

Most research on the gut microbiome in colon cancer focuses on taxonomic changes at the genus level using 16S rRNA gene sequencing. Here, we develop a new methodology to integrate DNA and RNA data sets to examine functional shifts at the species level that are important to tumor development. We uncover several metabolic pathways in the microbiome that, when perturbed by host genetics and H. hepaticus inoculation, contribute to colon cancer. The work presented here lays a foundation for improved bioinformatics methodologies to closely examine the cross talk between specific organisms and the host, important for the development of diagnostics and pre/probiotic treatment.
KEYWORDS: Helicobacter hepaticus, Smad3, bioinformatics, butyrate, colon cancer, gut inflammation, gut microbiome, host-pathogen interactions, metagenomics, metatranscriptomics, polyamines
ABSTRACT
Colorectal cancer (CRC) is one of the most treatable cancers, with a 5-year survival rate of ~64%, yet over 50,000 deaths occur yearly in the United States. In 15% of cases, deficiency in mismatch repair leads to null mutations in transforming growth factor β (TGF-β) type II receptor, yet genotype alone is not responsible for tumorigenesis. Previous work in mice shows that disruptions in TGF-β signaling combined with Helicobacter hepaticus cause tumorigenesis, indicating a synergistic effect between genotype and microbial environment. Here, we examine functional shifts in the gut microbiome in CRC using integrated -omics approaches to untangle the role of host genotype, inflammation, and microbial ecology. We profile the gut microbiome of 40 mice with/without deficiency in TGF-β signaling from a Smad3 (mothers against decapentaplegic homolog-3) knockout and with/without inoculation with H. hepaticus. Clear functional differences in the microbiome tied to specific bacterial species emerge from four pathways related to human colon cancer: lipopolysaccharide (LPS) production, polyamine synthesis, butyrate metabolism, and oxidative phosphorylation (OXPHOS). Specifically, an increase in Mucispirillum schaedleri drives LPS production, which is associated with an inflammatory response. We observe a commensurate decrease in butyrate production from Lachnospiraceae bacterium A4, which could promote tumor formation. H. hepaticus causes an increase in OXPHOS that may increase DNA-damaging free radicals. Finally, multiple bacterial species increase polyamines that are associated with colon cancer, implicating not just diet but also the microbiome in polyamine levels. These insights into cross talk between the microbiome, host genotype, and inflammation could promote the development of diagnostics and therapies for CRC.
IMPORTANCE Most research on the gut microbiome in colon cancer focuses on taxonomic changes at the genus level using 16S rRNA gene sequencing. Here, we develop a new methodology to integrate DNA and RNA data sets to examine functional shifts at the species level that are important to tumor development. We uncover several metabolic pathways in the microbiome that, when perturbed by host genetics and H. hepaticus inoculation, contribute to colon cancer. The work presented here lays a foundation for improved bioinformatics methodologies to closely examine the cross talk between specific organisms and the host, important for the development of diagnostics and pre/probiotic treatment.
Author Video: An author video summary of this article is available.
INTRODUCTION
In recent years, colorectal cancer (CRC) ranks as the third most deadly cancer with approximately ~50,000 deaths in the United States alone (1). Chronic intestinal inflammation plays a key role in CRC development, given that patients with inflammatory bowel disease (IBD), ulcerative colitis (UC), or Crohn’s disease (CD) have an increased risk of CRC (2–5). IBD-associated colorectal carcinogenesis is characterized by a sequence of inflammation > dysplasia > carcinoma (reviewed in reference 3). Transforming growth factor β (TGF-β) signaling is one of the key pathways altered in IBD-associated CRC (6–8). TGF-βs are multifunctional cytokines important in diverse biological processes, including development, differentiation, and immune regulation (reviewed in reference 9), yet it is unclear how these processes are involved in colon tumor suppression.
The human TGF-β type II receptor gene (TGFBR2) is one of the most frequently mutated genes in IBD-CRCs (10, 11). Previous studies in human CRC cell lines and tumors show that frameshift mutations in the poly(A)10 microsatellite region of TGFBR2 (10–13) result in the loss of TGFβR2 protein production and functional TGF-β signaling (14, 15). Like sporadic and hereditary nonpolyposis colorectal cancer (Lynch syndrome), IBD-CRCs with microsatellite instability have a higher frequency (57 to 76%) of mutations in TGFBR2. Consequently, mutations in TGFBR2 in dysplastic tissues that result in loss of TGF-β signaling play a role in the development of CRC (11).
Mutations in mothers against decapentaplegic (SMAD) genes at rates of 3.4% (SMAD2), 4.3% (SMAD3), and 8.6% (SMAD4) in sporadic CRC tumors also disrupt TGF-β signaling (16). SMAD2/3 are receptor activated and bind to SMAD4 (co-SMAD) to form a transcriptional complex. Mutations in SMAD3 can downregulate SMAD3 transcription. In inflamed intestinal mucosa samples from patients with active CD and UC, elevated production of SMAD7 (17) inhibits TGFβR1 kinase-mediated phosphorylation of SMAD3 protein and disrupts TGF-β signaling (18). Taken together, mutations in TGFBR2 or SMAD2/3/4 or elevated SMAD7 levels disrupt TGF-β signaling and accelerate CRC pathogenesis. Understanding how signaling pathways are perturbed in the absence of TGF-β signaling can offer novel strategies to decrease the incidence of CRC in IBD patients with TGF-β signaling deficiency. To investigate TGF-β signaling as it relates to colon cancer, several mouse models have been developed (8, 19–23).
Of the TGF-β-signaling-deficient colon cancer mouse models, the immunocompetent Smad3 knockout (Smad3−/−) mouse is the model of choice (23) because the Tgfb1−/− Rag2−/− model is immunodeficient (20), the TGFβR2-deficient mouse is embryonic lethal (24), and the intestine-specific Tgfbr2 knockout mouse must be combined with another colon tumor suppressor (8). In both the Tgfb1−/− Rag2−/− and Smad3−/− models, colon cancer develops only in conjunction with the presence of gut microbial Helicobacter species (21, 22). Interestingly, the potent inflammation-inducing agent dextran sodium sulfate (DSS) in the absence of Helicobacter does not induce colon cancer in the Tgfb1−/− Rag2−/− model (21) and induces only a few late-onset tumors in the Smad3−/− model (19). This suggests that inflammation alone in the absence of SMAD3 is not sufficient for tumor development. Consequently, the contribution of Helicobacter to tumor development in this model consists of more than just adding inflammatory stress to the colon. Aside from Helicobacter, there have been several species identified that are shown to be causative or correlative in the development of colon cancer (Table 1) (25–38). However, no study to date has investigated the functions affected by microbial ecology as a whole.
TABLE 1 .
Bacterial species associated with colon cancera
| Name(s) | Relationship | Reference(s) |
|---|---|---|
| Citrobacter rodentium | Min mice inoculated with this species had a 4-fold increase in colonic tumors. Found only in mice. | 25 |
| Enterococcus faecalis | Produces superoxide and hydrogen peroxide, both of which can damage DNA in epithelial cells | 26, 27 |
| Clostridium cluster XVIa (Clostridium scindens, C. hiranonis, and C. hylemonae) and Clostridium cluster XI (C. sordellii) | Can produce secondary bile acids, such as deoxycholic acid (DOC), that increase tumor burden in wild-type male B6.129PF2/J mice | 28, 29 |
| Acidovorax species | Associated with increased risk for colon cancer and may act as a pathogen by increased metabolism of nitroaromatic compounds | 30 |
| Enterotoxigenic Bacteroides fragilis | Produces a toxin that caused colitis and tumors in multiple intestinal neoplasia (MinApc716+/−) mice through an IL-17-dependent pathway | 31 |
| Streptococcus gallolyticus | Present in approximately 20 to 50% of colon tumors compared to less than 5% of normal tissue in CRC patients. Patients with high counts of S. gallolyticus have increased expression of proinflammatory cytokines, including interleukin-1 (IL-1) and cyclooxygenase-2 (COX-2). | 32 |
| Escherichia coli NC101 | Produces genotoxic colibactin that led to increased tumor burden in AOM/Il10−/− mice | 33 |
| Fusobacterium nucleatum | Induces hyperproliferation of human colon cancer cell lines through binding of its FadA adhesin to E-cadherin. Also, promotes immune evasion of tumors through binding of its Fap2 adhesin to TIGIT receptors on immune cells. | 34, 35, 37, 38 |
| Akkermansia muciniphila | Mucin-degrading species were found in 4-fold-larger amounts in CRC colons than in normal colons | 36 |
Species names, details of association, and references are listed in chronological order.
Additionally, most metagenomic studies of gut bacteria in colon cancer use 16S rRNA sequencing, which provides only an estimation of taxonomic composition to the genus level. As shown in Table 1, there are certain species that combine with genetic backgrounds to produce colon cancer. Because of this complexity, it is necessary to study gut microbial ecology at the species level and to identify functions causing or preventing CRC.
We hypothesize that a deficiency in TGF-β signaling through loss of SMAD3, combined with the presence of Helicobacter hepaticus, alters microbial ecology, leading to functional dysbiosis and colon cancer. To address this, we analyze the mouse gut microbiome in Smad3+/+ and Smad3−/− mice in the presence and absence of H. hepaticus using a novel approach of integrating metagenomics and metatranscriptomics.
Here, we report several novel findings related to the microbiome. First, we have identified the species, Lachnospiraceae bacterium A4, which has decreased RNA counts of butyrate kinase. The family Lachnospiraceae has previously been associated with a possible anti-inflammatory role (39–41), but the modality of that role has yet to be elucidated. Our results suggest that this species could be modulating inflammation via butyrate production. The second novelty is the change in RNA counts of polyamine genes arising from several bacterial species. Previous research shows changes in levels of polyamines to be associated with colon cancer (42, 43), but changes to prokaryotic polyamine genes in colon cancer have never been reported. Third, we observe increased RNA counts of lipopolysaccharide (LPS) genes in Mucispirillum schaedleri. Although it was previously just associated with inflammation (44–47), our results suggest that M. schaedleri is increasing inflammation through increased LPS production. Finally, H. hepaticus itself has increased RNA counts of genes involved in oxidative phosphorylation (OXPHOS). This suggests that besides its production of known inflammatory toxins, H. hepaticus exerts an oncogenic effect through oxidative damage.
RESULTS
Study overview.
Cecal samples from 40 mice were pooled into four comparison groups: (i) Smad3+/+/H. hepaticus negative (wild type; S+H−), (ii) Smad3−/−/H. hepaticus negative (Smad3−/− only; S−H−), (iii) Smad3+/+/H. hepaticus positive (H. hepaticus only; S+H+), and (iv) Smad3−/−/H. hepaticus positive (combined; S−H+). Figure 1A summarizes each comparison group where cecal samples from 10 mice were pooled per group (1:1 male/female ratio). Wild-type (S+H−), H. hepaticus-only (S+H+), and Smad3−/−-only (S−H−) mice show little histologically evident inflammation and no cancer or precancerous hyperplastic lesions. However, mice with the combined Smad3−/− and H. hepaticus inoculation (S−H+) show significant inflammation, and 40% of mice (2 males and 2 females) develop tumors in the cecum and proximal colon by 6 months of age. One animal had a large cecal tumor and was 9 months of age. The literature shows that, in animals with various combinations of Helicobacter species, Smad3−/− mice developed tumors with 22 to 66% penetrance over a 30-week period (22). Figure 1B shows a flow chart of bioinformatics methods. After quality control (QC) and filtering for host, 316 million reads remain, of which 120 million align to known bacterial genomes in the PATRIC database (48). After filtering based on positive and negative controls, 20 million reads align to 1,944 bacteria with high confidence, representing our “gold standard” genomes (details in Materials and Methods). Overall, ~60% of RNA reads (63 million of 106 million RNA reads) map to these gold standard genomes. Thus, although the DNA mapping to the gold standard genomes represents only ~7% of the total data set (20 million DNA reads of 316 million map to gold standard genomes), these species are functionally dominant based on RNA read recruitment.
FIG 1 .

Materials and methods flow chart. (A1) Four groups of mice were used for DNA/RNA extraction: a mouse that was Smad3+/+ and H. hepaticus negative (S+H−) (top left), a mouse that was Smad3−/− and H. hepaticus negative (S−H−) (top right), a mouse that was Smad3+/+ and H. hepaticus positive (S+H+) (bottom left), and a mouse that was Smad3−/− and H. hepaticus positive (S−H+) (bottom right, three panels from one histological section). These three panels display the types of lesions found in the S−H+ mice: hyperplasia, adenoma, and mucinous carcinoma. Inflammatory infiltrates are indicated by arrowheads. Four out of 10 mice in the S−H+ group had tumors at the time of sacrifice. The three double-headed red arrows indicate the Smad3−/−, H. hepaticus, and combined effects. (A2) Dissection of mouse large intestine from the S−H+ group showing tumor locations in dotted red lines and site of cecal matter removal. (B1) Flow chart showing number of reads at each step of DNA analysis. Reads were aligned to PATRIC (48) with Taxoner64 (95). The last step of the DNA analysis feeds into the second step of the RNA analysis. (B2) Flow chart showing number of reads at each step of RNA analysis. Reads were aligned with Bowtie2 (97) and quantified/normalized with cuffquant/cuffnorm, part of the Cufflinks suite of tools (99).
Functional shifts in the microbiome.
To guide our functional analysis, we examine the top 37 pathways (Fig. 2) ranked by overall estimated RNA counts, where each pathway contains total RNA counts above the pathway mean count (all 140 pathway counts are available at https://doi.org/10.6084/m9.figshare.5328700). Then, we examine pathways that either have previous links to CRC or have the greatest changes in RNA counts among groups. Previous literature shows that butyrate, polyamine, and OXPHOS pathways are important in colorectal cancer and/or inflammation (49–51), and they rank 8th, 9th, and 18th, respectively, in these 37 pathways (LPS is 73rd but is included because of its known role in inflammation [reviewed in reference 52]). In each case, we examine pathways gene by gene to highlight the greatest changes in RNA counts and potential points of enzymatic flux changes. Additionally, we focus on significant changes to RNA counts in genes by sample type using the following comparisons: S−H− versus S+H−, S+H+ versus S+H−, and S−H+ versus S+H− (here referred to as Smad3−/− effect, H. hepaticus effect, and combined effect, respectively). The combined effect represents the contributions of both the loss of SMAD3 and H. hepaticus inoculation. Results are split into two sections: first, pathways that are changed by the Smad3−/− and combined but not H. hepaticus effects, and second, pathways that are changed by the H. hepaticus and combined but not Smad3−/− effects.
FIG 2 .

Sum of estimated expression by KEGG pathway. Bubble chart of RNA count differences between sample groups among various KEGG pathways (102). The scale at bottom shows estimated RNA counts as outputted by cuffquant and cuffnorm (part of the Bowtie2/Cufflinks suite of tools). Labels at top show the sample groups. The heat map behind the bubbles shows log2 fold changes (log2 FC) of pathways for each of the effects. Fold changes are mapped to a blue-yellow color spectrum with bright yellow having the greatest increase in RNA counts and bright blue having the greatest decrease in RNA counts under the given condition versus control. Color behind the control bubbles represents no change and is provided for reference.
Smad3−/− and combined effects on bacterial pathways. (i) Lachnospiraceae bacterium A4 is responsible for decreased RNA counts of butyrate kinase.
In the normal colon, butyrate is a primary energy source for colon mucosal epithelial cell growth through oxidative rather than glucose metabolism (53, 54). In colon tumors, however, aerobic glycolysis of glucose is the primary source of energy, causing butyrate to accumulate in the nucleus, where it becomes a histone deacetylase (HDAC) inhibitor (55–58) under Warburg conditions (59). HDAC inhibition promotes cell cycle arrest and apoptosis through p21 expression (60) and inhibits NF-κB activation by decreasing the proteasome activity responsible for IκB degradation (61). In addition, butyrate also increases T-cell regulation (62). Butyrate, therefore, has both antitumor and anti-inflammatory activities, making it an effective anti-UC therapy (63). It seems reasonable, then, that in the context of colon cancer decreased colonic levels of butyrate would promote cancer cell growth and stimulate inflammation.
In the butyrate metabolism pathway, we see a decrease in RNA counts for butyrate kinase (buk) with a log2 fold chance (FC) of −1.1 in the combined effect (Fig. 3; also see https://doi.org/10.6084/m9.figshare.5047477 and https://doi.org/10.6084/m9.figshare.5325136). This enzyme is important because it is the last step in the production of butyrate. This decrease in buk may be a genotypic effect, given that the Smad3−/− effect shows a similar decrease. In contrast, the H. hepaticus effect shows a slight increase. Interestingly, the main bacterial species whose decrease in abundance contributes to changes in butyrate kinase is Lachnospiraceae bacterium A4, a relatively understudied species in the realm of bacterial butyrate producers. Other contributors include members of the Lachnospiraceae family, Lachnospiraceae bacterium 10-1 and Lachnospiraceae bacterium 28-4. Along with decreased RNA counts of buk, we see a decrease in the abundance of the Lachnospiraceae family and the Firmicutes phylum (Fig. 4; see also https://doi.org/10.6084/m9.figshare.5051722) but a surprising increase in the population of Lachnospiraceae bacterium A4 (2.92-fold) in the combined effect. However, despite the increase in population of Lachnospiraceae bacterium A4, the RNA counts show a decrease in buk expression by this bacterium, suggesting a downregulation of buk. Changes to the abundance of bacteria in the Lachnospiraceae family and Firmicutes phylum represent an avenue where host genotype may contribute more to microbial ecology than inoculation with H. hepaticus.
FIG 3 .

Species contribution to genes in butanoate (butyrate) metabolism. RNA count changes for buk (butyrate kinase) (EC 2.7.2.7), categorized by species/strain contribution. The y axis shows RNA counts, and the x axis shows sample groups. Species with majority contributions to count bars are numbered for clarity. Bottom right shows partial pathway with a red box to highlight gene. Partial pathway adapted from KEGG pathway database (102).
FIG 4 .

Taxonomic changes at phylum, family, and species levels. Changes in fractions of total bacterial counts at different taxonomic levels for the S+H− (Control), S−H− (Smad3−/−), S+H+ (H. hepaticus only), and S−H+ (Combined) groups. (A) Changes for the phyla of Bacteroidetes, Deferribacteres, Firmicutes, and Proteobacteria. (B) Changes for the families of Bacteroidaceae, Deferribacteraceae, Lachnospiraceae, Lactobacillaceae, and Helicobacteraceae. (C) Changes for the species of Mucispirillum schaedleri, Lachnospiraceae bacterium A4, Lactobacillus plantarum, Lactobacillus murinus, Parabacteroides distasonis, and Helicobacter hepaticus.
(ii) Multiple species have increased RNA counts for genes producing putrescine and spermidine.
Putrescine is known to be required for early development, as a lack of it causes cell apoptosis and prenatal death in mice (64). Spermidine was shown to be required for posttranslational modification of eukaryotic initiation factor 5A (eIF5A), which is required for growth in a range of species (65, 66). Large amounts of polyamines, thought to be derived from diets high in red meat (67), are associated with severity of colorectal cancer (43). Also, when patients are in remission, their polyamine levels decrease (43). While there have been numerous studies on the link between polyamines and colorectal cancer in eukaryotic cells, no study to date has shown a prokaryotic contribution.
Two genes that have higher RNA counts in the arginine and proline pathway are N-carbamoylputrescine amidase (aguB) and carboxynorspermidine decarboxylase (nspC), by a log2 FC of 2.07 and 2.24, respectively, in the combined effect (Fig. 5; see also https://doi.org/10.6084/m9.figshare.5051728 and https://doi.org/10.6084/m9.figshare.5325151). aguB and nspC are genes for enzymes responsible for one of the reactions that produce the polyamines putrescine and spermidine, respectively. Similarly to buk, these genes have different RNA counts in the Smad3−/− and combined effects but little difference in the H. hepaticus effect.
FIG 5 .

Species contribution to genes in arginine and proline metabolism. RNA count changes for aguB (N-carbamoylputrescine amidase) (EC 3.5.1.53) and nspC (carboxynorspermidine decarboxylase) (EC 4.1.1.96), categorized by species/strain contribution. The y axis shows RNA counts, and the x axis shows sample groups. Species with majority contributions to count bars are numbered for clarity. Bottom right shows partial pathway with a red box to highlight genes. Partial pathway adapted from KEGG pathway database (102).
For aguB, in the combined group (S−H+), the species Marinomonas mediterranea MMB-1, Desulfotomaculum ruminis DSM 2154, and Bacteroides uniformis dnLKV2 are responsible for the majority of the expression. For nspC, in the combined group, a majority of the RNA counts are represented by a diverse group of species from the genera Bacteroides, Clostridium, Ruminococcus, and Alistipes. Parabacteroides distasonis has a small contribution to the RNA counts but has a dramatic change in abundance. An interesting point is that there is no single dominant species that is responsible for the upregulation of these genes.
In terms of taxonomic shifts, P. distasonis has nearly a 10-fold increase in the combined effect. P. distasonis has been previously associated with inflammation in a DSS mouse model of colitis (68). The family Bacteroidaceae, of which Bacteroides uniformis is a member, is increased by 1.89-fold in the Smad3−/− effect and 1.20-fold in the H. hepaticus effect (Fig. 4; see also https://doi.org/10.6084/m9.figshare.5051722). In the combined effect, we see a synergistic effect in Bacteroidaceae with a 2.26-fold increase. We see similar fold changes at the phylum level of Bacteroidetes.
H. hepaticus and combined effects on bacterial pathways. (i) M. schaedleri, a core member of the mouse gut bacteria, is a major contributor to increased RNA counts for LPS genes in H. hepaticus-only mice.
Gram-negative bacteria use LPSs as structural molecules to make up their outer membrane, and they are released whenever the bacteria divide or die. In addition to lipopeptides and flagellins, LPSs are known to be signaling molecules for inflammatory pathways. In particular, LPSs have been shown to activate the cluster of differentiation 14/myeloid differentiation 2/Toll-like receptor 4 (CD14/MD2/TLR4) receptor pathway (69). This ultimately leads to increased transcription of proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6). As already discussed, there are strong correlations between the presence of inflammatory conditions and the progression of colorectal cancer; thus, we examined LPS gene signatures in our data set.
Several LPS genes have increased RNA counts in the H. hepaticus and combined effects: 3-deoxy-d-manno-octulosonic-acid transferase (kdtA), UDP-3-O-(3-hydroxymyristoyl) N-acetylglucosamine deacetylase (lpxD), and UDP-3-O-(3-hydroxymyristoyl) glucosamine N-acyltransferase (lpxC). In this same order, these genes have log2 FCs of 2.1, 1.8, and 1 in the combined effect (Fig. 6; see also https://doi.org/10.6084/m9.figshare.5051734 and https://doi.org/10.6084/m9.figshare.5325154). There is little change or a decrease in the Smad3−/− effect for these genes.
FIG 6 .

Species contribution to lpxC and lpxD in LPS biosynthesis. RNA count changes for lpxC [UDP-3-O-(3-hydroxymyristoyl) N-acetylglucosamine deacetylase] (EC 3.5.1.108) and lpxD [UDP-3-O-(3-hydroxymyristoyl) glucosamine N-acyltransferase] (EC 2.3.1.191), categorized by species/strain contribution. The y axis shows RNA counts, and the x axis shows sample groups. Species with majority contributions to count bars are numbered for clarity. Bottom right shows partial pathway with a red box to highlight genes. Partial pathway adapted from KEGG pathway database (102).
In terms of species contribution, a surprising finding is that M. schaedleri ASF457 is responsible for a majority of the RNA counts of lpxC and lpxD. This is interesting because M. schaedleri is associated with inflammatory pathways (44–47), but studies have not shown which genes may be involved. Mirroring the RNA count changes, M. schaedleri abundance is increased in the H. hepaticus-only and combined effects by 1.89-fold and 1.31-fold, respectively, while there is a decrease in the Smad3−/− effect (Fig. 4; see also https://doi.org/10.6084/m9.figshare.5051722).
(ii) Increased RNA counts of LPS genes in bacteria correlate significantly with host TLR gene expression.
As mentioned above, LPSs activate inflammation via the CD14/MD2/TLR4 receptor pathway. To determine whether TLR receptor activity of Smad3−/−/H. hepaticus-positive mice could correlate with increases in bacterial LPS production, we measured the mucosal epithelial expression of these genes using real-time quantitative reverse transcription PCR (qRT-PCR). We find that Tlr2 and Tlr4 expression both significantly correlate with the increased counts of bacterial lpxC and lpxD (Fig. 7).
FIG 7 .
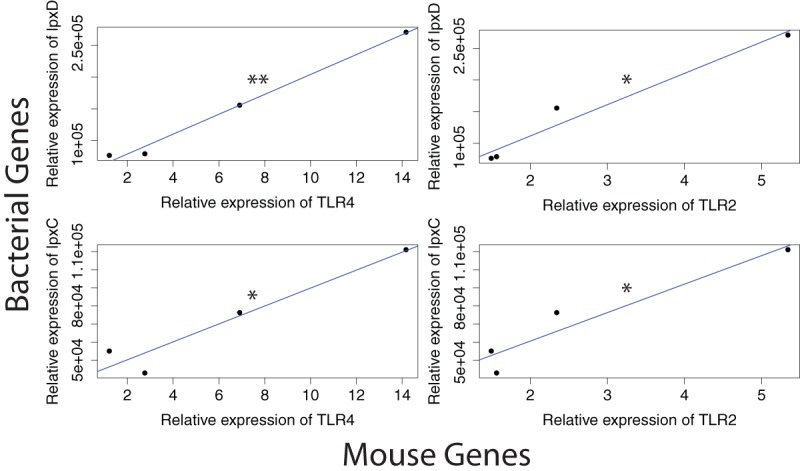
Correlation of mouse gene expression with bacterial gene expression. RNA expression correlation between mouse TLR genes and bacterial genes in the LPS pathway. Expression in arbitrary units due to normalization. Mouse gene expression based on qRT-PCR data; bacterial gene expression based on transcriptome sequencing from this study. P values: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
(iii) H. hepaticus has increased RNA counts of key genes in the OXPHOS pathway.
It has been established that cancer cells prefer aerobic glycolysis, but it has also been shown that they still contain active mitochondria to produce a portion of their ATP (70, 71). However, although oxidative phosphorylation (OXPHOS) may be taking place in the epithelial cells, less is known about the microbiome’s metabolic activities. It seems plausible that, as in cancer cells, a preference for aerobic glycolysis or OXPHOS in microbial cells will have an effect on the tumor microenvironment. OXPHOS, aside from being indicative of proliferating bacterial cells, increases the amount of reactive oxygen and nitrogen species (RONS), which are known to damage DNA, RNA, and proteins (72–74).
In our study, we see a log2 FC of 1 and 1.5 for nuo (NADH ubiquinone oxidoreductase [NADHuo]) in the combined and H. hepaticus effects, respectively (Fig. 8; see also https://doi.org/10.6084/m9.figshare.5051737 and https://doi.org/10.6084/m9.figshare.5325157). NADHuo is part of the first electron transport complex for the production of ATP. Likewise, in these same effects, we see a log2 FC of 1.3 and 1 for ppk (polyphosphate kinase), respectively. Polyphosphate kinase prepares inorganic triphosphate for feeding into ATP synthase. It is true that there are other genes in the pathway that are lowered in RNA counts, but the pathway overall has increased counts in the combined effect (Fig. 2 and https://doi.org/10.6084/m9.figshare.5328700).
FIG 8 .

Species contribution to genes in oxidative phosphorylation. RNA count changes for nuoA to nuoN (NADH ubiquinone oxidoreductase A to N) (EC 1.6.5.3) and ppk (polyphosphate kinase) (EC 2.7.4.1), categorized by species/strain contribution. The y axis shows RNA counts, and the x axis shows sample groups. Species with majority contributions to count bars are numbered for clarity. Dividing lines within species represent multiple subunits for that gene. Bottom right shows partial pathway with a red box to highlight genes. Partial pathway adapted from KEGG pathway database (102).
The species primarily responsible for the increase in oxidative phosphorylation is H. hepaticus. Not surprisingly, given that the S+H+ and S−H+ mice were inoculated with the species, we see the contribution of H. hepaticus to OXPHOS genes in those groups. There is a small contribution in the S−H− group for nuo, but this could be due to sequencing/alignment error of a closely related species.
DISCUSSION
Colon cancer is a multifactorial disease affected by host genetics, resident gut bacterial species, diet, and inflammation. In terms of host genetics, 10 to 15% of colon cancer patients have mutations in TGF-β signaling genes, but their function as tumor suppressors and interaction with gut bacteria is unclear. In mouse colon cancer models in which growth has been measured, no loss of growth control was attributable to the loss of TGF-β signaling under pretumor conditions (8, 20). However, a role for TGF-β signaling in both the differentiation and inflammatory states of colon tumors was revealed in a comparative microarray study of several mouse colon tumor models (75). Since TGF-β signaling is known to be an important regulator in immune tolerance and T-cell homeostasis (reviewed in reference 76), it is likely that its absence could function, at least in part, to exacerbate an inflammatory response in the mouse colon.
The propensity of colorectal cancer having an inflammatory component suggests that microbial dysbiosis may result from tumor-suppressing activities of TGF-β. Here, we report changes to microbial functions in a TGF-β signaling-deficient colon cancer model.
First, a key butyrate gene, buk, is found to have reduced RNA counts in the Smad3−/− and combined effects. This agrees with multiple studies that show butyrate to be crucial for proper maintenance of the colonic epithelium (54). It is well known that butyrate is the primary energy source of colonocytes; moreover, several studies have shown that in high concentrations (~5 mM), butyrate is a potent HDAC inhibitor, resulting in expression of several genes involved in cancer or inflammation: cyclin-dependent kinase inhibitor 1A (p21WAF1/Cip1), mucin 2 (MUC-2), testin LIM domain protein (TES), and hypoxia-inducible factor 1 (HIF-1) (55, 56, 58, 59). Additionally, it has been shown that it can stimulate histone acetyltransferase (HAT) activity at lower concentrations (~0.5 mM) (59). This happens when butyrate is converted to citrate in the tricarboxylic acid (TCA) cycle which combines with ATP citrate lyase (ACL) to produce acetyl coenzyme A (acetyl-CoA). This important coenzyme then acts as an acetyl group donor for various HATs. While we cannot conclude that the reduction in RNA counts of butyrate genes is a cause or effect of the proinflammatory, cancerous environment, the correlation is consistent with the literature.
In our study, Lachnospiraceae bacterium A4, a member of the Firmicutes phylum, decreases in abundance in the combined and Smad3−/− effects. This species, which has been little studied, belongs to the Lachnospiraceae family of bacteria, of which some literature suggests that it may play an anti-inflammatory role. For example, one study shows that inoculation with an isolate of Lachnospiraceae decreases disease severity of chronic Clostridium difficile infection in mice (39). Additionally, 16S rRNA studies of human fecal samples from IBD patients have revealed smaller amounts of Lachnospiraceae at the genus level (40, 41). One could easily hypothesize that butyrate supplements or probiotic Lachnospiraceae would slow cancer growth and reduce inflammation in our cancer model.
Second, we observe an upshift in RNA counts for genes involved in the production of putrescine and spermidine. Like the butyrate gene changes, this occurs in the SMAD3 and combined effects. Changes to polyamine genes are interesting because it has been known since the 1960s that polyamines are increased in rapidly proliferating tissues (77). More recently, it has been discovered that polyamines can also affect protein translation. Specifically, spermidine can be modified to the unique amino acid hypusine, which is the only known amino acid to modify the eukaryotic initiation factor EIF5A (66). This translation factor is not strictly required for translation in general, but it seems to prefer transcripts with polyproline motifs (78, 79). Additionally, it has been shown that blocking the production of hypusine or the modification of EIF5A by hypusine leads to reduced gene translation of growth promoters RhoA/ROCK1 (80). Also, increased polyamine levels in colon cancer patients correlate with severity of disease (42, 43). The novel finding here is that gut bacteria are at least possibly responsible for increased polyamines. It is plausible that the polyamines are being actively exported to the epithelial cells, as such transporters exist for both prokaryotes and eukaryotes (81–83). Further studies will need to be done to measure changes to polyamine levels as well as RNA counts of eukaryotic polyamine genes. Consequently, not just diet but also dysbiosis in the gut microbiome may result in increased polyamine uptake by colon mucosal epithelial cells.
Although not in the list of top pathways, our third focus was LPS biosynthesis because it has been known to produce an inflammatory response for more than a century (although LPSs were termed “endotoxins” before discovery of their structure) (84). Indeed, we find an increase in bacterial genes in the LPS pathway for both the H. hepaticus-only and combined effects. Intriguingly, it is not H. hepaticus itself that is responsible for the increased RNA counts, it is M. schaedleri. Though M. schaedleri has been associated with inflammation, its contribution is not known. It should be noted that this species is in the set of core bacteria given to mice in gnotobiotic models (85, 86). Our data show that inoculation with H. hepaticus correlates with an increased abundance of M. schaedleri that may result in a shift to a proinflammatory state.
In our fourth focus, we find a colon epithelial cell response consistent with increased bacterial LPS production. By qRT-PCR on colon mucosal epithelial mRNA, Tlr4 and Tlr2 are shown to be upregulated in all effects compared to control, and this significantly correlates with increased RNA counts of bacterial lpxC and lpxD. Accordingly, LPS has been shown to activate the proinflammatory NF-κB pathway through the CD14/MD2/TLR4 complex (69, 87). On the other hand, Helicobacter spp. have been shown to activate the TLR2 receptor, possibly explaining its upregulation in the mice (88).
Our fifth focus, OXPHOS, shows the most change in RNA counts. This is surprising given the normally anaerobic environment of the colon; increased rates of OXPHOS would imply an aerobic environment. Nevertheless, there are increased counts of nuo and ppk for the H. hepaticus and combined effects. Importantly, the species mainly responsible for this increase is H. hepaticus, which may be rapidly growing and depleting the environment of available oxygen. More importantly, an increase in OXPHOS points to an increase in RONS. It could be that the RONS produced by H. hepaticus and other species are causing oxidative damage to DNA, RNA, or proteins that leads to a cancerous state. Even though both SMAD3 deficiency and H. hepaticus inoculation are required for colon cancer in this model, the OXPHOS pathway is changed more by H. hepaticus than by SMAD3 deficiency.
It is important to note that Lactobacillus plantarum does not appear to be involved in altering butyrate, polyamine, LPS, or OXPHOS levels, yet it is undetectable in the mice with H. hepaticus (Fig. 4; see also https://doi.org/10.6084/m9.figshare.5051722). The dysbiotic environment produced by H. hepaticus is incompatible with L. plantarum through unknown mechanisms that may involve nutrient competition, susceptibility to toxins, or other environmental factors. Other studies have shown that adding L. plantarum reduces tumor size and burden in rats and inhibits survival of cancer stem cells (89, 90). On the other hand, Lactobacillus murinus increases in all effects compared to control. This is the first report that links L. murinus to inflammation or colon cancer.
In summary, loss of SMAD3 is associated with changes in bacterial RNA counts in the butyrate and polyamine synthesis pathways. And, with the addition of H. hepaticus, we see an increase in LPS and OXPHOS pathways’ RNA counts, suggesting an increase in the proinflammatory and free radical status of the colonic epithelium but without histological evidence of inflammation (Fig. 1A). Either of these changes alone is not sufficient to promote carcinogenesis. Rather, it takes their combination and possibly a reduction of probiotic species to reach the “tipping point” for tumorigenesis. The results of this study emphasize the multifactorial nature of colon cancer and how the microbiome may have a profound impact on the cancer microenvironment. This lends credence to the idea that changes in microbial ecology as well as in host genotype must be taken into consideration when examining the causes of colon cancer.
MATERIALS AND METHODS
Animal husbandry.
Smad3−/− mice (129/Sv) generated previously (23) were obtained from Jackson Laboratories and maintained in a specific-pathogen-free (SPF) facility under a University of Arizona IACUC protocol. Sentinel mice were routinely screened for pathogens. Homozygous Smad3−/− and Smad3+/+ mice were generated by breeding heterozygous animals.
PCR genotyping.
The genotype of newborn pups from double heterozygous mating was determined by PCR amplification of tail DNA and size fractionation on agarose gels (20).
Helicobacter culture, infection, and detection.
A pure culture of H. hepaticus was received from Craig Franklin (University of Missouri) and was suspended in brucella broth on tryptic soy agar supplemented with 5% sheep blood (Hardy Diagnostics) and incubated in a microaerophilic chamber at 37°C for 48 h. Later, the culture was resuspended in brucella broth and allowed to grow for another 48 h. Five breeding pairs of 1- to 3-month-old heterozygous Smad3+/− mice were inoculated with ~108 H. hepaticus organisms by direct introduction using a 1.5-in. feeding needle. Control animals of five breeding pairs, 1- to 3-month-old heterozygous Smad3+/− mice, were inoculated with equal amounts of brucella broth. A total of 3 inoculations for each mouse was completed at 24-h intervals. Animals were then checked for H. hepaticus infection by PCR analysis of fecal matter with H. hepaticus-specific primers as described earlier (91). Infected animals then were bred together. All animals in subsequent generations developed chronic infection by spontaneous parental/fecal contact without additional inoculation. To minimize cross contamination, uninfected and infected animals were housed in different buildings.
Tissue collection and staining.
Mice were euthanized by IACUC-approved cervical dislocation. The cecum and colon were dissected free from the mesenchyme. All tissue sections shown in the figure(s) were from the cecum, and staining was done with hematoxylin and eosin. The cecum and colon were opened longitudinally, and contents were collected according to location. Cecal content, proximal colon content, and distal colon content were placed in individual tubes and flash frozen in liquid nitrogen. All samples were stored at −80°C. Only cecal content was sent for sequencing.
DNA/RNA sequencing and quality control (including filtering).
Sequencing was done at the University of Arizona Genomics Core (UAGC). DNA was extracted using an in-house lysozyme extraction protocol. The libraries were built with Illumina TruSeq DNA kits (Illumina, San Diego, CA). RNA was ribodepleted using both eukaryotic and prokaryotic RiboMinus kits (Thermo Fisher, Waltham, MA). The RNA libraries were built with the Illumina TruSeq RNA kits. Ten mice from each group were pooled and run on two lanes using an Illumina HiSeq 2000/2500 machine. DNA/RNA reads were 2 × 100-bp paired-end reads, and the average insert size was ~325 bp for DNA and ~225 bp for RNA. After sequencing, adapter sequences were trimmed from raw data before being downloaded to the University of Arizona High-Performance Computing (UA HPC) center. Quality control (QC) was done using a custom pipeline using the programs SolexaQA++ (92) and fastx_clipper from the FASTX suite of tools (93) (https://github.com/hurwitzlab/fizkin). After QC, DNA reads were filtered for mouse and mouse chow (including yeast, barley, soy, wheat, and corn) using jellyfish (94), a kmer frequency counting tool. Filtering was done based on the assumption that reads coming from mouse or mouse chow will have similar kmer frequencies as the source genomes. Therefore, reads that had kmer modes of 2 or greater in comparison to the mouse or mouse chow were considered “rejected” and filtered from downstream analysis. Since quality control and filtering may have eliminated mate pairs, reads were reconstituted into new fastq files: two files for forward and reverse paired-end reads and two files for single-ended reads (those that lost their mates, either forward or reverse).
DNA alignment.
Alignment was done with Taxoner64 version 0.1.3 (95) against the ~30,000 bacterial and archaeal genomes in PATRIC (48) (genomes downloaded on 5 September 2015 from https://www.patricbrc.org/) using the parameters “-A --very-sensitive-local” (https://github.com/hurwitzlab/taxoner-patric). Results were then filtered with a minimum alignment score of 131 (the average alignment score for H. hepaticus, our positive control) and a minimum count of ~138 (the average count of Mycoplasma pulmonis, a pathogen that is a negative control since the facility is specific pathogen free [SPF]). A hierarchical pie chart of species composition (https://doi.org/10.6084/m9.figshare.5051722) was constructed using KronaTools (96).
RNA alignment.
Alignment was done with Bowtie2 version 2.2.6 (97) for aligning against bacterial genomes and TopHat version 2.1.1 (98) for aligning against the mouse genome (Mus_musculus GRCm38 dna_rm primary_assembly fa from Ensembl) (https://github.com/hurwitzlab/bacteria-bowtie). RNA coverage of mouse genome was ~3-fold on average (data not shown). Given this, we excluded mouse results from further analysis. Bacterial genomes were composed of the ~2,000 genomes that passed filtering criteria in the “DNA alignment” step. Additional filtering of RNA for mouse and mouse chow was not necessary due to the use of the selected bacterial genomes.
Differential gene expression.
To calculate bacterial gene expression, cuffquant was used with the parameters “-M rRNAGFF—no-length-correction,” and cuffnorm (99) was used with default parameters. No length correction was used in our study because we were interested in comparing genes across samples and not within samples (where gene length correction would be necessary [100]). Postprocessing of abundance counts and plotting/heat map generation was done with R version 3.2.2 (101) and Excel version 14.4.0 for Mac (Microsoft Corp., Redmond, WA).
Pathway mapping and species contribution.
To assign gene products to pathways, annotation information was downloaded from PATRIC (RefSeq.cds.tab files). Once each gene was annotated with pathways, sums were calculated for each gene and each pathway among species and samples. See the figure legends for more details. The bubble chart (Fig. 2) was created using custom R and perl scripts.
Isolation of RNA from tissue and cDNA synthesis (for qRT-PCR).
Total RNA from individual frozen tissue samples was isolated using TRI reagent (Molecular Research Center, Cincinnati, OH). RNA was treated with RNase-free DNase I (Qiagen, Valencia, CA) and purified using a Qiagen RNeasy minikit. RNA was reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA).
Primer design and SYBR green qRT-PCR.
qRT-PCRs were performed using a Light Cycler 480 (Roche, Basel, Switzerland) with 50 to 100 ng of cDNA template. At least one primer per pair was designed across exon-intron boundaries to prevent coamplification of genomic DNA; the sizes of the products range from 50 to 150 bp. For each gene, threshold cycle (CT) values were normalized to corresponding β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and relative expression was determined by the 2−ΔΔCT method.
Correlation of bacterial RNA count with mouse RNA count.
Since the bacterial RNA count had only a single data point for each group, median expression values were used from the mouse qRT-PCR data. Using these values, Pearson’s product-moment correlation tests were run using default parameters. The lm() command in R was used to construct linear models and plot regression lines in Fig. 7.
Data availability.
All raw DNA/RNA reads are available under BioProject number PRJNA379709.
ACKNOWLEDGMENTS
C.L.B. and T.D. designed and conducted molecular experiments. Bioinformatics and in silico experiments were conducted by B.L.H. and S.G.D. S.G.D., B.L.H., and T.D. analyzed data and wrote the manuscript.
This work was supported by the Gordon and Betty Moore Foundation (GBMF4491) to B.L.H.; a Cancer Biology training grant (CA09213) to C.L.B.; and HD026471, AI067903, and CA084291 to T.D.
We thank Kenneth Youens-Clark, Xiang Liu, James Eric Thornton, Jana U’Ren, and other members of the Hurwitz Lab for comments and suggestions on the manuscript; Constance Gard for managing the mouse colonies; the Vice President for Research of the University of Arizona for pilot funds for deep sequencing; and Ryan Sprissler of the University of Arizona Genetics Core for assistance with sequencing.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. 2016. Cancer statistics, 2016. CA Cancer J Clin 66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Eaden JA, Abrams KR, Mayberry JF. 2001. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 48:526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itzkowitz SH, Yio X. 2004. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol 287:G7–G17. doi: 10.1152/ajpgi.00079.2004. [DOI] [PubMed] [Google Scholar]
- 4.Lutgens MWMD, Vleggaar FP, Schipper MEI, Stokkers PCF, van der Woude CJ, Hommes DW, de Jong DJ, Dijkstra G, van Bodegraven AA, Oldenburg B, Samsom M. 2008. High frequency of early colorectal cancer in inflammatory bowel disease. Gut 57:1246–1251. doi: 10.1136/gut.2007.143453. [DOI] [PubMed] [Google Scholar]
- 5.Rogler G. 2014. Chronic ulcerative colitis and colorectal cancer. Cancer Lett 345:235–241. doi: 10.1016/j.canlet.2013.07.032. [DOI] [PubMed] [Google Scholar]
- 6.Rhodes JM, Campbell BJ. 2002. Inflammation and colorectal cancer: IBD-associated and sporadic cancer compared. Trends Mol Med 8:10–16. doi: 10.1016/S1471-4914(01)02194-3. [DOI] [PubMed] [Google Scholar]
- 7.Li F, Cao Y, Townsend CM Jr, Ko TC. 2005. TGF-beta signaling in colon cancer cells. World J Surg 29:306–311. doi: 10.1007/s00268-004-7813-6. [DOI] [PubMed] [Google Scholar]
- 8.Muñoz NM, Upton M, Rojas A, Washington MK, Lin L, Chytil A, Sozmen EG, Madison BB, Pozzi A, Moon RT, Moses HL, Grady WM. 2006. Transforming growth factor beta receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res 66:9837–9844. doi: 10.1158/0008-5472.CAN-06-0890. [DOI] [PubMed] [Google Scholar]
- 9.Massagué J. 2012. TGFβ signalling in context. Nat Rev Mol Cell Biol 13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Svrcek M, El-Bchiri J, Chalastanis A, Capel E, Dumont S, Buhard O, Oliveira C, Seruca R, Bossard C, Mosnier JF, Berger F, Leteurtre E, Lavergne-Slove A, Chenard MP, Hamelin R, Cosnes J, Beaugerie L, Tiret E, Duval A, Fléjou JF. 2007. Specific clinical and biological features characterize inflammatory bowel disease–associated colorectal cancers showing microsatellite instability. J Clin Oncol 25:4231–4238. doi: 10.1200/JCO.2007.10.9744. [DOI] [PubMed] [Google Scholar]
- 11.Fujiwara I, Yashiro M, Kubo N, Maeda K, Hirakawa K. 2008. Ulcerative colitis-associated colorectal cancer is frequently associated with the microsatellite instability pathway. Dis Colon Rectum 51:1387–1394. doi: 10.1007/s10350-008-9212-9. [DOI] [PubMed] [Google Scholar]
- 12.Schulmann K, Mori Y, Croog V, Yin J, Olaru A, Sterian A, Sato F, Wang S, Xu Y, Deacu E, Berki AT, Hamilton JP, Kan T, Abraham JM, Schmiegel W, Harpaz N, Meltzer SJ. 2005. Molecular phenotype of inflammatory bowel disease-associated neoplasms with microsatellite instability. Gastroenterology 129:74–85. doi: 10.1053/j.gastro.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 13.Svrcek M, Cosnes J, Beaugerie L, Parc R, Bennis M, Tiret E, Fléjou JF. 2007. Colorectal neoplasia in Crohn’s colitis: a retrospective comparative study with ulcerative colitis. Histopathology 50:574–583. doi: 10.1111/j.1365-2559.2007.02663.x. [DOI] [PubMed] [Google Scholar]
- 14.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B. 1995. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 15.Souza RF, Garrigue-Antar L, Lei J, Yin J, Appel R, Vellucci VF, Zou TT, Zhou X, Wang S, Rhyu MG, Cymes K, Chan O, Park WS, Krasna MJ, Greenwald BD, Cottrell J, Abraham JM, Simms L, Leggett B, Young J, Harpaz N, Reiss M, Meltzer SJ. 1996. Alterations of transforming growth factor-beta 1 receptor type II occur in ulcerative colitis-associated carcinomas, sporadic colorectal neoplasms, and esophageal carcinomas, but not in gastric neoplasms. Hum Cell 9:229–236. [PubMed] [Google Scholar]
- 16.Fleming NI, Jorissen RN, Mouradov D, Christie M, Sakthianandeswaren A, Palmieri M, Day F, Li S, Tsui C, Lipton L, Desai J, Jones IT, McLaughlin S, Ward RL, Hawkins NJ, Ruszkiewicz AR, Moore J, Zhu HJ, Mariadason JM, Burgess AW, Busam D, Zhao Q, Strausberg RL, Gibbs P, Sieber OM. 2013. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res 73:725–735. doi: 10.1158/0008-5472.CAN-12-2706. [DOI] [PubMed] [Google Scholar]
- 17.Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. 2001. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J Clin Invest 108:601–609. doi: 10.1172/JCI12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA Jr, Wrana JL, Falb D. 1997. The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 89:1165–1173. doi: 10.1016/S0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 19.Seamons A, Treuting PM, Brabb T, Maggio-Price L. 2013. Characterization of dextran sodium sulfate-induced inflammation and colonic tumorigenesis in Smad3(−/−) mice with dysregulated TGFβ. PLoS One 8:e79182. doi: 10.1371/journal.pone.0079182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. 1999. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res 59:3379–3386. [PubMed] [Google Scholar]
- 21.Engle SJ, Ormsby I, Pawlowski S, Boivin GP, Croft J, Balish E, Doetschman T. 2002. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res 62:6362–6366. [PubMed] [Google Scholar]
- 22.Maggio-Price L, Treuting P, Zeng W, Tsang M, Bielefeldt-Ohmann H, Iritani BM. 2006. Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res 66:828–838. doi: 10.1158/0008-5472.CAN-05-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Richardson JA, Parada LF, Graff JM. 1998. Smad3 mutant mice develop metastatic colorectal cancer. Cell 94:703–714. doi: 10.1016/S0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
- 24.Oshima M, Oshima H, Taketo MM. 1996. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol 179:297–302. doi: 10.1006/dbio.1996.0259. [DOI] [PubMed] [Google Scholar]
- 25.Newman JV, Kosaka T, Sheppard BJ, Fox JG, Schauer DB. 2001. Bacterial infection promotes colon tumorigenesis in Apc Min/+ mice. J Infect Dis 184:227–230. doi: 10.1086/321998. [DOI] [PubMed] [Google Scholar]
- 26.Huycke MM, Abrams V, Moore DR. 2002. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis 23:529–536. doi: 10.1093/carcin/23.3.529. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Huycke MM. 2007. Extracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology 132:551–561. doi: 10.1053/j.gastro.2006.11.040. [DOI] [PubMed] [Google Scholar]
- 28.Ridlon JM, Kang DJ, Hylemon PB. 2006. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, Bernstein H. 2011. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol 85:863–871. doi: 10.1007/s00204-011-0648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanapareddy N, Legge RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, Randall TA, Galanko J, Benson A, Sandler RS, Rawls JF, Abdo Z, Fodor AA, Keku TO. 2012. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J 6:1858–1868. doi: 10.1038/ismej.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, Housseau F, Pardoll DM, Sears CL. 2009. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdulamir AS, Hafidh RR, Bakar FA. 2010. Molecular detection, quantification, and isolation of Streptococcus gallolyticus bacteria colonizing colorectal tumors: inflammation-driven potential of carcinogenesis via IL-1, COX-2, and IL-8. Mol Cancer 9:249. doi: 10.1186/1476-4598-9-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C. 2012. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, El-Omar EM, Brenner D, Fuchs CS, Meyerson M, Garrett WS. 2013. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14:207–215. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. 2013. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 14:195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. 2013. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS One 8:e70803. doi: 10.1371/journal.pone.0070803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, Enk J, Bar-On Y, Stanietsky-Kaynan N, Coppenhagen-Glazer S, Shussman N, Almogy G, Cuapio A, Hofer E, Mevorach D, Tabib A, Ortenberg R, Markel G, Miklić K, Jonjic S, Brennan CA, Garrett WS, Bachrach G, Mandelboim O. 2015. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42:344–355. doi: 10.1016/j.immuni.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abed J, Emgård JEM, Zamir G, Faroja M, Almogy G, Grenov A, Sol A, Naor R, Pikarsky E, Atlan KA, Mellul A, Chaushu S, Manson AL, Earl AM, Ou N, Brennan CA, Garrett WS, Bachrach G. 2016. Fap2 mediates Fusobacterium nucleatum colorectal adenocarcinoma enrichment by binding to tumor-expressed Gal-GalNAc. Cell Host Microbe 20:215–225. doi: 10.1016/j.chom.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reeves AE, Koenigsknecht MJ, Bergin IL, Young VB. 2012. Suppression of Clostridium difficile in the gastrointestinal tracts of germfree mice inoculated with a murine isolate from the family Lachnospiraceae. Infect Immun 80:3786–3794. doi: 10.1128/IAI.00647-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. 2007. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, Roca J, Dore J. 2006. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dreyfuss F, Chayen R, Dreyfuss G, Dvir R, Ratan J. 1975. Polyamine excretion in the urine of cancer patients. Isr J Med Sci 11:785–795. [PubMed] [Google Scholar]
- 43.Löser C, Fölsch UR, Paprotny C, Creutzfeldt W. 1990. Polyamines in colorectal cancer. Evaluation of polyamine concentrations in the colon tissue, serum, and urine of 50 patients with colorectal cancer. Cancer 65:958–966. doi:. [DOI] [PubMed] [Google Scholar]
- 44.Hoffmann C, Hill DA, Minkah N, Kirn T, Troy A, Artis D, Bushman F. 2009. Community-wide response of the gut microbiota to enteropathogenic Citrobacter rodentium infection revealed by deep sequencing. Infect Immun 77:4668–4678. doi: 10.1128/IAI.00493-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berry D, Schwab C, Milinovich G, Reichert J, Ben Mahfoudh K, Decker T, Engel M, Hai B, Hainzl E, Heider S, Kenner L, Müller M, Rauch I, Strobl B, Wagner M, Schleper C, Urich T, Loy A. 2012. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J 6:2091–2106. doi: 10.1038/ismej.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El Aidy S, Derrien M, Aardema R, Hooiveld G, Richards SE, Dane A, Dekker J, Vreeken R, Levenez F, Doré J, Zoetendal EG, van Baarlen P, Kleerebezem M. 2014. Transient inflammatory-like state and microbial dysbiosis are pivotal in establishment of mucosal homeostasis during colonisation of germ-free mice. Benef Microbes 5:67–77. doi: 10.3920/BM2013.0018. [DOI] [PubMed] [Google Scholar]
- 47.Rooks MG, Veiga P, Wardwell-Scott LH, Tickle T, Segata N, Michaud M, Gallini CA, Beal C, van Hylckama-Vlieg JET, Ballal SA, Morgan XC, Glickman JN, Gevers D, Huttenhower C, Garrett WS. 2014. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J 8:1403–1417. doi: 10.1038/ismej.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, Gillespie JJ, Gough R, Hix D, Kenyon R, Machi D, Mao C, Nordberg EK, Olson R, Overbeek R, Pusch GD, Shukla M, Schulman J, Stevens RL, Sullivan DE, Vonstein V, Warren A, Will R, Wilson MJC, Yoo HS, Zhang C, Zhang Y, Sobral BW. 2014. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res 42:D581–D591. doi: 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Donohoe DR, Holley D, Collins LB, Montgomery SA, Whitmore AC, Hillhouse A, Curry KP, Renner SW, Greenwalt A, Ryan EP, Godfrey V, Heise MT, Threadgill DS, Han A, Swenberg JA, Threadgill DW, Bultman SJ. 2014. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov 4:1387–1397. doi: 10.1158/2159-8290.CD-14-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerner EW, Meyskens FL Jr, Goldschmid S, Lance P, Pelot D. 2007. Rationale for, and design of, a clinical trial targeting polyamine metabolism for colon cancer chemoprevention. Amino Acids 33:189–195. doi: 10.1007/s00726-007-0515-2. [DOI] [PubMed] [Google Scholar]
- 51.Wiseman H, Halliwell B. 1996. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J 313:17–29. doi: 10.1042/bj3130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller SI, Ernst RK, Bader MW. 2005. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol 3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 53.Ahmad MS, Krishnan S, Ramakrishna BS, Mathan M, Pulimood AB, Murthy SN. 2000. Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut 46:493–499. doi: 10.1136/gut.46.4.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. 2008. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther 27:104–119. doi: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- 55.Nakano K, Mizuno T, Sowa Y, Orita T, Yoshino T, Okuyama Y, Fujita T, Ohtani-Fujita N, Matsukawa Y, Tokino T, Yamagishi H, Oka T, Nomura H, Sakai T. 1997. Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53-negative human colon cancer cell line. J Biol Chem 272:22199–22206. doi: 10.1074/jbc.272.35.22199. [DOI] [PubMed] [Google Scholar]
- 56.Augenlicht L, Shi L, Mariadason J, Laboisse C, Velcich A. 2003. Repression of MUC2 gene expression by butyrate, a physiological regulator of intestinal cell maturation. Oncogene 22:4983–4992. doi: 10.1038/sj.onc.1206521. [DOI] [PubMed] [Google Scholar]
- 57.Takai N, Desmond JC, Kumagai T, Gui D, Said JW, Whittaker S, Miyakawa I, Koeffler HP. 2004. Histone deacetylase inhibitors have a profound antigrowth activity in endometrial cancer cells. Clin Cancer Res 10:1141–1149. doi: 10.1158/1078-0432.CCR-03-0100. [DOI] [PubMed] [Google Scholar]
- 58.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, Weir TL, Ehrentraut SF, Pickel C, Kuhn KA, Lanis JM, Nguyen V, Taylor CT, Colgan SP. 2015. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17:662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. 2012. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell 48:612–626. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Archer SY, Meng S, Shei A, Hodin RA. 1998. P21WAF1 is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc Natl Acad Sci U S A 95:6791–6796. doi: 10.1073/pnas.95.12.6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Place RF, Noonan EJ, Giardina C. 2005. HDAC inhibition prevents NF-κB activation by suppressing proteasome activity: down-regulation of proteasome subunit expression stabilizes IκBα. Biochem Pharmacol 70:394–406. doi: 10.1016/j.bcp.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 62.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY. 2013. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scheppach W, Sommer H, Kirchner T, Paganelli GM, Bartram P, Christl S, Richter F, Dusel G, Kasper H. 1992. Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology 103:51–56. doi: 10.1016/0016-5085(92)91094-K. [DOI] [PubMed] [Google Scholar]
- 64.Pendeville H, Carpino N, Marine JC, Takahashi Y, Muller M, Martial JA, Cleveland JL. 2001. The ornithine decarboxylase gene is essential for cell survival during early murine development. Mol Cell Biol 21:6549–6558. doi: 10.1128/MCB.21.19.6549-6558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Childs AC, Mehta DJ, Gerner EW. 2003. Polyamine-dependent gene expression. Cell Mol Life Sci 60:1394–1406. doi: 10.1007/s00018-003-2332-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park MH, Nishimura K, Zanelli CF, Valentini SR. 2010. Functional significance of eIF5A and its hypusine modification in eukaryotes. Amino Acids 38:491–500. doi: 10.1007/s00726-009-0408-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kalač P. 2006. Biologically active polyamines in beef, pork and meat products: a review. Meat Sci 73:1–11. doi: 10.1016/j.meatsci.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 68.Kverka M, Zakostelska Z, Klimesova K, Sokol D, Hudcovic T, Hrncir T, Rossmann P, Mrazek J, Kopecny J, Verdu EF, Tlaskalova-Hogenova H. 2011. Oral administration of Parabacteroides distasonis antigens attenuates experimental murine colitis through modulation of immunity and microbiota composition. Clin Exp Immunol 163:250–259. doi: 10.1111/j.1365-2249.2010.04286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mancuso G, Midiri A, Biondo C, Beninati C, Gambuzza M, Macrì D, Bellantoni A, Weintraub A, Espevik T, Teti G. 2005. Bacteroides fragilis-derived lipopolysaccharide produces cell activation and lethal toxicity via toll-like receptor 4. Infect Immun 73:5620–5627. doi: 10.1128/IAI.73.9.5620-5627.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ward PS, Thompson CB. 2012. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R. 2014. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 16:992–1003. doi: 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Newcomb TG, Loeb LA. 1998. Oxidative DNA damage and mutagenesis, p 65–84. In Nickoloff JA, Hoekstra MF (ed), DNA damage and repair. Humana Press, Totowa, NJ. [Google Scholar]
- 73.Cabiscol E, Tamarit J, Ros J. 2000. Oxidative stress in bacteria and protein damage by reactive oxygen species. Int Microbiol 3:3–8. [PubMed] [Google Scholar]
- 74.Liu M, Gong X, Alluri RK, Wu J, Sablo T, Li Z. 2012. Characterization of RNA damage under oxidative stress in Escherichia coli. Biol Chem 393:123–132. doi: 10.1515/hsz-2011-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaiser S, Park YK, Franklin JL, Halberg RB, Yu M, Jessen WJ, Freudenberg J, Chen X, Haigis K, Jegga AG, Kong S, Sakthivel B, Xu H, Reichling T, Azhar M, Boivin GP, Roberts RB, Bissahoyo AC, Gonzales F, Bloom GC, Eschrich S, Carter SL, Aronow JE, Kleimeyer J, Kleimeyer M, Ramaswamy V, Settle SH, Boone B, Levy S, Graff JM, Doetschman T, Groden J, Dove WF, Threadgill DW, Yeatman TJ, Coffey RJ Jr, Aronow BJ. 2007. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol 8:R131. doi: 10.1186/gb-2007-8-7-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bommireddy R, Doetschman T. 2007. TGFβ1 and T reg cells: alliance for tolerance. Trends Mol Med 13:492–501. doi: 10.1016/j.molmed.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Russell DH, Snyder SH. 1969. Amine synthesis in regenerating rat liver: extremely rapid turnover of ornithine decarboxylase. Mol Pharmacol 5:253–262. [PubMed] [Google Scholar]
- 78.Gutierrez E, Shin BS, Woolstenhulme CJ, Kim JR, Saini P, Buskirk AR, Dever TE. 2013. eIF5A promotes translation of polyproline motifs. Mol Cell 51:35–45. doi: 10.1016/j.molcel.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, Rodnina MV. 2013. EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 339:85–88. doi: 10.1126/science.1229017. [DOI] [PubMed] [Google Scholar]
- 80.Fujimura K, Choi S, Wyse M, Strnadel J, Wright T, Klemke R. 2015. Eukaryotic translation initiation factor 5A (EIF5A) regulates pancreatic cancer metastasis by modulating RhoA and Rho-associated kinase (ROCK) protein expression levels. J Biol Chem 290:29907–29919. doi: 10.1074/jbc.M115.687418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sakata K, Kashiwagi K, Igarashi K. 2000. Properties of a polyamine transporter regulated by antizyme. Biochem J 347:297–303. [PMC free article] [PubMed] [Google Scholar]
- 82.Uemura T, Tachihara K, Tomitori H, Kashiwagi K, Igarashi K. 2005. Characteristics of the polyamine transporter TPO1 and regulation of its activity and cellular localization by phosphorylation. J Biol Chem 280:9646–9652. doi: 10.1074/jbc.M410274200. [DOI] [PubMed] [Google Scholar]
- 83.Rai AN, Thornton JA, Stokes J, Sunesara I, Swiatlo E, Nanduri B. 2016. Polyamine transporter in Streptococcus pneumoniae is essential for evading early innate immune responses in pneumococcal pneumonia. Sci Rep 6:26964. doi: 10.1038/srep26964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Howard JG, Rowley D, Wardlaw AC. 1958. Investigations on the mechanism of stimulation of non-specific immunity by bacterial lipopolysaccharides. Immunology 1:181–203. [PMC free article] [PubMed] [Google Scholar]
- 85.Orcutt RP, Gianni FJ, Judge RJ. 1987. Development of an ‘altered Schaedler flora’ for NCI gnotobiotic rodents. Microecol Ther 17:59. [Google Scholar]
- 86.Wymore Brand M, Wannemuehler MJ, Phillips GJ, Proctor A, Overstreet AM, Jergens AE, Orcutt RP, Fox JG. 2015. The altered Schaedler flora: continued applications of a defined murine microbial community. ILAR J 56:169–178. doi: 10.1093/ilar/ilv012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. 1999. Cutting edge: toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162:3749–3752. [PubMed] [Google Scholar]
- 88.Mandell L, Moran AP, Cocchiarella A, Houghton J, Taylor N, Fox JG, Wang TC, Kurt-Jones EA. 2004. Intact Gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect Immun 72:6446–6454. doi: 10.1128/IAI.72.11.6446-6454.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Walia S, Kamal R, Kanwar SS, Dhawan DK. 2015. Cyclooxygenase as a target in chemoprevention by probiotics during 1,2-dimethylhydrazine induced colon carcinogenesis in rats. Nutr Cancer 67:603–611. doi: 10.1080/01635581.2015.1011788. [DOI] [PubMed] [Google Scholar]
- 90.An J, Ha EM. 2016. Combination therapy of Lactobacillus plantarum supernatant and 5-fluouracil increases chemosensitivity in colorectal cancer cells. J Microbiol Biotechnol 26:1490–1503. doi: 10.4014/jmb.1605.05024. [DOI] [PubMed] [Google Scholar]
- 91.Drazenovich NL, Franklin CL, Livingston RS, Besselsen DG. 2002. Detection of rodent Helicobacter spp. by use of fluorogenic nuclease polymerase chain reaction assays. Comp Med 52:347–353. [PubMed] [Google Scholar]
- 92.Cox MP, Peterson DA, Biggs PJ. 2010. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gordon A. 2010. FASTX toolkit. [Google Scholar]
- 94.Marçais G, Kingsford C. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pongor LS, Vera R, Ligeti B. 2014. Fast and sensitive alignment of microbial whole genome sequencing reads to large sequence datasets on a desktop PC: application to metagenomic datasets and pathogen identification. PLoS One 9:e103441. doi: 10.1371/journal.pone.0103441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ondov BD, Bergman NH, Phillippy AM. 2011. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, Mortazavi A. 2016. A survey of best practices for RNA-seq data analysis. Genome Biol 17:13. doi: 10.1186/s13059-016-0881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Team RC. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 102.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. 2017. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All raw DNA/RNA reads are available under BioProject number PRJNA379709.