

Abstract
The tyrosine kinase inhibitor sunitinib is used as first‐line therapy in patients with metastasized renal cell carcinoma (mRCC), given in fixed‐dose regimens despite its high variability in pharmacokinetics (PKs). Interindividual variability of drug exposure may be responsible for differences in response. Therefore, dosing strategies based on pharmacokinetic/pharmacodynamic (PK/PD) models may be useful to optimize treatment. Plasma concentrations of sunitinib, its active metabolite SU12662, and the soluble vascular endothelial growth factor receptors sVEGFR‐2 and sVEGFR‐3, were measured in 26 patients with mRCC within the EuroTARGET project and 21 patients with metastasized colorectal cancer (mCRC) from the C‐II‐005 study. Based on these observations, PK/PD models with potential influence of genetic predictors were developed and linked to time‐to‐event (TTE) models. Baseline sVEGFR‐2 levels were associated with clinical outcome in patients with mRCC, whereas active drug PKs seemed to be more predictive in patients with mCRC. The models provide the basis of PK/PD‐guided strategies for the individualization of anti‐angiogenic therapies.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ There is a high interindividual variability (IIV) in response to sunitinib. Hence, predictive biomarkers are needed in order to maximize efficacy and minimize toxicity.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The objective of this study was the development of PK models, linking sunitinib plasma concentrations to PD response and clinical outcome, including the identification of potential genetic predictors for patients with mRCC and patients with mCRC.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ The developed PK/PD models adequately describe plasma concentration‐time profiles of sunitinib, SU12662, sVEGFR‐2, sVEGFR‐3, and clinical outcome showing the strength of an integrated modeling approach. Clinical response in patients with mRCC is best predicted by baseline sVEGFR‐2 levels, whereas in patients with mCRC, active drug PKs is more predictive.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ The PK/PD models presented in this study provide a better understanding of the relationship between sunitinib exposure, pharmacological response, and clinical outcome, and, hence, are an important step toward finding predictive biomarkers for the clinical outcome of sunitinib.
Sunitinib is a multitarget tyrosine kinase inhibitor, which is successfully used in the treatment of metastasized renal cell carcinomas (mRCCs), gastrointestinal stromal tumors (GISTs), and other solid tumor types. Sunitinib inhibits the vascular endothelial growth factor receptors (VEGFR‐1, 2, and 3), the platelet‐derived growth factor receptors α and β, among other tyrosine kinases.1, 2 CYP3A4 converts sunitinib into its active N‐desethyl metabolite (SU12662) and subsequently into inactive metabolites. The elimination half‐life of sunitinib is 40–60 hours and 80–110 hours for SU12662. An increased exposure to sunitinib is associated with improved survival but also with an increased risk for adverse events.3, 4
The individual response to sunitinib is highly variable: some patients experience severe toxicity and need dose reductions or even cessation of therapy, whereas others show no response at all when using the same dose. Biomarker testing prior to the start or during therapy may help provide the individual patient with the most effective treatment and the lowest possible risk of adverse effects. Whereas several potential biomarkers have been identified, they are not applied in clinical routine yet. However, sunitinib meets the requirements for therapeutic drug monitoring enabling dose adjustment based on measured plasma drug concentrations.3, 4, 5
Soluble VEGFR‐3 (sVEGFR‐3) was observed to be a potential predictive biomarker for overall survival (OS) on sunitinib treatment in a study of 303 patients diagnosed with GIST.6 Furthermore, vascular endothelial growth factor (VEGF)‐A and VEGFR‐3 protein expression were associated with OS and progression‐free survival (PFS), respectively, in 67 sunitinib‐treated patients with mRCC.7 Likewise, levels of VEGF, sVEGFR‐2, and sVEGFR‐3 were associated with objective responses in 63 patients with mRCC.8 With regard to genetic predictors, previous studies associated single nucleotide polymorphisms (SNPs) in genes encoding metabolizing enzymes or transporters related to pharmacokinetics (PKs) and pharmacodynamics (PDs) of sunitinib with efficacy and toxicity.9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22
In order to find predictive biomarkers for the clinical outcome of sunitinib, a better understanding of the relationships between sunitinib exposure, the pharmacological response, and the clinical outcomes is vital. This is part of the objectives of the European collaborative project EuroTARGET.23 Several PK models for sunitinib have previously been published. Here, we used a nonlinear mixed‐effects PK model for analyzing data of both patients with mRCC and patients with metastasized colorectal cancer (mCRC) in a pooled dataset.23, 24, 25, 26 This model was linked to PD models for sVEGFR‐2 and sVEGFR‐3, which were previously developed by our group.27 The purpose of our study was the development of PK models, linking sunitinib plasma concentrations to PD response, and clinical outcome in a model‐based time‐to‐event (TTE) analysis, including the identification of potential genetic predictors.
METHODS
Patient population
For the underlying PK/PD analysis, data were used from two PK studies, which focused on sunitinib treatment in patients with mRCC and patients with mCRC.23, 25 Both studies were designed as prospective, open label, single arm, multicenter, nonrandomized studies and performed in accordance with the Declaration of Helsinki. Patients gave written informed consent to give venous blood for PK/PD analysis and genotyping taken in the course of routine blood draw, and allowed the study sites to document clinical data.
The C‐IV‐001 study (EudraCT‐No: 2012‐001415‐23) was a phase IV PK/PD substudy of the noninterventional EuroTARGET project.23 Patients with mRCC were recruited in 9 medical centers in Germany and The Netherlands. Sunitinib doses ranged from 37.5–50 mg daily in the 4‐week on/2‐week off scheme. A patient was eligible for this study with a minimum age of 18 years, a diagnosis of mRCC, and a first‐line treatment with sunitinib. Within the EuroTARGET project, PFS was evaluated as the primary endpoint.23
The C‐II‐005 study (EudraCT‐No: 2008‐00151537) was performed to investigate the beneficial effect of sunitinib as add‐on to biweekly folinate, fluorouracil, and irinotecan in patients with mCRC and liver metastases.25 Patients received a daily dose of 37.5‐mg sunitinib on a 4‐week on/2‐week off treatment schedule. Primary endpoints were the reduction of tumor vessel permeability and blood flow determined by imaging techniques. Time to progression (TTP) was defined as a secondary endpoint. In case of toxicity, sunitinib therapy was interrupted or continued after dose reduction to 25 mg per day until the symptoms disappeared.25
Data collection and sampling
Clinical information was collected, especially demographic characteristics, concomitant medication, clinical response to the treatment, and toxicity. Serial blood samples were drawn, immediately centrifuged (1000 g, 4°C, 15 minutes) and stored at –80°C. In the C‐IV‐001 study, up to 12 plasma samples were collected within 3 cycles during routine checkups. Except for a mandatory baseline sample before treatment start, each center was free to develop a schedule according to their specific clinical routine. In the C‐II‐005 study, plasma samples were collected within 2 cycles at baseline, day 2 of each cycle, and afterward approximately every 2 weeks, always before sunitinib intake.
Plasma concentrations of sunitinib and SU12662 were determined using high‐performance liquid chromatography tandem mass spectrometry (MDS SCIEX API 5000 triple quadrupole mass spectrometer; Applied Biosystems/MDS SCIEX, Thornhill, Ontario, Canada). Between‐run precision and accuracy ranged from 1.6–6.1% and 0.2–9.1% for sunitinib and from 1.1–5.3% and −0.1 to 6.2% for SU12662, respectively.28 The sVEGFR‐2 concentrations were determined by commercially available enzyme‐linked immunosorbent assay kits (R&D Systems, Minneapolis, MN). The sVEGFR‐3 was measured using a validated immunoassay.26 Within‐laboratory precision and accuracy of all assays were within the acceptance criteria of the European Medicines Agency29 with 2.2–4.3% and 6.2–14.3% for sVEGFR‐2, and 0.4–14.7% and −3.8 to +16.2% for sVEGFR‐3, respectively. Quality control samples were analyzed in all assays and runs to determine run acceptance.
SNP selection and genotyping
The selection of SNPs was based on previously reported SNP associations (P < 0.05) with sunitinib treatment outcome with regard to efficacy and toxicity. Herein, we have focused on SNPs that were very likely to have an effect on VEGF or VEGFRs, or SNPs that have a high biomarker potential because of confirmatory findings in large cohorts. Thirteen SNPs were selected located in CYP3A5, ABCB1, VEGF‐A, VEGFR‐2, VEGFR‐3, and interleukin‐8 9–22 (details are provided in Supplementary Material S1).
Germline DNA was isolated from whole blood samples taken at baseline (before treatment initiation), using the Chemagic blood kit (PerkinElmer), and genotyping was performed using the LightCycler 480 Real‐Time polymerase chain reaction Instrument (Roche Applied Science, Almere, The Netherlands).
Pharmacokinetic/pharmacodynamic modeling
Data from all patients were analyzed together using the first‐order conditional estimation method with interaction implemented in NONMEM, version 7.3.30 The PK/PD models were built in a sequential manner. The structure of the models is shown in Figure 1.
Figure 1.
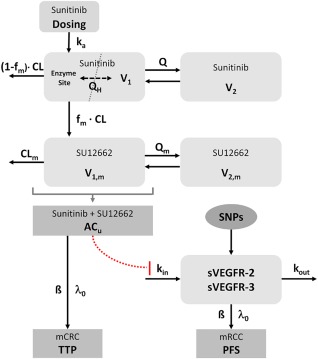
Model structure. ACU, unbound active concentration (sunitinib + SU12662); β, regression coefficient; CLm, clearance of the metabolite SU12262; fm, fraction metabolized to SU12662; ka, absorption rate constant; kin, zero‐order release rate constant; kout, first‐order elimination rate constant; λ0, baseline hazard; Q, inter‐compartmental clearance of sunitinib; QH, liver blood flow; Qm, intercompartmental clearance of the metabolite SU12662; V1, volume of the central compartment of sunitinib; V1,m, volume of the central compartment of the metabolite SU12662; V2, volume of the peripheral compartment of sunitinib; V2,m volume of the peripheral compartment of the metabolite SU12662.
Pharmacokinetic model
The PK model was partially based on a semiphysiological model published by Yu et al.24 This model features a one‐compartment model for sunitinib and a biphasic distribution for SU12662. Presystemic formation of SU12662 is handled via a hypothetical enzyme compartment incorporated into the central compartment of sunitinib. The central compartment and the enzyme compartment are connected by an intercompartmental clearance, which was fixed to the liver blood flow. The addition of a peripheral compartment for sunitinib was tested because other published models featured this structure and the underlying data indicated a similar distribution as the active metabolite.3, 4, 27 Interindividual variability (IIV) was initially included for all parameters and removed if their exclusion did not significantly worsen the model fit (P < 0.05). A proportional, additive, and a combined (additive + proportional) error model were tested for the parent drug and metabolite separately to describe the residual unexplained variability.
Pharmacodynamic models
The concentration‐time profiles of sVEGFR‐2 and sVEGFR‐3 were described using models developed previously by our group for healthy volunteers.27 The concentration‐effect relationship was described by a simple hyperbolic function (fractional tyrosine kinase inhibition (INH); Eq. (1)) using the unbound concentration of the total active drug including SU12662 (ACu) with a dissociation constant (kd) fixed to 4 ng/mL obtained in vitro in a tyrosine kinase phosphorylation assay.27, 31 Unbound concentrations were computed by assuming a protein binding of 95% for sunitinib and 90% for SU12662.32 Decreasing concentrations of the soluble receptors were described by an indirect response model with zero‐order production (kin) and first‐order elimination (kout). The inhibitory drug effect on kin was included using an inverse‐linear model with α as the intrinsic activity:
| (1) |
| (2) |
As for the PK model, IIV was initially included for all model parameters and removed in case the model did not significantly worsen after exclusion.
Covariate analysis
Covariates were tested on the final models using the automated covariate search provided in PsN (Pearl speaks NONMEM, version 4.4.0).33 In the forward inclusion step, a potential covariate was significant when the objective function value (OFV) decreased by at least 3.84 (1 degree of freedom (DF), P = 0.05) and was kept in the model if it showed in the backward exclusion step an OFV increase of at least 5.99 (1 degree of freedom, P = 0.01). Covariates were tested in the included demographic parameters, tumor type, and the preselected SNPs (see Supplementary Material S1).
Model qualification
Nested models were compared using the likelihood ratio test. Goodness‐of‐fit plots showing population predicted concentrations (PREDs) and individual predicted concentrations (IPREDs) vs. observed concentrations and conditional weighted residuals vs. IPREDs or time were used to evaluate the models visually. For the final models, precision of all model parameters was given as 90% confidence interval (CI) calculated by the nonparametric bootstrap approach (n = 1,000). Prediction‐corrected visual predictive checks (VPCs) were generated for the patients using 1,000 simulations.34 Both procedures, bootstrap and prediction‐corrected VPCs, were performed using the PsN software.33
Sensitivity analysis
The effect of fixed parameters on the model predictions was tested by varying the respective parameters between +50 and −50% in 10% steps of the base value derived from literature. As time of drug intake or sampling time was missing in some patients, administration time was set to 8:00 am, assuming that an intake in the morning is the most likely scenario. A similar approach was used for missing sampling times. Here, 12:00 pm was chosen, because most checkups across study centers were scheduled around mid‐day. Moreover, the influence of dosing time on parameter estimates was tested randomly varying the time of drug intake between −3 and +3 hours of the documented or imputed value.
The deviation from the original estimate was quantified by calculating the relative prediction error (RPE) and the root squared mean prediction error (RSME, %), which are defined as:
| (3) |
| (4) |
where θbase denotes the parameter estimate of the original model and θnew the new estimate under changed conditions.
Outcome modeling
Outcome analysis was performed separately for patients with mRCC and patients with mCRC using TTE models based on a proportional hazard model allowing the analysis of continuous and time‐dependent covariates. Different TTE distributions were tested using NONMEM.35 Although a constant hazard is usually a viable assumption in patients with cancer due to the short survival and progression times, models with time‐dependent hazards were also tested for comparison:
| (5) |
| (6) |
| (7) |
Dichotomous covariates were divided by their characteristic values, whereas continuous covariates were grouped. Statistical significance of the difference between groups was determined using the log‐rank test.
In addition, the Kaplan‐Meier analysis as the classical nonparametric method was used to determine the median PFS in patients with mRCC and the TTP in patients with mCRC.36, 37 Kaplan‐Meier analysis and Cox regression were performed using the survival package in R.36
RESULTS
Patients
Clinical characteristics of the included patients are presented in Table 1. Twenty‐seven patients with mRCC treated with sunitinib were recruited of which one patient was excluded from the analysis due to lack of PK data. Twenty‐eight patients with mCRC were recruited of which seven patients were excluded because of missing drug administration (n = 5), missing data (n = 1), or uncertainty in the documentation of sunitinib intake (n = 1). Thus, 26 patients with mRCC and 21 patients with mCRC treated with sunitinib were included into the combined PK/PD analysis.
Table 1.
Patient characteristics (median and range)
| Patients with mRCC (n = 26) | Patients with mCRC (n = 21) | |
|---|---|---|
| Age, years (range) | 64 (43–75) | 61 (33–85) |
| Gender, M/F | 25/1 | 12/9 |
| Weight, kg (range) | 83 (65–106) | 73 (57–106) |
| Height, cm (range) | 180 (155–186) | 172 (149–184) |
| BMI, kg/m2 (range) | 25.7 (22.5–34.5) | 26.0 (13.3–39.3) |
BMI, body mass index; mCRC, metastasized colorectal cancer; mRCC, metastasized renal cell carcinoma.
Outcome analysis was performed for each tumor entity separately with regard to the different end points of each study using data of 24 patients with mRCC and 21 patients with mCRC. Two of the 26 patients with mRCC were excluded from the outcome analysis as both received sunitinib as second‐line therapy.
Moreover, 25 patients with mRCC and 14 patients with mCRC could be genotyped on the 13 selected SNPs. Here, we observed SNP call rates of 94–100% and 10 of 13 SNPs were in the Hardy‐Weinberg equilibrium with P values > 0.05. Only the SNPs ABCB1 rs1045642, and VEGFA rs699947 and rs2010963 were not in the Hardy‐Weinberg equilibrium with P = 0.009, P = 0.002, and P = 0.030, respectively.
Pharmacokinetic model
A PK model previously published by Yu et al.24 was adapted as the basis for the structural model. To allow comparison of the estimated parameters, volume and clearance parameters were, as in the reference model, allometrically scaled to a standard weight of 70 kg. Values for liver blood flow (QH) and the fraction metabolized (fm) were fixed to their respective literature values as in the original model.24 In contrast to the original model, a peripheral compartment for sunitinib was introduced, which improved the model significantly (objective function value difference (dOFV) = −123.98, P < 0.0001). However, the volume of the peripheral compartment (V2) could not be estimated with enough precision, hence, the value was fixed to 588 L, which was previously reported by Houk et al.3, 4 Compared to the base model, this still improved the model fit significantly (dOFV = −112.37; P < 0.0001). The model fit significantly worsened without IIV for sunitinib CL (dOFV = 90.58; P < 0.0001), V1 of sunitinib (dOFV = 41.97; P < 0.0001), fm (dOFV = 134.67; P < 0.0001), as well as V1 of SU12662 (dOFV = 18.47; P < 0.0001). Therefore, the IIV was kept in the final model for these parameters. The estimation of covariances improved the model further (dOFV = −20.34; P < 0.005).
The sensitivity analysis did not reveal major effects on parameter estimates when the fixed parameters QH and V2 (sunitinib) were varied between +50% and −50%. However, variation of fm resulted in a high variation (RSME of up to 50%) for clearance, intercompartmental clearance, as well as the central and peripheral volume of distribution of SU12662, which could be expected by their definition. Randomly varying dosing time between +3 and −3 hours relative to the reported or imputed value had primarily an effect on the absorption rate constant (ka). The RSME was relatively high with 36.9% for this parameter. As expected, the residual error for sunitinib was also highly affected with an RSME of 25.2%.
Forward inclusion and backward elimination of potential covariates did not reveal any significant effects on the tested model parameters. In addition, no statistically significant differences of PK parameters between both tumor entities were found, confirming that the underlying model can be used across different tumor types. A complete list of covariates tested is provided in Supplementary Material S1. Final parameter estimates are shown in Table 2. VPCs indicated that central tendency and variability of both active compounds could be described adequately with the underlying model (Figures 2 a and 2b).
Table 2.
Population parameter estimates of the final PK model in comparison to the model of Yu et al.24
| Our study | Yu et al.24 | ||||||
|---|---|---|---|---|---|---|---|
| Parameter | Unit | Estimate (RSE, %) | IIV% (RSE, %) | Bootstrap mean | Bootstrap 90% CI | Estimate (RSE, %) | IIV% (RSE, %) |
| Sunitinib (parent drug) | |||||||
| ka | 1/h | 0.133 (34.6) | ‐ | 0.149 | 0.01–0.25 | 0.34 (10.8) | ‐ |
| CL | L/h | 33.9 (6.0) | 30.3 (29.0) | 33.92 | 30.76–37.53 | 35.7 (5.7) | 33.9 (12.0) |
| V1 | L | 1820 (6.6) | 25.3 (30.3) | 1812.1 | 1607.8–1812.2 | 1360 (6.0) | 32.4 (10.6) |
| V2 | L | 588a | ‐ | 588a | ‐ | ‐ | ‐ |
| Q | L/h | 0.371 (18.9) | ‐ | 0.373 | 0.263–0.494 | ‐ | ‐ |
| QH | L/h | 80a | ‐ | 80a | ‐ | 80a | ‐ |
| Residual error (proportional) | ‐ | −0.367 (14.1) | ‐ | −0.361 | −0.450 to −0.283 | 0.06 (13.5) | |
| SU12662 (metabolite) | |||||||
| CLm | L/h | 16.5 (5.4) | ‐ | 16.5 | 15.0–17.9 | 17.1 (7.4) | 42.1 (7.0) |
| V1,m | L | 730 (14.1) | 42.9 (54.8) | 713.6 | 545.9–872.9 | 635 (13.1) | 57.9 (8.8) |
| V2,m | L | 592 (13.2) | ‐ | 604.9 | 481.0–737.4 | 388 (14.9) | ‐ |
| Qm | L/h | 2.75 (24.6) | ‐ | 2.90 | 1.96–4.27 | 20.1 (32.6) | ‐ |
| fm | ‐ | 0.21a | 34.6 (20.5) | 0.21a | ‐ | 0.21a | ‐ |
| Residual error (proportional) | ‐ | −0.281 (10.8) | ‐ | −0.276 | −0.326 to −0.229 | 0.03 (14.1) | ‐ |
| Correlations | |||||||
| ρ (CL,V1) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| ρ (CL, CLm) | ‐ | ‐ | ‐ | ‐ | ‐ | 0.53 | ‐ |
| ρ (CL, V 1,m) | ‐ | −0.0607 (48.3) | ‐ | −0.0685 | −0.127 to −0.019 | ‐ | ‐ |
| ρ (V1, V1,m) | ‐ | 0.0481 (51.8) | ‐ | 0.0534 | 0.0091 to −0.0996 | 0.48 | ‐ |
| ρ (CLm, V1,m) | ‐ | ‐ | ‐ | ‐ | 0.45 | ‐ | |
| ρ (CL, fm) | ‐ | −0.0425 (40.8) | ‐ | −0.0392 | −0.0671 to −0.0130 | ‐ | ‐ |
| ρ (V1, fm) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| ρ (fm, V1,m) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
CI, confidence interval; CL, clearance of sunitinib; CLm, clearance of the metabolite SU12262; fm, fraction metabolized to SU12662; IIV, interindividual variability; ka, absorption rate constant; prop, proportional error model; PK, pharmacokinetic; Q, intercompartmental clearance of sunitinib; QH, liver blood flow; Qm, intercompartmental clearance of the metabolite SU12662; ρ, correlation coefficient; RSE, relative standard error; V1, volume of the central compartment of sunitinib; V1,m, volume of the central compartment of the metabolite SU12662; V2, volume of the peripheral compartment of sunitinib; V2,m, volume of the peripheral compartment of the metabolite SU12662.
Parameter fixed to literature value.
Figure 2.
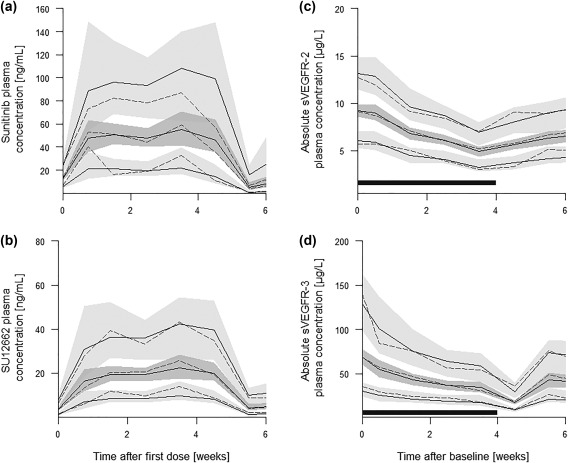
Prediction‐corrected visual predictive checks of (a) the final pharmacokinetic (PK) model of sunitinib, (b) the final PK model of SU12662, (c) the soluble vascular endothelial growth factor receptor (sVEGFR)‐2 model, and (d) the sVEGFR‐3 model for one treatment cycle. Solid lines indicate the estimated mean as well as the 90% prediction interval. Dashed lines show the respective observed mean and interval. Shaded gray areas represent the 90% confidence band of the predictions. The dark gray rectangle indicates the time of treatment.
Pharmacokinetic/pharmacodynamic models
The inverse‐linear model previously developed for healthy volunteers was also applicable to describe the concentration‐time profile of both sVEGFRs in patients with mRCC and patients with mCRC after sunitinib therapy.27 The shape of the concentration‐time curves of both soluble receptors was comparable and their response was highly correlated in patients with mRCC (r2 = 0.594; P < 0.0001) and also patients with mCRC (r2 = 0.635; P < 0.0001). However, the covariate analysis performed on both models revealed PD differences between the tumor entities. Addition of a proportional covariate effect of “tumor type” on the intrinsic activity (α) of sunitinib on sVEGFR‐2 levels improved the model significantly (dOFV = −7.45; P = 0.006). It was shown that intrinsic activity (α) was 32.8% lower in patients with mCRC compared to patients with mRCC.
Intrinsic activity on sVEGFR‐2 levels was also influenced by VEGF‐R3 rs6877011 genotype (1 = CG/GG; 0 = CC). Presence of the G‐allele (CG and GG genotypes) showed a decreased α compared to the wildtype CC (2.31 vs. 1.00 in case of patients with mRCC and 1.55 vs. 0.65 for patients with mCRC). A decreased intrinsic activity was also observed for patients with presence of a T‐allele in ABCB1 rs2032582 (2.31 vs. 1.59 in case of patients with mRCC and 1.55 vs. 1.07 for patients with mCRC).
Final parameter estimates of both models are shown in Table 3. Visual predictive checks indicated that central tendency and variability of both proteins could be described adequately with the underlying models (Figure 2 c and 2d).
Table 3.
Population parameter estimates of the final PD models (sVEGFR‐2 and sVEGFR‐3)
| Parameter | Unit | Estimate (RSE, %) | IIV (RSE, %) | Bootstrap mean | Bootstrap 90% CI |
|---|---|---|---|---|---|
| sVEGFR‐2 | |||||
| Baseline | µg/L | 9.0 (2.9) | 19.9 (21.4) | 9.0 | 8.6–9.5 |
| α | ‐ | 2.31 (8.8) | ‐ | 2.31 | 1.98–2.64 |
| kout | 1/h | 0.0043 (7.6) | ‐ | 0.0043 | 0.0038 to −0.0049 |
| Kd | µg/mL | 4a | ‐ | 4a | ‐ |
| Residual error | ‐ | 0.124 (6.8) | ‐ | 0.122 | 0.108–0.136 |
| Tumor type on α (proportional) | ‐ | −0.328 (24.6) | ‐ | −0.322 | −0.440 to −0.186 |
| VEGFR‐3 rs6877011 on α (proportional) | ‐ | −0.565 (25.4) | ‐ | −0.557 | −0.787 to −0.319 |
| ABCB1 rs2032582 on α (proportional) | ‐ | −0.311 (37.9) | ‐ | −0.307 | −0.497 to −0.117 |
| sVEGFR‐3 | |||||
| Baseline value | µg/L | 63.5 (5.9) | 42.6 (24.4) | 63.7 | 57.3–69.8 |
| α | ‐ | 1.74 (9.8) | 54.3 (43.5) | 1.76 | 1.49–2.05 |
| kout | 1/h | 0.0053 (7.2) | ‐ | 0.0054 | 0.0047 to −0.0060 |
| Kd | µg/mL | 4a | ‐ | 4a | ‐ |
| Residual error | ‐ | 0.15 (6.9) | ‐ | 0.15 | 0.13–0.17 |
| Tumor type on baseline value (proportional) | ‐ | −0.642 (6.5) | ‐ | −0.640 | −0.703 to −0.569 |
| Correlations | |||||
| ρ (baseline value, α) | ‐ | 0.124 | ‐ | 0.123 | 0.045–0.209 |
α, intrinsic activity; CI, confidence interval; IIV, interindividual variability; kd, dissociation constant; kout, elimination rate constant; ρ, correlation coefficient; PD, pharmacodynamic; RSE, relative standard error; sVEGFR, soluble vascular endothelial growth factor receptor; VEGFR, vascular endothelial growth factor receptor.
Parameter fixed to literature value.
Outcome model for patients with mRCC
Median PFS for patients with mRCC was calculated with 6.9 months (n = 24). The PFS could be described by a parametric TTE model assuming exponentially distributed data with a baseline hazard function λ0 of 0.0252 weeks‐1 (90% CI = 0.0168–0.0336). The inclusion of the measured and estimated sVEGFR‐2 baseline value led to a decrease of the OFV by 4.14 or 4.67 (P < 0.05), respectively. However, the dichotomized covariate, dividing patients into two groups with baseline values above and below the population median of 8.8 µg/L, had a stronger effect with a decrease of the OFV by −6.40 (P < 0.025). The β was estimated with 1.45 corresponding to a hazard ratio (HR) of 4.26 (with β defined as the natural logarithm of the HR). Inclusion of the active, unbound sunitinib/SU12662 concentrations resulted in an estimated β of −0.14 mL/ng indicating that a higher plasma level reduces the hazard and, hence, the probability of progression during treatment. However, the effect was not statistically significant (dOFV = −1.1; P = 0.29). Likewise, plasma concentrations of sVEGFR‐2 and sVEGFR‐3 over time were not statistically significant predictors of PFS either (dOFV = −3.7 and −0.99, respectively). Besides absolute plasma levels of both proteins, also the relative decrease with respect to individual baseline values predicted by the PK/PD models was tested as a potential covariate. However, no significant improvement of the model fit could be observed either (dOFV = −0.31 and −0.98).
Best prediction of PFS in patients with mRCC was achieved by a hazard function h(t), including the dichotomized baseline value of sVEGFR‐2, which was independent of the developed PK/PD models:
| (8) |
The observed Kaplan‐Meier curve describing the PFS function of the patients with mRCC was within the predicted 90% prediction interval of 1,000 simulations and could sufficiently be described by the TTE model except for later time points as a result of censored data (Figure 3 a). Final parameter estimates are shown in Table 4.
Figure 3.
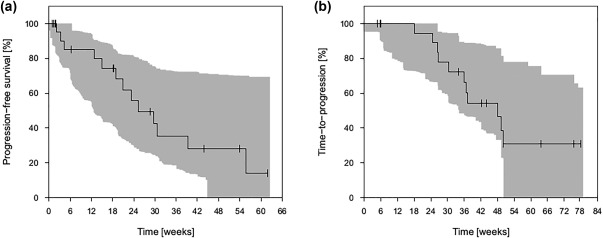
Prediction‐corrected visual predictive checks of (a) the final survival model for patients with metastasized renal cell carcinoma, and (b) the final survival model for patients with metastasized colorectal cancer. Shaded gray areas represent the 90% prediction interval.
Table 4.
Population parameter estimates of the final time‐to‐event models
| Parameter | Covariate | Unit | Estimate (RSE, %) | Bootstrap mean | Bootstrap median | Bootstrap 90% CI |
|---|---|---|---|---|---|---|
| Patients with mRCC | ||||||
| λ0 | Weeks‐1 | 0.0118 (46.3) | 0.0121 | 0.0117 | 0.0038–0.0220 | |
| ß | sVEGFR‐2 baseline valuea | ‐ | 1.45 (43.3) | 1.57 | 1.49 | 0.71–2.68 |
| HR | ‐ | 4.26 | 4.81 | 4.44 | 2.03–14.59 | |
| Patients with mCRC | ||||||
| λ0 | Weeks‐1 | 0.0234 | 0.0256 | 0.0241 | 0.0120–0.0447 | |
| ß | ACu | mL/ng | −0.758 | −0.919 | −0.836 | −0.366 to −1.736 |
| HR | ‐ | 0.47 | 0.40 | 0.43 | 0.18–0.69 | |
ACU, unbound active concentration (sunitinib + SU12662); ß, regression coefficient; CI, confidence interval; HR, hazard ratio; λ0, baseline hazard; mCRC, metastasized colorectal cancer; mRCC, metastasized renal cell carcinoma; RSE, relative standard error; sVEGFR, soluble vascular endothelial growth factor receptor.
Dichotomized above (1) and below (0) population median.
These findings were confirmed in a multivariate Cox regression analysis. The only covariates exhibiting a significant influence were the dichotomized baseline values of both soluble proteins (data not shown).
Outcome model for patients with mCRC
Median TTP for patients with mCRC was 8.4 months (n = 21). Analogous to the patients with mRCC, the TTP could be described by a parametric TTE model assuming exponentially distributed data. The baseline hazard function λ0 was estimated with 0.0234 weeks‐1 (90% CI = 0.012–0.042 weeks‐1). The inclusion of the current concentration of the unbound, active drug (ACu) reduced the OFV by 6.07 (P <0.05). The β was estimated to be −0.758 mL/ng corresponding to an HR of 0.47. None of the other variables describing the individual PK or biomarker response were identified to be predictive for TTP. Therefore, TTP in patients with mCRC was best predicted by the PKs of sunitinib and SU12262 with an appropriate hazard function h(t) dependent on the current ACu(t):
| (9) |
The observed Kaplan‐Meier curve describing the PFS function of the patients with mCRC was within the predicted 90% prediction interval of 1,000 simulations and could sufficiently be described by the TTE model. However, TTP was difficult to predict for the time from 1 year onward due to censored data (Figure 3 b). Final parameter estimates are shown in Table 4.
A multivariate Cox regression analysis confirmed these results exhibiting the area under the curve (AUC) at steady‐state of the unbound, active drug in combination with age as positive predictive covariates (data not shown).
DISCUSSION
In this study, we successfully integrated distinct models for sunitinib in a modeling framework, including PK, PD, pharmacogenetic, and outcome data. The developed models adequately describe plasma concentration‐time profiles of sunitinib, its active metabolite SU12662, sVEGFR‐2, and s‐VEGFR‐3, as well as clinical outcome in both tumor types. Similar models (but without pharmacogenetics) were published in patients with GIST6 and recently hepatocellular carcinoma,38 but there is no model with integrated outcome data yet published for the tumor entities investigated here.
Covariate analysis on the PK parameters did not reveal any significant findings. The significant increase of sunitinib clearance in patients with ABCB1 rs2032582 TT (18%) found in previous studies could not be confirmed.17 Presumably, this is due to the small and, with two tumor entities, relatively heterogeneous cohort. Biomarker response of sVEGFR‐2 and sVEGFR‐3 was highly associated in each tumor entity, which suggested a comparable predictive value of both soluble receptors. As previously reported, decreasing plasma concentrations were observed for both receptors after sunitinib administration with a subsequent increase after stop of treatment.8, 27, 39 Independent of tumor entity and dosing scheme, baseline levels are not fully recovered after a 2‐week off phase.
A difference in sVEGFR‐2 response to sunitinib between patients with mRCC and patients with mCRC could be identified. However, decrease of sVEGFR‐2 plasma levels relative to the individual baseline did not have a significant impact on PFS or TTP in both studies, hence, the clinical relevance of this effect might be negligible. Observed baseline values of sVEGFR‐3 were in the same magnitude previously reported by Motzer et al.40 ranging between 22.3 and 129.2 µg/L for patients with mRCC. However, they were significantly higher compared with patients with mCRC. This finding might indicate a higher expression of this protein in patients with mRCC. However, data regarding the baseline values of sVEGFR‐3 in patients with mCRC is sparse, because the first‐line and second‐line treatment usually does not involve tyrosine kinase inhibitors targeting sVEGFR.41
In this study, we found that the presence of the variant G‐allele in SNP rs6877011 in VEGFR‐3 was associated with a 56.5% decrease in intrinsic activity on sVEGFR‐2 compared to the wild‐type CC. The same VEGFR‐3 SNP was associated with a decreased PFS in an earlier study.12 Maitland et al.42 associated variant G‐allele carriers of VEGFR‐2 (KDR) rs34231037 with sVEGFR2 baseline levels and a decline in sVEGFR‐2 in response to treatment with pazopanib. We have recently found that rs34231037 variant G‐allele carriers have a tendency toward a better response to sunitinib.43 VEGFR‐1, 2, and 3 have similar binding domains.44 A SNP in any of the genes encoding these VEGFRs could result in a conformation change and prevent or stimulate binding of the drug ligand to VEGFRs, and change the ability of sunitinib to decrease sVEGFR‐2 and sVEGFR‐3. It is remarkable that the SNP effect of G‐allele carriers of rs6877011 in VEGFR‐3 was not found on the intrinsic activity of sunitinib on sVEGFR‐3 but on sVEGFR‐2. Possibly, a lower activity of sunitinib on sVEGFR‐3 could also affect sVEGFR‐2. The conformation change may have more impact on VEGFR‐2 binding affinity than VEGFR‐3.
In both patient groups, we succeeded in linking clinical outcome data to either PDs (mRCC) or PKs (mCRC). In patients with mRCC, baseline levels of sVEGFR‐3 and sVEGFR‐2 as well as the decrease in sVEGFR‐2 plasma levels over the treatment duration were previously reported to be related to clinical outcome.7, 45 These findings could be further confirmed by this study. Although the effect of sVEGFR‐2 decrease over time was not significant in the TTE analysis, patients with a substantially higher baseline value of sVEGFR‐2 showed a significantly worse PFS with an estimated HR of 4.26. The baseline value of sVEGFR‐3 had a lower influence on PFS: for patients with an sVEGFR‐3 baseline above the population median, the HR was 2.38 without statistical significance (P = 0.2). An effect of similar magnitude (HR = 2.4; 95% CI = 1.13–5.11) was reported by Harmon et al.46 for the same covariate. In contrast, in patients with mCRC, the TTE model showed an effect of the PKs on TTP with precise parameter estimates. Higher exposure to sunitinib and SU12662 included as active drug concentration over time was associated with a longer TTP. Similarly, a meta‐analysis with 443 patients with cancer, including advanced GIST, mRCC, and other solid tumors, suggested that an increased AUC at steady‐state is associated with a longer TTP and a longer OS.4
It is not surprising that plasma concentrations of proteins related to the VEGF pathway seem to be more predictive for clinical outcome in patients with mRCC, as most RCC cells overexpress VEGF due to mutations in the von‐Hippel Lindau gene.47 Furthermore, sunitinib showed no additional effects in patients with colorectal cancer,25 which is consistent with our findings that sVEGFR‐2 and sVEGFR‐3 levels were not correlated to outcome. The lower intrinsic activity of sunitinib on sVEGFR‐2 baseline levels and the overall lower plasma concentrations of sVEGFR‐3 may also underline the lower dependency of colorectal carcinomas on angiogenesis, especially via VEGF signaling.
In conclusion, a semimechanistic PK model for sunitinib could be successfully linked to PD models for sVEGFR‐2 and sVEGFR‐3, including various genotypes. Although we could show that sunitinib PK does not differ between the two tumor entities, we found differences in PD response with respect to the decrease of sVEGFR‐2 and sVEGFR‐3 plasma concentrations during therapy. Furthermore, sVEGFR‐2 baseline levels seemed to be more predictive for clinical outcome in patients with mRCC in contrast to patients with mCRC where active drug PKs showed the highest impact. Nevertheless, our study provides the basis of PK/PD‐guided individualization strategies for the optimization of anti‐angiogenic therapies and underlines that it is quite unlikely to identify a general, tumor entity‐independent biomarker for sunitinib therapy response.
Supporting information
Supplementary Material S1 List of covariates tested with the respective models.
Supplementary Material S2 NONMEM codes.
Acknowledgments
The research leading to these results has received funding from the European Union's Seventh Framework Programme (FP7/2007–2013) under grant agreement no 259939. We thank Danielle Klootwijk for her assistance in SNP genotyping.
Conflict of Interest
The authors declared no conflict of interest.
Author Contributions
M.H.D., A.F., F.K., H.J.G., and U.J. wrote the manuscript. E.O., L.A.K., H.J.G., and U.J. designed the research. M.H.D., A.F., F.K., J.J.S., D.J.A.R.M., F.S., M.K., C.S., D.S., T.G., S.H., D.H., M.R., B.M., K.M., and L.B. performed the research. M.H.D., A.F., F.K., and D.J.A.R.M. analyzed the data. A.F., F.K., F.S., M.K., M.H.D., D.J.A.R.M., and C.S. contributed new reagents/analytical tools.
References
- 1. Motzer, R.J. et al Sunitinib in patients with metastatic renal cell carcinoma. JAMA 295, 2516–2524 (2006). [DOI] [PubMed] [Google Scholar]
- 2. Motzer, R.J. et al Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 27, 3584–3590 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Houk, B.E. , Bello, C.L. , Kang, D. & Amantea, M. A population pharmacokinetic meta‐analysis of sunitinib malate (SU11248) and its primary metabolite (SU12662) in healthy volunteers and oncology patients. Clin. Cancer Res. 15, 2497–2506 (2009). [DOI] [PubMed] [Google Scholar]
- 4. Houk, B.E. , Bello, C.L ., Poland, B ., Rosen, L.S ., Demetri, G.D . & Motzer, R.J . Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta‐analysis. Cancer Chemother. Pharmacol. 66, 357–371 (2010). [DOI] [PubMed] [Google Scholar]
- 5. Lankheet, N.A. et al Pharmacokinetically guided sunitinib dosing: a feasibility study in patients with advanced solid tumours. Br. J. Cancer 110, 2441–2449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hansson, E.K. et al PKPD modeling of VEGF, sVEGFR‐2, sVEGFR‐3, and sKIT as predictors of tumor dynamics and overall survival following sunitinib treatment in GIST. CPT Pharmacometrics Syst. Pharmacol. 2, e84 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garcia‐Donas, J. et al Prospective study assessing hypoxia‐related proteins as markers for the outcome of treatment with sunitinib in advanced clear‐cell renal cell carcinoma. Ann. Oncol. 24, 2409–2414 (2013). [DOI] [PubMed] [Google Scholar]
- 8. Deprimo, S.E. et al Circulating protein biomarkers of pharmacodynamic activity of sunitinib in patients with metastatic renal cell carcinoma: modulation of VEGF and VEGF‐related proteins. J. Transl. Med. 5, 32 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eechoute, K. et al Polymorphisms in endothelial nitric oxide synthase (eNOS) and vascular endothelial growth factor (VEGF) predict sunitinib‐induced hypertension. Clin. Pharmacol. Ther. 92, 503–510 (2012). [DOI] [PubMed] [Google Scholar]
- 10. Xu, C.F. et al Pazopanib efficacy in renal cell carcinoma: evidence for predictive genetic markers in angiogenesis‐related and exposure‐related genes. J. Clin. Oncol. 29, 2557–2564 (2011). [DOI] [PubMed] [Google Scholar]
- 11. Kim, J.J. et al Association of VEGF and VEGFR2 single nucleotide polymorphisms with hypertension and clinical outcome in metastatic clear cell renal cell carcinoma patients treated with sunitinib. Cancer 118, 1946–1954 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scartozzi, M. et al VEGF and VEGFR polymorphisms affect clinical outcome in advanced renal cell carcinoma patients receiving first‐line sunitinib. Br. J. Cancer 108, 1126–1132 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garcia‐Donas, J. et al Single nucleotide polymorphism associations with response and toxic effects in patients with advanced renal‐cell carcinoma treated with first‐line sunitinib: a multicentre, observational, prospective study. Lancet Oncol. 12, 1143–1150 (2011). [DOI] [PubMed] [Google Scholar]
- 14. Beuselinck, B. et al Single‐nucleotide polymorphisms associated with outcome in metastatic renal cell carcinoma treated with sunitinib. Br. J. Cancer 108, 887–900 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beuselinck, B. et al Efflux pump ABCB1 single nucleotide polymorphisms and dose reductions in patients with metastatic renal cell carcinoma treated with sunitinib. Acta Oncol. 53, 1413–1422 (2014). [DOI] [PubMed] [Google Scholar]
- 16. van der Veldt, A.A. et al Genetic polymorphisms associated with a prolonged progression‐free survival in patients with metastatic renal cell cancer treated with sunitinib. Clin. Cancer Res. 17, 620–629 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Teo, Y.L. et al Effect of the CYP3A5 and ABCB1 genotype on exposure, clinical response and manifestation of toxicities from sunitinib in Asian patients. Pharmacogenomics J. 16, 47–53 (2016). [DOI] [PubMed] [Google Scholar]
- 18. Diekstra, M.H. et al Association analysis of genetic polymorphisms in genes related to sunitinib pharmacokinetics, specifically clearance of sunitinib and SU12662. Clin. Pharmacol. Ther. 96, 81–89 (2014). [DOI] [PubMed] [Google Scholar]
- 19. Diekstra, M.H. et al CYP3A5 and ABCB1 polymorphisms as predictors for sunitinib outcome in metastatic renal cell carcinoma. Eur. Urol. 68, 621–629 (2015). [DOI] [PubMed] [Google Scholar]
- 20. van Erp, N.P. et al Pharmacogenetic pathway analysis for determination of sunitinib‐induced toxicity. J. Clin. Oncol. 27, 4406–4412 (2009). [DOI] [PubMed] [Google Scholar]
- 21. Xu, C.F. et al IL8 polymorphisms and overall survival in pazopanib‐ or sunitinib‐treated patients with renal cell carcinoma. Br. J. Cancer 112, 1190–1198 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Diekstra, M.H. et al Association of single nucleotide polymorphisms in IL8 and IL13 with sunitinib‐induced toxicity in patients with metastatic renal cell carcinoma. Eur. J. Clin. Pharmacol. 71, 1477–1484 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van der Zanden, L.F . et al Description of the EuroTARGET cohort: a European collaborative project on TArgeted therapy in renal cell cancer‐ GEnetic‐ and tumor‐related biomarkers for response and toxicity. Urol. Oncol. (2017). [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 24. Yu, H. et al Integrated semi‐physiological pharmacokinetic model for both sunitinib and its active metabolite SU12662. Br. J. Clin. Pharmacol. 79, 809–819 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mross, K. et al FOLFIRI and sunitinib as first‐line treatment in metastatic colorectal cancer patients with liver metastases‐‐a CESAR phase II study including pharmacokinetic, biomarker, and imaging data. Int. J. Clin. Pharmacol. Ther. 52, 642–652 (2014). [DOI] [PubMed] [Google Scholar]
- 26. Kanefendt, F. , Lindauer, A. , Mross, K. , Fuhr, U. & Jaehde, U. Determination of soluble vascular endothelial growth factor receptor 3 (sVEGFR‐3) in plasma as pharmacodynamic biomarker. J. Pharm. Biomed. Anal. 70, 485–491 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Lindauer, A. et al Pharmacokinetic/pharmacodynamic modeling of biomarker response to sunitinib in healthy volunteers. Clin. Pharmacol. Ther. 87, 601–608 (2010). [DOI] [PubMed] [Google Scholar]
- 28. Rodamer, M. , Elsinghorst, P.W. , Kinzig, M. , Gütschow, M. & Sörgel, F. Development and validation of a liquid chromatography/tandem mass spectrometry procedure for the quantification of sunitinib (SU11248) and its active metabolite, N‐desethyl sunitinib (SU12662), in human plasma: application to an explorative study. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 879, 695–706 (2011). [DOI] [PubMed] [Google Scholar]
- 29. European Medicines Agency . Guideline on Bioanalytical Method Validation. EMEA/CHMP/EWP/192217/2009. <http://www.ema.europa.eu/> (2011).
- 30. Beal S.L., Sheiner L.B., Boeckmann A.J. & Bauer R.J. eds. NONMEM 7.3.0 Users Guide (ICON Development Solutions, Hanover, MD, 1989. –2013). [Google Scholar]
- 31. Mendel, D.B. et al In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 9, 327–337 (2003). [PubMed] [Google Scholar]
- 32. Rini, B.I. Sunitinib. Expert Opin. Pharmacother. 8, 2359–2369 (2007). [DOI] [PubMed] [Google Scholar]
- 33. Lindbom, L. , Ribbing, J. & Jonsson, E.N. Perl‐speaks‐NONMEM (PsN)‐‐a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75, 85–94 (2004). [DOI] [PubMed] [Google Scholar]
- 34. Bergstrand, M. , Hooker, A.C. , Wallin, J.E. & Karlsson, M.O. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 13, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holford, N. A time to event tutorial for pharmacometricians. CPT Pharmacometrics Syst. Pharmacol. 2, e43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Therneau, T. A Package for Survival Analysis in S. R package version 2.38. <http://CRAN.R-project.org/package=survival> (2015).
- 37. Venkatraman, E. Seshan. clinfun: Clinical Trial Design and Data Analysis Functions. R package version 1.0.11. <https://CRAN.R-project.org/package=clinfun> (2015).
- 38. Ait‐Oudhia, S. et al Bridging sunitinib exposure to time‐to‐tumor progression in hepatocellular carcinoma patients with mathematical modeling of an angiogenic biomarker. CPT Pharmacometrics Syst. Pharmacol. 5, 297–304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Norden‐Zfoni, A. et al Blood‐based biomarkers of SU11248 activity and clinical outcome in patients with metastatic imatinib‐resistant gastrointestinal stromal tumor. Clin. Cancer Res. 13, 2643–2650 (2007). [DOI] [PubMed] [Google Scholar]
- 40. Motzer, R.J. et al Activity of SU11248, a multitargeted inihibitor of vascular endothelial growth factor receptor and platelet‐derived growth factor receptor, in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 24, 16–24 (2006). [DOI] [PubMed] [Google Scholar]
- 41. Van Cutsem, E. , Cervantes, A. , Nordlinger, B. , Arnold, D. & ESMO Guidelines Working Group . Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann. Oncol. 25(suppl. 3):iii1–9 (2014). [DOI] [PubMed] [Google Scholar]
- 42. Maitland, M.L. et al Identification of a variant in KDR associated with serum VEGFR2 and pharmacodynamics of Pazopanib. Clin. Cancer. Res. 21, 365–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Apellániz‐Ruiz, M. et al Evaluation of KDR rs34231037 as predictor of sunitinib efficacy in patients with metastatic renal cell carcinoma. Pharmacogenet. Genomics 27, 227–231 (2017). [DOI] [PubMed] [Google Scholar]
- 44. Leppänen, V.M. et al Structural and mechanistic insights into VEGF receptor 3 ligand binding and activation. Proc. Natl. Acad. Sci. USA 110, 12960–12965 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Funakoshi, R. , Lee, C.H. & Hsieh, J.J. A systematic review of predictive and prognostic biomarkers for VEGF‐targeted therapy in renal cell carcinoma. Cancer Treat. Rev. 40, 533–547 (2014). [DOI] [PubMed] [Google Scholar]
- 46. Harmon, C.S. et al Circulating proteins as potential biomarkers of sunitinib and interferon‐ɑ efficacy in treatment‐naïve patients with metastatic renal cell carcinoma. Cancer Chemother. Pharmacol. 73, 151–161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rini, B.I. , Campbell, S.C. & Escudier, B. Renal cell carcinoma. Lancet 373, 1119–1132 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material S1 List of covariates tested with the respective models.
Supplementary Material S2 NONMEM codes.