we characterized the genome-wide gene expression dynamics upon activation of the TLR4/LPS pathway in control and mcircRasGEF1B-deficient backgrounds.
To determine how knockdown of mcircRasGEF1B alters the transcriptomic profile of murine macrophage upon LPS stimulation, we knocked down the expression of mcircRasGEF1B in RAW264.7 cells using two different antisense oligonucleotides (ASOs), ASO I and II, both of them targeting the back-splice junction unique to mcircRasGEF1B (Fig. 1). A sense-strand version of ASO I was used as a control22. We observed robust knockdown effciency, with ASO I reducing mcircRasGEF1B levels by 76% and ASO II depleting mcircRasGEF1B by 85% (Supplementary Fig. S1a). In agreement with our previous findings, we also observed reduction of ICAM-1 expression in both ASO I, and ASO II-treated cells (Supplementary Fig. S1b).
Figure 1.
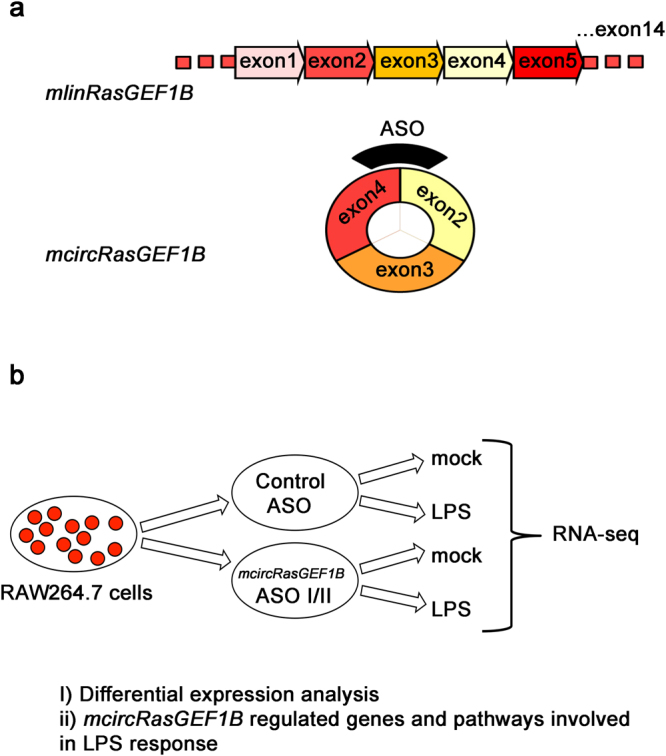
Characterization of the role of mcircRasGEF1B in LPS response. (a) mcircRasGEF1B is produced by the RasGEF1B locus in mouse through backsplicing. Antisense oligos (ASO) were designed specifically targeting the backsplice junction for the purpose of depleting mcircRasGEF1B. (b) RAW264.7 cells were treated with the mcircRasGEF1B targeting ASOI and ASOII oligos as well as with a control oligo, then subjected to LPS treatment (n = 3). Gene expression changes were then characterized at the global level using RNA-seq, thus identifying the genes and pathways that appear to be regulated by mcircRasGEF1B.
We collected total RNA from three biological replicates of RAW264.7 cells of all three (control, ASO I and ASO II) groups, with and without LPS stimulation, and carried out RNA-seq experiments after rRNA removal. After mapping reads to the genome (Supplementary Table S1), quantification at the gene level, and extraction of read counts for each gene, genes differentially expressed upon LPS stimulation in each group, and genes differentially expressed between control and mcircRasGEF1B-knockdown cells were identified using DESeq223 (see the Methods section for details).
Gene expression changes during LPS response
After correcting for multiple hypothesis testing and setting an adjusted p-value threshold of 0.05, we identified 2,714 upregulated, and 2,590 downregulated genes in control cells after LPS stimulation (Fig. 2a and d). In the ASO I-transfected cells, 3,155 and 2,754 genes were upregulated and downregulated, respectively (Fig. 2b and d), while 2,352 and 2,115 genes were upregulated and downregulated in ASO II-treated cells (Fig. 2c and d). To confirm the specificity of the observed transcriptional response, we examined the top upregulated genes upon LPS stimulation in control cells, and found that most are immune-related genes such as IL23a, CXCL10, CCL5, IL6, IL1B, and IFNB1 (Supplementary Fig. S2). We also compared the LPS-responsive genes between control, ASO I-, and ASO II-treated cells, and observed that 1,793 upegulated and 1,522 downregulated genes were common to all three conditions (Fig. 2e). However, a number of genes were only up or downregulated in control or in control and ASO I-treated cells, with ASO II-treated cells exhibiting the fewest LPS-responsive genes. As noted above and in Supplementary Fig. S1, ASO I decreased mcircRasGEF1B levels by 76% compared to the 85% knockdown observed in ASO II treatments. These observations are therefore consistent with the greater depletion of mcircRasGEF1B in ASO II-treated cells affecting LPS response to a greater extent than the more moderate knockdown in ASO I-treated cells.
Figure 2.

Gene expression changes upon LPS stimulation. (a–c) Scatter plots show gene expression changes relative to resting cells after LPS treatment in (a) control cells, (b) ASO I-treated cells; and (c) ASO II-treated cells. (d) Number of differentially expressed genes upon LPS stimulation in all conditions. (e) Overlap between the sets of differentially expressed genes upon LPS treatment in control, ASO I- and ASO II- treated cells.
Gene expression changes upon mcircRasGEF1B depletion
To directly examine the role of mcircRasGEF1B in the cellular response to TLR4/LPS pathway activation, we compared differentially expressed genes between ASO I-treated, ASO II-treated and control cells upon LPS stimulation (Fig. 3). We observed 558 upregulated and 409 downregulated genes after LPS stimulation in ASO I-treated cells relative to control cells (Fig. 3a and c). The transcriptome profiles of ASO II-treated cells were considerably more different, with 1,916 upregulated and 1,870 downregulated genes (Fig. 3b and c), again consistent with the higher efficiency of ASO II-mediated mcircRasGEF1B knockdown. We also compared the LPS-responsive genes between ASO I- and ASO-II-treated cells, and observed that 166 upregulated and 262 downregulated genes were common to both conditions (Fig. 3d). These results show that perturbation of mcircRasGEF1B affects the transcriptional or post-transcriptional regulation of hundreds to thousands of genes in response to LPS stimulation.
Figure 3.

Gene expression changes upon mcircRasGEF1B depletion. (a and b) Scatter plots show gene expression changes in (a) LPS-stimulated ASOI-treated cells; (b) LPS-stimulated ASO II-treated cells; relative to LPS-stimulated control cells. (c) Number of differentially expressed genes in ASOI- or ASO II-treated, and LPS-stimulated cells. (d) Overlap between differentially expressed genes in ASO I- or ASO II-treated, and LPS-stimulated cells.
Genes affected by mcircRasGEF1B depletion are enriched for functional categories related to LPS response
In order to understand the biological roles of genes misregulated upon mcircRasGEF1B depletion, we identified significantly enriched (p ≤ 0.05 after correcting for multiple hypothesis testing) gene ontology (GO) functional categories of genes in the sets of genes up- and downregulated relative to control in LPS-stimulated ASO-treated cells (Fig. 4; Supplementary Tables S2–S5). We focused on genes up- and downregulated in the ASO II background due to the higher magnitude of the effect of ASO II on the macrophage transcriptome profile. The GO analysis revealed that genes upregulated in mcircRasGEF1B knockdown cells are enriched for categories involved in metabolic activity, autophagy, DNA replication and mitotic division, macrophage activation too, and immune response, specifically the regulation of IκB/NFκB signaling and the LPS response pathway. The set of downregulated genes also revealed a number of coherent functional categories, specifically genes involved in chromatin remodeling, RNA splicing, cell adhesion, as well as, common with upregulated genes, mitochondrial respiratory function and macrophage activation. Detailed examination of the lists of downregulated genes corroborates these global observations. For example, among the top downregulated genes was IFNB1, a member of the type I interferons, which play key roles in the defense against viral infections and in the innate immune responses to pathogens; production of IFNB1 is dependent on the LPS-induced TRIF-dependent pathway24. The LPS-mediated activation of RAW264.7 cells is known to be associated with the regulation of cell cycle progression25, and the NF-κB and TLR4/LPS signaling pathways are the mechanism through which LPS response is mediated, thus the observations of global misregulation of genes involved in these pathways underscore the functional importance of mcircRasGEF1B during LPS response.
Figure 4.

Functional categories enriched among differentially expressed genes in ASO II- and LPS-stimulated cells relative to control LPS-stimulated cells. Representative enriched functional categories are shown for (a) downregulated genes; (b) upregulated genes, with the x-axis indicating the statistical significance of the observed enrichment.
Conclusion
In this study, we identified a broad spectrum of genes involved in the cellular response to LPS activation whose proper expression dynamics is dependent on the LPS-inducible cytoplasmic circular RNA mcircRasGEF1B. We knocked down mcircRasGEF1B and studied the effects of its depletion on the transcriptome in resting and LPS-stimulated cells.
We observed that depletion of mcircRasGEF1B leads to the misregulation of a plethora of genes, among them functional modules involved in cell cycle progression, macrophage activation and LPS response signaling, cell adhesion and metabolic activity. Normal levels of mcircRasGEF1B thus appear to be important for the proper progression of macrophage activation and LPS signaling, and mcircRasGEF1B likely plays an important role in the process, confirming its functional significance. Further experiments should reveal in detail the precise mechanisms through which mcircRasGEF1B exercises its function.
Materials and Methods
Cell culture and reagents
RAW264.7 cells were cultured in Rosewell Park Memorial Institute medium (RPMI), supplemented with 10% FBS, 20 U/ml penicillin and 100 µg/ml streptomycin (GIBCO). LPS was purchased from Sigma.
ASOs transfection
ASOs (Supplementary Table S6) were synthesized by IDT technologies. ASOs (20 nM) were transfected into RAW264.7 cells with the X-tremeGENE HP DNA (Roche) according to the manufacturer’s protocol. To maximize knockdown efficiency, ASO transfection was repeated 24 hours after the initial transfection. Cells were then treated with LPS for 2 hours.
Quantitative RT-PCR
Total RNA was isolated with the Thermo Scientific GeneJET RNA Purification Kit. Complementary DNAs were synthesized and quantitative RT-PCR was performed with 2X SYBR Green PCR Master mix (Thermo Scientific) and run on a Bio-Rad Connect Real-Time PCR System. Expression levels of circular RNAs described in this study were measured by qPCR using gene specific divergent primers (Supplementary Table S6). The relative expression levels of circular versus linear isoforms were normalized to the L32 gene.
RNA extraction, library preparation, and sequencing
Total RNA was isolated with the Thermo Scientific GeneJET RNA Purification Kit. The RNA samples were checked for quality using Bio-Analyzer 2100 (Agilent Technologies, San Diego, CA, USA) and Qubit RNA assay kit. 1.5 μg of total RNA from each sample was used to prepare the library using ScriptSeq Complete Kit (Epicentre Inc, Madison, WI, USA) according to the manufacturer’s protocol.
RNA-seq data processing and analysis
Except where otherwise indicated, all analysis were carried out using custom-written Python scripts.
Paired-end (2 × 75 bp) RNA-seq reads were aligned against the mm9 version of the mouse genome using TopHat226 (version 2.0.8, run with Bowtie27 version 0.12.9) and the Ensembl 66 annotation with the following parameters:–no-discordant–no-mixed–read-realign-edit-dist 0–read-edit-dist 4–read-mismatches 4–min-segment-intron 10–min-coverage-intron 10. Read mapping statistics can be found in the Supplementary Table S1. Raw sequencing reads are available from the Gene Expression Omnibus under GEO accession number GSE99811.
Gene-level quantification in FPKM (Fragments Per Kilobase per Million mapped fragments) units was carried out using Cufflink28 (version 2.0.2).
For differential expression analysis, sequencing counts at the gene level were obtained using HTSeq29 (version 0.6.1p1). DESeq223 was then used to identify differential expressed genes between different conditions. We note that one of the three replicates of unstimulated ASO II treated cells exhibited a globally discordant transcriptomic profile, and was accordingly excluded from the differential expression analysis.
Statistically enriched functional categories of genes were identified using FuncAssociate 2.030.
Electronic supplementary material
Acknowledgements
This work was supported by the University of Malaya through High Impact Research Grant (HIR) [UM.C/625/1/HIR/MOHE/CHAN/14/2]; and the Post Graduate Research Grant [PG055-2014B]. The RNA-seq processing and analysis described in the paper is based upon work supported by the National Science Foundation under Grant No. CNS-0521433.
Author Contributions
W.L.N., performed the experiments. G.K.M., performed bioinformatics analysis. W.L.N., Y.-Y.L., and C.-K.E. designed experiments. W.L.N., G.K.M., Y.-M.C., Y.-Y.L., and C.-K.E. analyzed the data. W.L.N., G.K.M., Y.-Y.L., and C.-K.E. wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-12550-w.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yat-Yuen Lim, Email: yatyuen.lim@um.edu.my.
Chee-Kwee Ea, Email: Chee-Kwee.Ea@utsouthwestern.edu.
References
- 1.Nigro JM, et al. Scrambled exons. Cell. 1991;64:607–613. doi: 10.1016/0092-8674(91)90244-S. [DOI] [PubMed] [Google Scholar]
- 2.Cocquerelle C, Mascrez B, Hetuin D, Bailleul B. Mis-splicing yields circular RNA molecules. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1993;7:155–160. doi: 10.1096/fasebj.7.1.7678559. [DOI] [PubMed] [Google Scholar]
- 3.Hansen TB, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 4.Jeck WR, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. Rna. 2013;19:141–157. doi: 10.1261/rna.035667.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Memczak S, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 6.Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PloS one. 2012;7:e30733. doi: 10.1371/journal.pone.0030733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang XO, et al. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome research. 2016;26:1277–1287. doi: 10.1101/gr.202895.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome biology. 2015;16:4. doi: 10.1186/s13059-014-0571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song X, et al. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic acids research. 2016;44:e87. doi: 10.1093/nar/gkw075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li F, et al. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/beta-catenin pathway. Oncotarget. 2015;6:6001–6013. doi: 10.18632/oncotarget.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang, K. et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. European heart journal, doi:10.1093/eurheartj/ehv713 (2016). [DOI] [PubMed]
- 12.Qu S, et al. Circular RNA: A new star of noncoding RNAs. Cancer letters. 2015;365:141–148. doi: 10.1016/j.canlet.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 13.Wu, Y., Zhang, Y., Zhang, Y. & Wang, J. J. CircRNA hsa_circ_0005105 up regulates NAMPT expression and promotes chondrocyte extracellular matrix degradation by sponging miR-26a. Cell biology international, doi:10.1002/cbin.10761 (2017). [DOI] [PubMed]
- 14.Tang CM, et al. CircRNA_000203 enhances the expression of fibrosis-associated genes by derepressing targets of miR-26b-5p, Col1a2 and CTGF, in cardiac fibroblasts. Scientific reports. 2017;7:40342. doi: 10.1038/srep40342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng L, et al. Circular RNA ZNF609 functions as a competitive endogenous RNA to regulate AKT3 expression by sponging miR-150-5p in Hirschsprung’s disease. Oncotarget. 2017;8:808–818. doi: 10.18632/oncotarget.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nature structural & molecular biology. 2015;22:256–264. doi: 10.1038/nsmb.2959. [DOI] [PubMed] [Google Scholar]
- 17.Ashwal-Fluss R, et al. circRNA biogenesis competes with pre-mRNA splicing. Molecular cell. 2014;56:55–66. doi: 10.1016/j.molcel.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 18.Legnini I, et al. Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Molecular cell. 2017;66(22–37):e29. doi: 10.1016/j.molcel.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luster MI, Germolec DR, Yoshida T, Kayama F, Thompson M. Hepatology. 1994. Endotoxin-Induced Cytokine Gene-Expression and Excretion in the Liver; pp. 480–488. [PubMed] [Google Scholar]
- 20.West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annual review of cell and developmental biology. 2006;22:409–437. doi: 10.1146/annurev.cellbio.21.122303.115827. [DOI] [PubMed] [Google Scholar]
- 21.Luster AD. The role of chemokines in linking innate and adaptive immunity. Current opinion in immunology. 2002;14:129–135. doi: 10.1016/S0952-7915(01)00308-9. [DOI] [PubMed] [Google Scholar]
- 22.Ng WL, et al. Inducible RasGEF1B circular RNA is a positive regulator of ICAM-1 in the TLR4/LPS pathway. RNA biology. 2016;13:861–871. doi: 10.1080/15476286.2016.1207036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toshchakov V, et al. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nature immunology. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 25.Zhuang JC, Wogan GN. Growth and viability of macrophages continuously stimulated to produce nitric oxide. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:11875–11880. doi: 10.1073/pnas.94.22.11875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berriz GF, Beaver JE, Cenik C, Tasan M, Roth FP. Next generation software for functional trend analysis. Bioinformatics. 2009;25:3043–3044. doi: 10.1093/bioinformatics/btp498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.