

Abstract
IgG antibodies mediate a diversity of immune functions by coupling of antigen specificity through the Fab domain to signal transduction via Fc-Fc receptor interactions. Indeed, balanced IgG signaling through Type I and Type II Fc receptors is required for the control of pro-inflammatory, anti-inflammatory, and immunomodulatory processes. In this review, we discuss the mechanisms that govern IgG-Fc receptor interactions, highlighting the diversity of Fc receptor-mediated effector functions that regulate immunity and inflammation, as well as determine susceptibility to infection and autoimmunity, and responsiveness to antibody-based therapeutics, and vaccine responses.
Keywords: IgG, Fc receptors, effector function, Immunomodulation, Immunity, Inflammation
Introduction
IgG is the primary antibody type secreted by memory B cells and plasma cells and is the dominant isotype involved in adaptive immune responses. The activity of IgG antibodies is determined by the Fab domain, which binds to antigen, and the Fc domain, which signals through distinct types of receptors (Fcγ receptors; FcγRs) to mediate cellular effector functions that are required for in vivo protection against many infections and malignancies. In addition to directly mediating protective immune functions, IgG immune complexes signal through FcγRs to modulate adaptive immune responses, enhancing T-cell activation and selection of high affinity B cells. Here, we discuss IgG signaling pathways that are involved in mediating immunity and review recent studies demonstrating the critical role of signaling by antibodies in vivo.
Structural and Functional Heterogeneity of IgG
Because the majority of FcγRs exhibit low affinity for monomeric IgG, signaling by IgG molecules is mediated by multivalent IgG-antigen immune complexes that enable avidity-based Fc-FcγR interactions and, in turn, FcγR aggregation and signaling. The specific FcγR engaged by a given Fc domain is dictated by Fc structure, which is determined by the IgG subclass and composition of an N-linked glycan present at Asn297 within the CH2 domain of the Fc (Figure 1A) (1, 2). On the basis of binding either an open or closed conformation taken by the Fc domain, FcγRs can be distinguished as Type I or II, respectively. These two receptor families transduce distinct effector and modulatory pathways and thus it is the structure of the Fc domain that dictates IgG signaling.
Figure 1. Overview of the structural characteristics of IgG (A) and FcγRs (B).
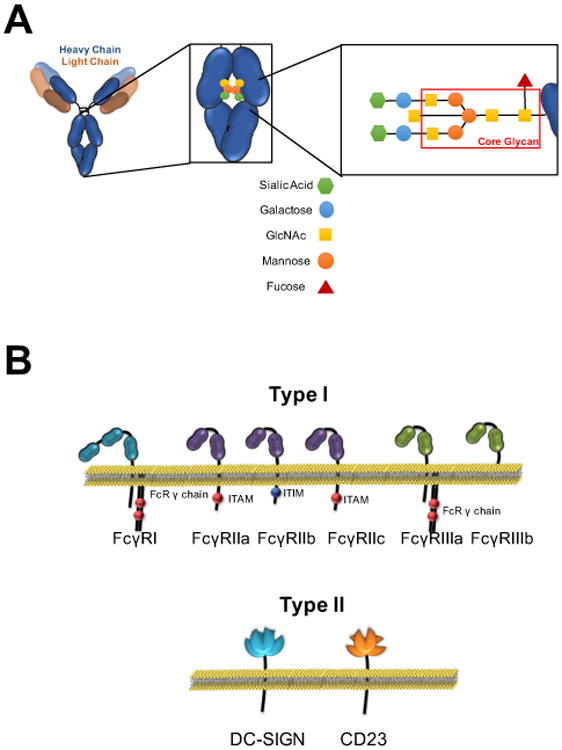
IgG antibodies comprise two identical heavy and light chains linked by inter-chain disulfide bonds. At the core of the Fc domain, an N-linked glycan structure is conjugated at a highly-conserved site at the CH2 domain (Asn297 for human IgG1), which regulates the conformation of the Fc domain and determines binding to Type I and Type II FcγRs. The structure and composition of the Fc-associated glycan can be dynamically regulated through the addition of specific sugar moieties to the core glycan structure (highlighted in the red box), which in turn determines the binding affinity of the Fc domain for Type I and Type II FcγRs. Despite their common property to interact with the IgG Fc, Type I and Type II FcγRs present distinct structural and functional differences and have differential capacity to induce diverse immunomodulatory consequences that affect several aspects of innate and adaptive immunity.
IgG is composed of two identical heavy chains and two identical light chains, linked by inter-chain disulfide bridges. The subclass of IgG is determined by protein sequence of the heavy chain, with all subclasses pairing with either κ or λ light chains. A basic Y-shaped structure is common to all IgG subclasses and results from a hinge that separates the CH1 and light chains, comprising the Fab domain, from the CH2 and CH3 domains that constitute the Fc domain (Figure 1A). While elaboration of a κ vs. λ light chain is not known to modulate IgG activity, the heavy chain subclass can have a profound impact on the in vivo activity of IgG antibodies, as it is one of the two determinants of Fc-FcγR engagement and thus IgG-mediated signaling. Humans and mice produce four different IgG heavy chain subclasses, termed IgG1-4 in humans and IgG1, 2a (or 2c in some inbred strains), 2b, and 3 in mice. Protein homology between subclasses of human or mouse IgG is over 90%, but differences that exist at the hinge-proximal CH2 domain, the interface of Fc-FcγR interactions, confer distinct biological activities to IgG subclasses by determining FcγR binding affinity. Each subclass is distinct in its ratio of binding to activating over inhibitory FcγRs (A/I ratio), with human IgG1 and IgG3 or mouse IgG2a/2c having the highest A/I ratios. In addition to distinct FcγR binding affinities, the abundance of circulating IgG subclasses varies with IgG1 in highest abundance in humans, followed by IgG2>IgG3>IgG4.
Along with the heavy chain subclass, composition of the biantennary N-linked glycan attached at Asn297 of human CH2 domains is the second determinant of Fc domain structure. A core heptasaccharide glycan is required to maintain the Fc domain in a conformational state that is permissive for Type I or Type II FcγR binding. The core structure of this glycan can be modified by additional saccharide units that determine specific Fc-FcγR interactions (Figure 1A). For example, sialylated glycoforms are thought to destabilize the quaternary structure of the Fc domain, conferring binding capacity of Fc to Type II FcγRs (3). Glycan structures lacking a core fucose residue enhance affinity for the activating Type I FcγRIIIa through stabilized glycan-glycan interactions between the Fc domain and FcγRIIIa (4). Fc glycan modifications are dynamic, with major changes reported to occur during pregnancy (5), in some malignancies (6), in response to vaccination (7), and during viral infection in select individuals (TTW and JVR, unpublished observation). Specific inflammatory diseases are also associated with Fc glycan modifications, such as diminished galactosylation and sialylation on disease-associated IgG specificities in rheumatoid arthritis (8) and in Wegener's granulomatosis (9).
Type I and Type II Fcγ receptors
Type I FcγRs belong to the immunoglobulin superfamily and share many structural and functional properties (Figure 1B). The extracellular domain of Type I FcγRs is comprised of either two or three Ig domains and interacts with the hinge proximal region of the CH2 domain of IgG in a 1:1 stoichiometric complex. Type I FcγRs share highly conserved intracellular motifs that mediate signaling upon receptor crosslinking by IgG immune complexes, yet distinct functional outcomes of signaling define each receptor. Type I FcγRs can be classified as activating or inhibitory based on the presence of intracellular activating (immunoreceptor tyrosine activation motif (ITAM)) or inhibitory (immunoreceptor tyrosine inhibition motif (ITIM)) signaling motifs. Three activating FcγRs are expressed in mice (FcγRI, FcγRIII, and FcγRIV) and humans (FcγRI, FcγRIIa, and FcγRIIIa), and a single inhibitory FcγR, FcγRIIb, is expressed in both species. Each FcγR has a distinct pattern of cellular expression (Table 1) with engagement of activating FcγRs inducing positive mechanisms such as antibody-dependent cellular cytotoxicity (ADCC), phagocytosis, and pro-inflammatory cytokine production. Signaling through the inhibitory receptor, FcγRIIb, results in negative functions such as inhibition of inflammatory immune responses. FcγRI is the only high affinity FcγR, capable of binding monomeric IgG (KD 10−9–10−10 M for human IgG1); all other Type I FcγRs exhibit lower affinity for IgGs, with precise affinities varying by subclass (KD for human IgG1: 10−5–10−7 M). Whether Fc domains within IgG immune complexes engage activating or inhibitory Type I FcγRs is determined by the IgG subclass and the Fc glycan composition, as described above (1).
Table 1. Expression pattern of Type I and Type II FcγRs among the various leukocyte types.
| Type I | Type II | |||||||
|---|---|---|---|---|---|---|---|---|
| FcγRI | FcγRIIa | FcγRIIb | FcγRIIc | FcγRIIIa | FcγRIIIb | DC-SIGN | CD23 | |
| B cells | - | - | + | - | - | - | - | + |
| T cells | - | - | - | - | - | - | - | # |
| NK cells | - | - | -^ | +^ | + | - | - | - |
| Monocytes | + | + | + | - | +/- | - | +/- | # |
| Neutrophils | # | + | + | - | - | + | - | # |
| Eosinophils | # | + | + | - | - | # | - | # |
| Macrophages | +/- | + | + | - | +/- | - | +/- | # |
| Dendritic Cells | # | + | + | - | # | - | + | - |
constitutive expression;
no expression;
inducible expression;
expression restricted to certain subsets;
expression depends on FcγR genetic variation.
Type II FcγRs are C-type lectins that have been studied in various biological contexts for some time, but their identification as receptors for IgG antibodies occurred relatively recently (3, 10). Type II FcγRs with known functional activities include DC-SIGN (mouse orthologue SIGN-R1) and CD23, which share an oligomeric structure that is stabilized through an α-helical coiled-coil stalk motif at the extracellular receptor domain. The primary determinant of IgG binding to Type II FcγRs is the presence of sialylated Fc glycoforms, which destabilize the Fc structure enabling assumption of a conformation that is the ligand for Type II FcγRs. As with the majority of Type I FcγRs, Type II FcγRs are low affinity for their Fc ligand (KD for hIgG1: 10−5–10−7 M) and they have distinct immunomodulatory activities: DC-SIGN signaling is involved in the anti-inflammatory activity of IgG through the production of cytokines such as IL-33 that are involved in setting thresholds for inflammatory myeloid cell activation (11), while CD23 mediates selection of high affinity B cells during maturation of antibody responses (7); both receptors may be involved in additional modulatory functions that have yet to be characterized. Specific Type I and Type II FcγR signaling pathways and downstream functions are described throughout this chapter.
Species specificity in IgG signaling
All known animal models, including non-human primates, express species-specific IgG subclasses and FcγRs that are unique in structure, function and cellular distribution. Such inter-species variation limits interpretation of data from mixed-species systems, i.e. in experiments using human IgGs in non-human primates or in mice. While affinities between human IgGs and FcγRs from other species can be determined and may guide interpretation of mixed-species systems, use of data from these systems to reliably inform development of antibody therapeutics is, at this time, nearly impossible. Models with IgGs and FcγRs that are species-matched can be easier to interpret when the animal system has been well-defined, as with C57BL/6 and BALB/c mice. Still, major hurdles exist. One example of discord that can prevent extrapolation of in vivo mouse data is the difference in receptor expression on human and mouse monocyte-derived dendritic cells. For example, human monocyte-derived dendritic cells express one activating Type I receptor, FcγRIIa, along with the inhibitory FcγRIIb, whereas mouse monocyte-derived dendritic cells express all murine Type I FcγRs. Another example from non-human primates is found in neutrophil FcγR expression: while human neutrophils express FcγRIIa, FcγRIIb and FcγRIIIb, neutrophils from non-human primate species like rhesus macaques do not express FcγRIIIb but do express FcγRI in addition to FcγRIIa and FcγRIIb. These differences in receptor expression can result in major differences in cellular functions – one example is mouse ADCC, which is mediated primarily by mouse IgG2a engagement of murine FcγRIV on macrophages; mouse NK cells do not express FcγRIV and thus do not significantly contribute to antibody mediated cellular depletion in vivo by IgGa (12). In contrast, ADCC in humans is mediated through the human IgG1 subclass engaging FcγRIIIa, which is expressed by both macrophages and NK cells; both cell types have been suggested to contribute to ADCC in humans and are influenced by the allele of FcγRIIIa expressed and the fucosylation state of the IgG1 Fc. The best model currently available for the in vivo study of human FcγRs is a mouse model in which endogenous mouse FcγR genes were deleted and human FcγRs are instead expressed under the control of their endogenous human promoters. This FcγR humanized mouse strain recapitulates the human Type I FcγRs in form, function and cellular expression (13).
Interplay Between Type I and Type II FcγRs in Regulating Immune Responses
Interactions between FcγRs and the Fc domain of IgG antibodies activate signaling pathways that vary depending on the FcγR and cell type that is engaged; each Fc-FcγR interaction has the potential to modulate a diverse set of immunoregulatory processes (Table 2) (1). As discussed in the previous section, the affinity of IgG for different FcγRs is regulated by the Fc domain structure, which is determined by the primary amino acid sequence of the Fc domain (IgG subclass) and the associated N-linked glycan. While Fc structure regulates Fc-FcγR interactions and subsequent signaling events, several factors influence the capacity of FcγR-mediated signaling to modulate immune processes. For example, each FcγR exhibits a distinct expression pattern among leukocyte populations, with different cell subsets commonly co-expressing more than one FcγR type (Table 1) (1, 13). Expression levels and, to a degree, the pattern of FcγR expression can fluctuate during leukocyte differentiation and activation, with each cell type generally co-expressing more than one FcγR type at any time (Table 1). Additionally, several FcγR genes can be induced acutely by chemokines and/or cytokines present at sites of inflammation, infection, or tissue injury, thereby influencing FcγR-mediated expression and, in turn, effector activities (11, 14, 15). The specific FcγR expression pattern on a cell determines whether receptor crosslinking will lead to cellular activation, ADCC and phagocytosis, release of cytokines and chemokines, leukocyte differentiation and survival, and/or modulation of T- and B-cell responses (16). In this section, we will focus on the key role of IgG antibodies in modulating immune signaling through selective engagement of different FcγRs (Table 2).
Table 2. Overview of FcγR-mediated effector functions in different leukocyte types.
| Cell type | Role of FcγR | Short-term Effects | Long-term Effects | ||
|---|---|---|---|---|---|
| + | - | ||||
| Myeloid | Granulocytes | FcγRIIa FcγRIIIb* |
FcγRIIb | Degranulation ROI production Phagocytosis |
Release of pro-inflammatory molecules Cell survival and motility Impact on other myeloid leukocytes |
| Monocytes | FcγRIIa FcγRIIIa# |
FcγRIIb | Phagocytosis Cytokine & chemokine expression | Monocyte recruitment and differentiation, cell survival, stimulation of pro-inflammatory pathways | |
| Macrophages | Fc γRIIa FcRIIIa DC-SIGN |
FcγRIIb | Phagocytosis Cytokine & chemokine expression | Influence macrophage polarization and responsiveness to IgG-mediated inflammation | |
| Platelets | FcγRIIa | Degranulation, platelet activation | Platelet binding to leukocytes, impact on leukocyte functional activity | ||
| Dendritic cells | FcγRIIa | FcγRIIb | Cell maturation, upregulation of co-stimulatory molecules | Enhanced antigen processing and presentation, impact on T-cell responses | |
| Lymphoid | B cells | CD23 FcγRIIb |
B cell selection | Generation of high-affinity IgG responses | |
| Plasma cells | FcγRIIb | B cell survival | Regulation of plasma cell apoptosis and control of IgG production | ||
| NK cells | FcγRIIIa | Cell activation Degranulation |
Cytotoxicity, cell survival, impact on other effector leukocytes, generation of immune complexes | ||
activates downstream effects;
inhibits downstream effects
function unknown, possible activating;
expressed on certain subsets.
Cytotoxicity of IgG-coated targets
All cytotoxic effector functions induced upon FcγR-IgG interactions are mediated through activating Type I FcγRs. One of the first activities described for FcγRs is their capacity to mediate clearance or cytotoxicity of IgG-coated targets, ranging from small antigens (toxins, infectious pathogens, etc.), to whole cells. Crosslinking of activating Type I FcγRs by IgG immune complexes triggers a cascade of signaling events that culminate in the elimination of the IgG-opsonized target. Following receptor clustering and oligomerization by IgG immune complexes, phosphorylation of receptor ITAM domains occurs, promoting the recruitment and activation of several tyrosine kinases of the Src and Syk families. This leads to activation of the PI3K-PKC pathway and to Ca2+ mobilization and consequently cellular activation (17, 18). In addition to these early signaling events, several other late signaling pathways are activated, including the MEK and MAP family kinases and the Ras pathway, which in turn induce the transcription of pro-inflammatory cytokines and chemokines with divergent effects on cellular survival and differentiation (19).
Although intracellular signals transduced upon receptor crosslinking are common to all leukocytes expressing activating Type I FcγRs, the precise biological responses following FcγR engagement vary greatly between different leukocyte types. For example, granulocytes mediate biological effects within minutes following activation of FcγR-mediated pathways by IgG immune complexes (20). Granulocyte effector functions aim to destroy invading pathogens, often by releasing microbicidal molecules that are either preformed and stored in specialized cytoplasmic granules, or that are generated, de novo, upon FcγR engagement. For example, FcγR crosslinking leading to the activation of Syk and Src family kinases, triggers the production of reactive oxygen intermediates (ROI) through formation and activation of the NADPH-dependent oxidase complex (21, 22). Additionally, FcγR crosslinking induces activation of the PKC pathway and subsequent increase in intracellular Ca2+ stimulates mobilization and release of pre-formed cytotoxic or pro-inflammatory molecules (20). These molecules vary among granulocyte subtypes, but generally include serine proteases (elastase, cathepsins, and collagenases) and other enzymes (peroxidase, alkaline phosphatase), antimicrobial peptides and proteins (lysozyme, defensins, lactoferrin) as well as lipid mediators (leukotrienes) with potent pro-inflammatory activity (23-25). In addition, binding of multimeric IgG immune complexes to FcγRs triggers receptor crosslinking and oligomerization, which induces receptor internalization and activation of downstream signaling pathways that facilitate actin remodeling, endosomal uptake and sorting (26, 27). Indeed, granulocytes are among the most potent phagocytic leukocytes and can efficiently engulf and eliminate IgG-opsonized bacteria, fungi and other cellular antigens (28). These effects, along with the production and release of ROI and cytotoxic proteolytic compounds represent the two main strategies that granulocyte employ to achieve cytotoxicity of IgG-coated targets.
Similar signaling processes are also initiated upon engagement of the activating FcγRIIIa on NK cells, which triggers cellular activation and degranulation. In an analogy to granulocyte activation and degranulation following FcγR crosslinking, engagement of FcγRIIIa on NK cells is associated with ITAM phosphorylation and rapid increase in the intracellular Ca2+ levels that triggers the release of granular content, which include perforin and granzymes (29, 30). The release of these potent enzymes in close proximity to IgG-coated targets induces the formation of pores at the target cell membrane and stimulation of pro-apoptotic pathways that ultimately lead to cell death.
In contrast to granulocytes and NK cells, elimination of IgG-opsonized targets by macrophages is achieved primarily through phagocytic processes, and not through the immediate release of cytotoxic and cytolytic compounds. Indeed, several studies have shown that FcγR-expressing macrophages mediate cytotoxic activity in vivo through phagocytic clearance of IgG-coated cells. In seminal experiments using IgG-opsonized erythrocytes, their uptake was observed to be mediated exclusively by splenic macrophages in an FcγR-dependent process (31, 32). Similarly, in mice humanized for FcγRs, specific depletion of macrophages by clodronate liposomes completely abrogated the cytotoxic activity of antibodies against various surface antigens, further confirming the central role of macrophages in IgG-mediated cytotoxicity in vivo (12).
With the exception of NK cells that only express FcγRIIIa, activating Type I FcγR signaling is always balanced on effector leukocytes by signaling through co-expressed inhibitory FcγRIIb; thus, FcγR-mediated effector functions are the result of cumulative signaling from activating and inhibitory Type I FcγRs (2). For example, the in vivo activity of cytotoxic therapeutic antibodies depends on a signaling balance that is weighted towards activating FcγRs (2, 12, 33-36). This was demonstrated in early experiments utilizing an antibody (TA99) against a melanosome protein, gp75, in which it was evident that tumor clearance required activating FcR engagement and IgG subclass or glycovariants of the antibodies with enhanced capacity to engage activating FcγRs exhibited improved in vivo activity in a mouse model of metastatic melanoma (2).
Additional evidence on the importance of the balanced activity of activating and inhibitory FcγRs in regulating IgG-mediated inflammation comes from genetic association studies of FcγR genetic variants. For several autoimmune and chronic inflammatory pathologies, disease susceptibility and/or severity is associated with genetic variants of FcγR-encoding genes in the form of single nucleotide polymorphisms or copy number variants that influence receptor expression, activity, or IgG binding affinity (37). For example, SLE patients are enriched for the low binding alleles for FcγRIIa, FcγRIIIa, and FcγRIIIb, as well as lower copy number of the FCGR3B gene, which is characterized by reduced FcγRIIIb expression levels (37-40). These variants exhibit reduced clearance of IgG immune complexes, likely increasing tissue immune complex deposition and inflammation. In addition, low copy number of FCGR3B is associated with the induction of ectopic expression of the inhibitory FcγRIIb in NK cells, likely reducing cytotoxic responsiveness (40). In contrast, high affinity variants of FcγRIIIa (V158) and FcγRIIIb (NA1) are commonly associated with inflammatory diseases, such as autoantibody-induced vasculitis, idiopathic thrombocytopenia purpura, and rheumatoid arthritis (37). These cumulative studies demonstrate the key role of FcγR-IgG interactions in mediating cytotoxic effector responses, and highlight that balanced activating and inhibitory FcγR signaling is required for optimal effector activity without pathogenic immune activation.
Antigen Processing and Modulation of antigen-presenting cell function
Despite structural and functional differences, the different Type I FcγRs follow an identical pattern of receptor crosslinking and oligomerization that ultimately leads to sequestration of IgG-FcγR complexes in intracellular vesicles for lysosomal degradation and antigen processing (26). Indeed, all Type I FcγRs (with the exception of the FcγRIIb1 splice variant) have the capacity to mediate phagocytosis of IgG-opsonized targets (41). The efficiency of FcγR-mediated phagocytosis varies greatly between leukocyte types and is dependent on differentiation stage, cytoskeletal motility, cellular size, and expression levels of Type I FcγRs and other receptors such as pattern recognition receptors. Although both activating and inhibitory Type I FcγRs can mediate phagocytosis in myeloid phagocytes, internalization of IgG-opsonized targets through activating FcγRs mediates more potent effector responses (42-44). For example, phosphorylation of ITAM domains after FcγR crosslinking leads to pro-inflammatory signaling that mediate cellular activation and pro-inflammatory cytokine and chemokine expression, which modulates leukocyte differentiation, functional activity, and survival. Uptake of IgG-coated targets through activating FcγRs is associated with enhanced endosomal maturation and lysosomal fusion, leading to more efficient antigen processing and presentation on MHC class-II molecules (42, 45). Antigen processing and presentation resulting from FcγR-mediated internalization of immune complexes results in enhanced T-cell activation (36, 46-48).
Apart from the direct effects on antigen uptake, processing and presentation to MHC class-II molecules, pro-inflammatory signals following Type I FcγR engagement by IgG immune complexes also influence several aspects of the activity and function of antigen-presenting cells. For example, signaling through FcγRs can influence macrophage differentiation and polarization, as well as dendritic cell maturation and activity, which can affect T-cell activation and tolerance. In steady-state conditions, human monocytic dendritic cells express two Type I FcγRs: the inhibitory FcγRIIb and the activating FcγRIIa (1). Since these receptors are co-expressed on the surface of dendritic cells, pro-inflammatory signals from FcγRIIa are antagonized by the inhibitory signaling activity of FcγRIIb. This balance between activating and inhibitory FcγRs represents a key regulatory process of dendritic cell function, necessary to prevent uncontrolled dendritic cell maturation and maintain peripheral tolerance (49). Indeed, the expression levels of these two FcγRs are tightly regulated under homeostatic conditions, whereas certain mediators present at the inflammatory milieu can influence the expression of FcγRIIb or induce expression of additional activating Type I FcγRs, altering thereby the threshold for IgG-mediated dendritic cell maturation. For example, IFN-γ induces the expression of the activating Type I FcγR, FcγRI and downregulates FcγRIIb expression. In contrast, IL-4 stimulates the expression of the inhibitory FcγRIIb (11, 14, 15, 48, 50). This finely-tuned interplay between activating and inhibitory FcγR expression determines the threshold for FcγR-mediated dendritic cell maturation and responsiveness to other stimuli (e.g. TLR ligands). In the absence of co-stimulatory signals, stimulation of dendritic cells with IgG immune complexes often fails to induce robust cell maturation. Activation of pro-inflammatory signaling pathways through innate immune receptors is generally required to overcome the inhibitory activity of FcγRIIb and induce dendritic cell activation (12, 36, 46, 48, 50).
Skewing the balance of activating and inhibitory FcγR signaling has profound consequences to the maturation of antigen-presenting cells (APC) and subsequent generation of T-cell responses. For example, preferential engagement of dendritic cell FcγRIIa through Fc domain engineering of anti-CD20 antibodies resulted in improved antigen-specific T-cell responses in a model of CD20+ lymphoma (12). Likewise, blocking FcγRIIb ligand binding activity using function blocking antibodies or genetic deletion of the Fcgr2b gene greatly augments IgG immune complex-mediated cell maturation, resulting in upregulation of co-stimulatory molecules as well as enhanced antigen presentation and T-cell activation (36, 48, 50). These studies further highlight the critical balance in signaling between different types of FcγRs that must be achieved to regulate dendritic cell activity associated with generation of robust adaptive immune responses.
As with dendritic cells, FcγR-mediated signaling can also influence the differentiation status and function of macrophages. Human macrophages exist as a continuum of diverse differentiation states with distinct functional and phenotypic characteristics (51). Although macrophage polarization was originally classified into two broad phenotypes – M1 and M2, that are induced by Th1 (IFN-γ) and Th2 (IL-4) cytokines, respectively, stimulation through FcγR-mediated pathways represents a distinct determinant for macrophage polarization, resulting in the induction of a characteristic activation phenotype (51). Engagement of activating Type I FcγRs and stimulation of downstream signaling pathways in macrophages is associated with induction of pro-inflammatory cytokine and chemokine expression that alter their differentiation, function, and survival (16). However, when activating FcγR signals are coupled with TLR signaling, like TLR4 in non-polarized macrophages, this synergistic signaling activity triggers induction of a specific polarization state that resembles the M2 phenotype (52, 53). This phenotype, also termed “regulatory” or M2b, is characterized by production of IL-10, IL-1 and IL-6 as well as by increased migratory and phagocytic capacity (52, 53). Apart from Type I FcγRs, signaling through Type II FcγRs on macrophages can influence their functional activity and responsiveness. For example, studies designed to identify the mechanistic basis of the anti-inflammatory activity of high dose intravenous immunoglobulin (IVIG) treatment showed that engagement of the Type II FcγR, DC-SIGN, by sialylated Fc domains of IgG antibodies triggers the expression of IL-33 by regulatory macrophages. This secreted IL-33, a potent Th2-polarizing cytokine, stimulates production and release of IL-4 by basophils, which, in turn, upregulates FcγRIIb on effector macrophages (11). Increased FcγRIIb expression alters the balance between activating and inhibitory Type I FcγR signaling, raising the threshold for IgG-mediated inflammation in macrophages.
In addition to macrophage polarization and differentiation, several studies utilizing mouse strains with genetic deletion of the different types of FcγRs provide evidence for the contribution of FcγR-mediated signaling to macrophage function and in vivo activity. For example, mice lacking activating Type I FcγRs are non-responsive to IgG-mediated inflammation (32, 52), whereas FcγRIIb-/- mice exhibit lower macrophage activation threshold upon challenge with immune complexes and present a more severe phenotype in models of immune complex-induced anaphylaxis, arthritis, and alveolitis (32, 52, 54). Additionally, in mouse strains deficient for either FcγRIIb or SIGNR1, the mouse orthologue for human DC-SIGN, administration of high dose IVIG or sialylated Fcs fails to provide anti-inflammatory activity in models of IgG-mediated inflammation (10, 11). These studies clearly highlight the importance of Type I and Type II FcγR signaling in the regulation of the activity and differentiation of antigen-presenting cells, with diverse consequences for immunity and inflammation (Table 2).
B cell selection and regulation of IgG production
In the majority of leukocyte populations, expression of activating FcγRs is coupled to expression of the inhibitory FcγRIIb. In contrast, B cells constitutively express FcγRIIb throughout their development, without co-expressing any activating Type I FcγRs. FcγRIIb on B cells functions to balance the activating signals triggered by binding of antigen to the B-cell receptor (BCR). In the absence of BCR signaling, exclusive engagement of B cell FcγRIIb induces pro-apoptotic signals, whereas FcγRIIb – BCR co-engagement attenuates these apoptotic signals, leading to B cell survival (55, 56). This regulatory mechanism controls B cell selection, as B cells with low or no affinity for the antigen are eliminated through apoptosis, whereas those with high affinity receptors receive pro-survival signals (56). Transition of B cells to plasma cells is associated with loss of BCR expression. In the absence of opposing BCR signaling, FcγRIIb engagement by IgG immune complexes leads to plasma cell apoptosis; this negative feedback mechanism regulates plasma cell survival to prevent uncontrolled IgG production (56-58).
As described above, pro-apoptotic signaling activated through engagement of B-cell FcγRIIb alone is attenuated by co-aggregation with BCR. At the same time, thresholds for B cell activation through BCR are determined by cis-signaling though FcγRIIb, which can occur when antigen within an IgG immune complex binds BCR and Fc domains within the same complex co-engage FcγRIIb. Immune complex engagement of FcγRIIb leads to phosphorylation of the ITIM domains of FcγRIIb, in turn recruiting SHIP and SHP2 phosphatases (55, 56, 58). These phosphatases directly interfere with BCR signaling by hydrolyzing phosphoinositide intermediates, such as phosphatidylinositol-3,4,5-triphosphate, thus preventing recruitment and activation of PH-domain containing kinases such as PLCγ and BTK and blocking induction of downstream signaling pathways (55, 56, 58). Studies using FcγRIIb-deficient mice have revealed how critical the contribution of FcγRIIb signaling is in setting the activation threshold for B cells, enabling high affinity antibody responses and maintaining peripheral tolerance. Indeed, in the absence of FcγRIIb, antibody responses are characterized by high titer and low affinity due to absence of selection of B cells based on affinity of BCR (49, 59). In addition, genetic variants of FCGR2B that influence receptor activity or expression are associated with increased susceptibility to autoimmunity. For example, promoter haplotypes of the FCGR2B gene that decrease FcγRIIb expression levels are associated with SLE susceptibility and likely contribute to disease pathogenesis (60, 61). Additionally, several studies have reported an association between a single nucleotide polymorphism (I232T) at the transmembrane region of FcγRIIb with SLE (62). This polymorphism affects receptor partitioning to lipid raft membrane microdomains, thus diminishing FcγRIIb signaling activity. This central role of FcγRIIb in regulating BCR signaling activity represents a key determinant in the generation of antibody responses, B-cell selection and affinity maturation, as well as maintenance of peripheral tolerance (49, 55, 56, 59).
Apart from the Type I FcγRIIb, B cells express the Type II FcγR CD23, which also participates in the regulation of B-cell activity and generation of high affinity antibody responses. Early studies on the role of CD23 showed that CD23 regulates IgE production and mediates the capture of IgE-containing immune complexes and transfer to CD11c+ APCs, leading to enhanced CD4+ T-cell immunity (63). As CD23 has also the capacity to interact with sialylated IgG Fcs, more recent studies have demonstrated that IgG-CD23 interactions can also modulate B-cell function. For example, studies have shown that IgG immune complexes containing sialylated Fc domains can drive the development of high affinity IgG responses (3, 7). A recent study identified the mechanisms by which CD23 influences affinity of IgG responses; engagement of CD23 by sialylated IgG immune complexes was shown to upregulate B-cell expression of FcγRIIb, thus elevating the threshold for B-cell selection, resulting thereby in high affinity IgG responses. This mechanism was tested using a model of influenza hemagglutinin (HA) immunization, in which it was shown that vaccination with sialylated Fc anti-HA immune complexes induced higher affinity anti-HA IgG responses with increased breadth of in vivo protective activity. This effect was lost in CD23-/- mice or when wild-type mice were vaccinated with asialylated anti-HA immune complexes (7). These studies highlight the synergistic activity of Type I and Type II FcγRs to shape humoral immune responses by directly regulating B-cell selection, IgG affinity, and plasma cell survival.
In the next sections, using specific examples of antibodies against neoplastic or infectious diseases, we will review the role and function of Type I and Type II FcγRs in the in vivo protective activities of these antibodies. Numerous monoclonal antibodies (mAbs) with antitumor or antimicrobial activity are currently in development or clinical use, representing promising therapeutic modalities for the treatment of several infectious diseases and tumors. Unravelling the complexity of Fc-FcγR interactions and the requirements for activation of specific FcγR-mediated pathways by these therapeutic mAbs to achieve full protective activity has led to the development of Fc-optimized IgG therapeutics with improved clinical efficacy.
Signaling and Function of Anti-Tumor Antibodies
IgG antibodies are now routinely used for the treatment of a variety of human malignancies. The relative success of rituximab, the first FDA-approved mAb developed to treat cancer led to the evaluation of numerous other IgG cancer therapeutics, resulting in many mAbs being brought to the clinic over the last two decades. Cancer-targeting therapeutic antibodies can be mechanistically distinguished by the type of cell targeted by their Fab domains. They target either the tumor cells to directly mediate cytotoxicity, or immune cells to potentiate anti-tumor immunity. It is now clear that, independent of the cell type being targeted by the Fab domain, the Fc domain of anti-tumor therapeutic antibodies must be optimized to engage the appropriate class of FcγRs on effector cells to achieve potent clinical activity. Therefore, the IgG isotype and specific Fc scaffold of these antibodies are critical for mediating full therapeutic efficacy.
Cytotoxic anti-tumor antibodies
The development of hybridoma technology along with identification of antigens that were specific or over-expressed on tumor, but not normal cells, enabled the exploitation of the fine specificity of antibodies to precisely target tumor cells. The first mAbs that emerged as cancer therapeutics targeting tumor-associated antigens were rituximab, an anti-CD20 mAb that targets malignant B cells in non-Hodgkin's lymphoma and chronic lymphocytic leukemia, trastuzumab, an anti-human epidermal growth receptor 2 (HER2)/neu mAb that targets HER2+ tumor cells in breast cancers and cetuximab, an anti-epidermal growth factor receptor (EGFR) mAb for the treatment of metastatic colorectal cancer, metastatic non-small cell lung cancer and head and neck cancer. Multiple mechanisms of action were proposed for these antibodies: Fab-mediated pathways, such as the induction of cell death signaling pathways (rituximab) or receptor downregulating and blocking (trastuzumab and cetuximab), and Fc-mediated pathways, such as complement-dependent cytotoxicity (CDC) or ADCC, were proposed based primarily on in vitro studies in a variety of experimental systems. Among these mechanisms, only engagement of activating Type I FcγRs for depletion of target cells was found to be an absolute requirement for the in vivo activity of tumor-targeting mAbs, including rituximab, trastuzumab, and cetuximab.
The most striking evidence for FcγR-mediated mechanisms determining the anti-tumor activity of cytotoxic antibodies in vivo comes from work showing that anti-tumor mAbs can be therapeutically potent in wild-type mice, but not in mice lacking the FcR γ chain, which is required for the expression and signaling of all murine activating FcγRs (64). Moreover, the anti-tumor effect of mAb therapy was enhanced in mice lacking the inhibitory FcγRIIb (47), implicating the signaling balance between activating and inhibitory FcγRs in determining the potency of anti-tumor cytotoxic mAbs. This was further demonstrated through characterizing cytotoxic activities of anti-tumor mAbs of different murine IgG subclasses (2). When the variable region of the anti-gp75 TA99 mAb clone was produced with different IgG subclass scaffolds, the cytotoxic effect of the mAb was found to directly correlate with the binding affinity ratio of activating FcγR to inhibitory FcγR, or the A/I ratio. The mouse IgG2a subclass that has the highest A/I ratio demonstrated the most potent in vivo activity. In preclinical studies, activating FcγR-expressing monocytes and/or macrophages, but not NK cells, were shown to be the primary effector cell types mediating elimination of tumor cells by therapeutic mAbs (65-68). A similar role for macrophages in humans is still a matter of debate and needs to be elucidated. The requirement of Fc-FcγR interactions and the impact of the binding affinity of the IgG Fc to activating FcγRs has been confirmed in humans by comparing the response to these therapeutics in patients expressing high or low binding alleles of FcγRIIa and FcγRIIIa. Patients who express higher affinity FcγRIIa and FcγRIIIa alleles have enhanced response rate in lymphoma patients treated with rituximab (69, 70), breast cancer patients treated with trastuzumab (71), and metastatic colorectal cancer patients treated with cetuximab (72).
These animal and clinical studies provided motivation for the development of a second generation of cytotoxic antibodies that exploits Fc-FcγR pathways to improve clinical efficacy. Indeed, Fc engineering to increase the A/I ratio of therapeutic IgG1 antibodies, either by point mutations at the Fc domain (13) or by modifying the composition of the Fc-associated glycan (73), represents an effective strategy to yield therapeutic antibodies with increased potency. Indeed, in preclinical animal studies enhanced in vivo activity was observed when mAbs targeting melanoma associated differentiation antigen gp75, CD19, and CD20 were Fc-engineered though point mutations to enhance affinity for activating FcγRs (12, 13, 74). Similarly, a bisected, afucosylated, Fc-glycoengineered anti-EGFR mAb demonstrated enhanced ADCC activity and resulted in superior in vivo efficacy compared to cetuximab, a non-glycoengineered anti-EGFR mAb (75). This approach has been advanced to the clinic with the FDA approval of obinutuzumab, an afucosylated anti-CD20 mAb that, when combined with chemotherapy, extended progression-free survival of chronic lymphocytic leukemia patients compared to the wild-type IgG1 rituximab plus chemotherapy. Based on these preclinical and clinical studies, many next-generation, Fc-optimized anti-tumor mAbs have been developed and are currently being evaluated for clinical use.
While the clearance of tumor cells is readily achieved by anti-tumor targeting mAbs, evidence has accumulated suggesting that a long-term cellular memory “vaccinal” response can also be induced as a consequence of cytotoxic mAb therapy. Studies using mouse models have demonstrated that CD20 mAb treatment induces anti-tumor T-cell responses (12, 76). Similar effects in humans are supported by the long-lasting responses observed in some patients after a single course treatment with rituximab and the presence of tumor antigen-specific T-cell responses in some B-cell lymphoma patients treated with rituximab (77, 78). A vaccinal effect has also been characterized in patients with HER2- and MUC1-expressing tumors who can develop specific T cell responses after treatment with anti-HER2 and anti-Mucin-1 (MUC1) mAbs (79, 80). Recent studies have elucidated the involvement of FcγR pathways in the induction of anti-tumor vaccinal effects. Using human CD20 as a tumor neoantigen, anti-human CD20 mAb treatment in wild-type and in FcγR knock-out mouse strains revealed that both the primary clearance of the CD20-expressing tumor cells, as well as the generation of memory T-cell responses were dependent on FcγR interactions (12, 76). Studies in FcγR-humanized mice showed that the initial depletion of tumors by cytotoxic effector cells was mediated by FcγRIIIa on monocytes and macrophages, while the formation of a memory T-cell response was dependent on FcγRIIa expression on dendritic cells. Overall, this work suggested a mechanism whereby administration of an anti-CD20 mAb triggered macrophage-mediated killing of tumor cells through FcγRIIIa, leading to IgG immune complex formation and engagement of activating FcγRIIa on dendritic cells; this engagement stimulated dendritic cell activation, maturation, and cross presentation of tumor neoantigens to T cells, leading to the induction of anti-tumor T cell memory. These studies suggest novel strategies for third generation anti-tumor antibodies in which Fc engineering is designed to enhance not only cytotoxicity but also T-cell memory responses by appropriate FcR engagement.
Immunomodulatory Antibodies
Cancer therapy has been revolutionized with the development of immune checkpoint-blocking mAbs targeting the inhibitory T-cell signaling receptors CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) and PD-1 (programmed death-1) (81). Clinical studies have supported use of these mAbs either as monotherapies or in combination, thus validating the approach of targeting immune cells with specific mAbs (82-84). These mAbs act by targeting co-regulatory receptors on the surface of immune cells in order to enhance anti-tumor immune responses. Mechanistically, immunomodulatory mAbs act as agonists or antagonists to stimulate or block, respectively, a target antigen. It is now clear that many of these therapeutic mAbs depend on Fc-FcγR interactions to achieve full therapeutic activity. Animal studies have highlighted the importance of activating Type I FcγRs for the anti-tumor activity of several immunomodulatory mAbs, such as anti-CTLA-4 (85-87), GITR (glucocorticoid-induced TNFR-related protein) (87), OX40 (88), and PD-L1 (programmed death-ligand 1)(89). These studies elucidated unexpected FcγR-dependent depletion of specific regulatory immune cell populations in the tumor microenvironment (TME), providing therapeutic modulation of the immune cell composition at tumor sites. Thus, independent of their Fab-mediated mechanism aiming to either block or activate inhibitory or stimulating T-cell signals, these mAbs act to deplete target immune cells in an activity similar to that of cytotoxic tumor-targeting mAbs described above. More specifically, the anti-tumor activity of anti-CTLA-4, GITR, and OX40 mAbs is significantly augmented by the depletion of regulatory T cells (Treg) within the TME (90). Despite the wide distribution and expression of these receptors on different T-cell compartments, mAb-mediated targeting and elimination is preferentially directed to intratumoral Treg cells. Treg cell depletion increases the ratio of Teff/Treg cells in the TME, which favors efficient tumor clearance. Simpson et al. (85) demonstrated that this specific effect of anti-CTLA-4 mAb depends on two main factors that distinguish the TME from lymphoid organs. First, Treg cells have higher expression density of surface CTLA-4 compared to effector T cells (Teff) and CTLA-4 expression level is elevated in the tumors compared to lymphoid organs. Thus, higher receptor expression density preferentially targets Treg for ADCC. Secondly, the depletion of the Tregs by CTLA-4 mAbs is dependent on FcγRIV-expressing macrophages which are more abundant and express higher levels of FcγRIV at the tumor site, compared to the draining lymph nodes. By characterizing the activity of different mouse IgG subclasses, Selby et al. (86) further demonstrated the requirement for an Fc scaffold with high A/I ratio in order to mediate the therapeutic activity of CTLA-4 mAbs. Similar requirements for receptor over-expression by intratumoral Treg cells and the presence of myeloid cells expressing activating FcγRs in the TME have been characterized as a general mechanism for additional immunomodulatory mAbs. The therapeutic activity of mAbs targeting the activating receptors GITR and OX40 depends on their ability to modulate the TME by depleting intratumoral Treg cells. An alternative mechanism to immunomodulate the TME has been described for anti-PD-L1 mAbs, which target the ligand for PD-1 (89). PD-L1 mAbs were found to have superior anti-tumor activity when engaged to activating FcγRs to deplete intratumoral myeloid cells, and thereby modulating myeloid cell populations in the TME. This FcγR-dependent mechanism synergized with the FcγR-independent blocking of the inhibitory PD-1/PD-L1 axis for optimal therapeutic efficacy.
Evidence for Treg depletion in patients was recently demonstrated in small groups of melanoma patients treated with ipilimumab, the FDA-approved anti-CTLA4 IgG1 mAb. In these patients, a higher baseline frequency of peripheral non-classical monocytes and tumor-associated macrophages that expressed FcγRIIIa was associated with treatment responsiveness. Furthermore, these non-classical monocytes were shown to mediate Treg cell depletion by ipilimumab ex vivo (91). A second human anti-CTLA-4 mAb that has been used clinically is tremelimumab, which is an IgG2, and therefore has significantly reduced FcγRIIIa binding affinity relative to IgG1 and would be expected to mediate reduced ADCC activity. Despite failing to show any benefit over chemotherapy in a phase III study, tremelimumab treatment exhibits some therapeutic activity in combinational trials assessing tremelimumab in combination with other anticancer drugs (92). This activity may represent the multiple mechanisms that can be induced by CTLA-4 mAbs in humans. Further studies on these therapeutic mAbs are needed to fully exploit their mechanism of action in patients. Interestingly, PD-L1 mAbs that demonstrate clinical activity are either human IgG4, which have low FcγR binding and ADCC activity, or an Fc-null version mutated to abrogate FcγR binding. One PD-L1 mAb that has recently completed phase I trials, avelumab, is a human IgG1 isotype and would therefore be predicted to mediate ADCC in vivo. It remains to be evaluated in advanced human trials whether therapeutic activity of avelumab is enhanced through engagement of FcγRs, as has been observed in preclinical models using other anti-PD-L1 antibodies.
Recent studies revealed an unexpected requirement for engagement of the inhibitory FcγRIIb for optimal agonistic activity of mAbs targeting members of the TNFR superfamily, including CD40, DR5, CD30, and Fas (93-96). Contrary to antitumor cytotoxic and immunomodulatory mAbs that require activating FcγR engagement and signaling to mediate specific elimination of target cell populations, engagement of the inhibitory FcγRIIb by anti-TNFR mAbs does not require FcγR intracellular signaling to mediate IgG agonistic activity. FcγRIIb potentiates the activity of agonistic mAbs by solely providing a scaffold, in trans, for antibody crosslinking, enhancing thereby target receptor clustering on the cell surface. FcγRIIb engagement is mainly required for the activity of mAbs targeting receptors that depend on multimerization for their activation, such as members of the TNFR family. Whereas soluble, non-crosslinked mAbs fail to mimic the trimeric TNFR-TNF complex needed for activation, FcγRIIb-crosslinked mAbs have the capacity to confer potent agonistic activity in vivo. A similar requirement for FcγRIIb engagement was observed for the agonistic activity of mAbs targeting a member of the class A scavenger receptor family (MARCO; macrophage receptor with collagenous structure) (97). Anti-MARCO mAbs mediate reprogramming of MARCO-expressing tumor associated macrophages (TAMs) from a pro-tumoral, immunosuppressive M2 phenotype to an M1-like one, modulating the TME into more inflammatory and anti-tumoral state. Similar to anti-TNFR mAbs, FcγRIIb engagement mediates oligomerization of the MARCO collagenous domains, inducing thereby intracellular signaling and downstream effects on macrophage polarization.
Given the requirement of these antibodies for FcγRIIb engagement, FcγRIIb-targeted Fc engineering could enhance their in vivo antitumor activity. CD40 is perhaps the most studied therapeutic target for the FcγR-dependent crosslinking pathway. Agonistic anti-CD40 mAbs represent physiologic adjuvants, which function by potently ligating CD40 to induce APC maturation and subsequent expansion of tumor-specific cytotoxic T cells that have the capacity to efficiently eradicate of tumors in multiple models (98). Indeed, studies in FcγR-humanized mice revealed that agonistic anti-CD40 antibodies optimized for enhanced human FcγRIIb binding displayed a remarkable (>10-fold) improvement in their immunostimulatory and anti-tumor activity (13, 93). Likewise, further studies on agonistic antibodies targeting other TNFR superfamily members, such as DR5 also showed that their activity can be enhanced through FcγRIIb-targeted Fc engineering (94, 95). Several anti-human CD40 antibodies have been developed, and among them, CP-870,893 represents the most potent agonistic anti-human CD40 mAb reported to date (99). Despite its potent antitumor activity in preclinical animal models, CP-870,893 displayed only modest antitumor activity in patients with tumors in a series of phase I clinical trials (99-101). Using mice fully humanized for both CD40 and FcγRs, the modest activity of CP-870,893 was shown to be a function of the limited capacity of human IgG2 to interact with human FcγRIIb (102). Indeed, subclass switching of CP-870,893 to human IgG1 significantly improved its in vivo activity, due to the increased affinity of human IgG1 for FcγRIIb. Likewise, Fc domain engineering to selectively enhance affinity if the IgG1 Fc for FcγRIIb further improved the in vivo antitumor activity of CP-870,893, indicating that Fc variants specifically enhanced for FcγRIIb binding may be the optimal IgG framework for agonistic anti-CD40 mAbs. This work further highlights the utility of animal models humanized for both the target antigen and FcγRs to evaluate the contributions of Fab- and Fc-mediated mechanisms to the in vivo activity of human therapeutic antibodies and to guide the development of Fc-optimized clinical candidates.
IgG signaling in infection
The critical role of antibodies in host protection against infectious diseases is manifest by recurrent infections in patients with X-linked agammaglobulinemia (XLA), a primary immunodeficiency caused by a tyrosine kinase mutation that disrupts B-cell differentiation, maturation and signaling. XLA patients are typically healthy in infancy until maternal antibodies wane, after which time they become agammaglobulinemic and suffer tremendous infectious morbidity in the absence of replacement Ig therapy. Lifelong IVIG treatment complements the immune deficiency and enables most XLA patients to lead healthy, full lives.
Because IgG-mediated protection is often linked to recruitment of FcγR-mediated effector functions, genetic variation in FcγRs can underlie differences in susceptibility to infectious diseases. For example, the low affinity variant of FcγRIIa (R131) confers increased susceptibility to severe bacterial infections and sepsis, likely due to impaired microbial clearance (37, 103, 104). In contrast, in dengue infection, where antibody-mediated enhancement (ADE) of virus infectivity can occur, this low binding FcγRIIa polymorphism is associated with asymptomatic infection and improved clinical outcomes (105, 106). IgG-mediated immunity against infection occurs through numerous mechanisms involving FcγR-dependent and independent mechanisms. In vitro assays have been useful in defining the various IgG-mediated mechanisms of protection, such as virus neutralization (107, 108), opsonophagocytic killing of bacteria (109) and killing of infected cells (110), but mechanisms that actually play a role in in vivo protection cannot be reliably predicted from in vitro experiments. Here, we will review the mechanisms of IgG-mediated protection in vivo using studies of immunity to influenza viruses and to HIV as model systems.
Influenza
Neutralizing antibodies (nAb) against influenza viruses bind the HA, which is responsible for mediating viral entry into host cells. Anti-HA nAbs work by blocking one of the two key functions of the HA that result in viral entry - attachment to host cells or fusion with the endosome, which is required for release of viral RNA into the cytoplasm (111, 112). The two types of anti-HA nAbs (those that prevent attachment or fusion) can be distinguished in vitro using the classic hemagglutination inhibition (HI) assay, which detects nAbs that act by preventing attachment of HA to sialic acids, or, in cell-based microneutralization assays, which detect nAbs that act by either mechanism. NAbs that prevent attachment bind to specific antigenic sites surrounding the receptor binding pocket in HA and are characterized by potency and prompt selection of viral escape variants. Escape in the presence of these nAbs occurs for three reasons: 1) the influenza virus polymerase is a low fidelity complex lacking exonuclease activity that generates a high rate of transcriptional mutation relative to other RNA polymerases, 2) the rapid kinetics of influenza virus replication results in efficient selection of escape variants when selective pressure of a nAb is applied, and 3) the ability of antigenic sites in the HA to accommodate tremendous structural diversity means that the virus can nearly always escape nAbs that bind to those regions. Because the nAbs that prevent viral attachment readily select for escape variants, they are, by nature, strain-specific (113). In vivo, nAbs that prevent attachment are generally protective at very low concentrations and those that have been studied protect without a requirement for FcγR engagement (35).
In contrast to nAbs that prevent viral attachment, those that prevent fusion are characterized by binding to highly conserved domains of the HA such as the stalk domain, and they are are usually of lower protective potency in vivo. Because fusion-inhibiting nAbs bind regions of the HA that cannot accommodate as much antigenic change as antigenic sites, they can demonstrate breadth of protective activity against different influenza viruses that is not seen with nAbs that prevent viral attachment (107). New influenza vaccine candidates designed to elicit broadly neutralizing antibodies, often aim to elicit these fusion-inhibiting nAbs because of the breadth of protection that they can mediate in vivo. Several fusion-inhibiting nAbs have been studied for FcγR requirements and were shown to protect without a requirement for FcγR engagement at relatively high concentrations, but activating Type I FcγRs were required for their activity at lower concentrations (114, 115). Thus, potency of broadly-neutralizing nAbs is dependent on Type I FcγRs in vivo.
In contrast to nAbs, which can confer sterilizing immunity against influenza viruses, non-neutralizing antibodies specific for the influenza HA, neuraminidase, nucleoprotein or M2 proteins can also contribute to protection by limiting virus replication and spread. Antibodies against the viral neuraminidase, for example, can function in several ways including inhibiting neuraminidase activity that is required for release of new virus particles from the host cell, or, they can mediate cytotoxic activity of infected cells (116). Like the fusion-inhibiting nAbs, non-neutralizing antibodies can be broadly reactive and broadly protective in vivo; they are generally of lower protective potency than the nAbs that prevent viral attachment and several broadly reactive anti-HA or neuraminidase mAbs have been shown to be dependent on Type I FcγRs for potent in vivo protection (114). The cumulative data support a model where strain-specific nAbs do not require recruitment of FcγR-mediated effector functions, while broadly protective antibodies depend on activating Type I FcγR interactions for potent activity in vivo.
Interestingly, in addition to the varying requirements of different anti-influenza IgGs for FcγRs to mediate protection in vivo, different categories of protective IgGs also appear to have distinct capacities for recruiting cellular effector functions. For example, a few nAbs that have been studied that prevent viral attachment do not readily engage FcγRIIIa without Fc engineering to increase Fc-FcγRIIIa binding affinity and therefore cannot mediate ADCC in in vitro assays (115). This appears to be inconsequential in vivo as they protect, potently, without a requirement for FcγRs. In contrast however, nAbs and mAbs with broad protective activity in vivo do generally require FcγR engagement and their Fcs are able to engage FcγRIIIa to mediate ADCC in vitro (114, 115). Determinants of FcγR engagement by bound IgGs are not fully understood but they include binding density, which is dictated by nature and expression of antigen, and may also be impacted by affinity of the Fab for binding epitope; IgGs with moderate-affinity Fab domains have been shown to recruit effector function more effectively when compared to IgGs with higher affinity Fab domains. This has been hypothesized to result from the increased off-rates with lower affinity Fab-antigen interactions, leading to more monovalent Fab-antigen interactions and increased potential for dense IgG opsonization (1 Fc domain per 1 bound Fab domain). In contrast, higher affinity IgGs are more likely to form bivalent interactions with antigen (1 Fc domain per 2 bound Fab domains), limiting the potential for dense IgG binding (117). ADCC mediated by broadly reactive anti-HA mAbs may also be supported through increased affinity between target and effector cell due to broadly neutralizing mAb Fc-FcγRIIIa engagement in conjunction with binding by the viral HA to sialic acids on the effector cell (118). Yet another mechanism that may impact the ability of a bound IgG to engage FcγRs is conformational change in the Fc domain induced upon binding of the Fab domain to antigen (119-121). In this model, some epitope-paratope interactions induce structural change in the Fc domain that affects FcγR interactions. A correlate to this is that IgG constant domains can impact fine specificity of the variable domain; this has been demonstrated in studies showing that identical variable domains in IgGs of distinct subclass can have different binding specificities (122).
An area for further investigation in FcγR signaling and influenza biology is the extent to which effector functions mediated by activating Type I FcγRs can exert selective pressure on influenza viruses in vivo. The seasonal antigenic drift that dictates constant updating of virus strains in current influenza vaccines occurs under FcγR-independent selective pressure by nAbs that block virus attachment. Because approved influenza virus vaccines do not aim to elicit broadly reactive antibodies, it is not clear how a new generation of vaccines that may provide broad immunity will impact the antigenic landscape of circulating influenza viruses. Vaccines that elicit broadly reactive HA or neuraminidase IgGs will likely protect in vivo largely through FcγR-dependent mechanisms, meaning cellular effector functions will become the driving force behind seasonal antigenic drift. The rate of antigenic drift and effects on viral fitness that will be observed in viruses that are selected under pressure of broadly-protective IgGs will be important to understand as new influenza vaccines are studied and brought to market.
HIV
Neutralizing antibodies against HIV-1 are specific for the major envelope (Env) glycoprotein that mediates viral entry. As with the influenza HA, epitopes of the HIV envelope protein that can mediate potent virus neutralization are often able to accommodate tremendous structural diversity, making broadly neutralizing anti-HIV IgGs extremely rare. A small fraction of HIV-infected individuals however, do develop affinity-matured IgGs with broad protective activity against distinct virus isolates (123), demonstrating potential for broad in vivo protection against HIV-1 through passively transferred anti-Env IgGs. Advances in B-cell cloning technology have enabled screening of single B cells from these HIV patients and rare cells producing broadly neutralizing IgG specificities have been isolated and the human antibodies have been cloned and characterized (123). In vivo studies have demonstrated that sterilizing immunity against SHIV infection could be achieved through passive administration of broadly neutralizing anti-Env mAbs in macaques and against HIV-1 infection in humanized mice (124-126). Recent landmark clinical studies have shown that administration of a broadly neutralizing anti-Env mAb which disrupts interaction with CD4 can suppress viremia in chronically infected HIV-1 patients (127).
Mechanistic studies have demonstrated increased in vivo activity of anti-Env nAbs with capacity for enhanced binding to activating Type I FcγRs in preventing HIV-1 entry, suggesting a role for activating FcγRs in in vivo neutralization (33). Similarly, activating FcγRs have been shown to mediate in vivo clearance of HIV-infected cells, further contributing to the antiviral activity of anti-Env nAbs (128). Further assessment of the protective activity of Fc variants of anti-Env mAbs with differential FcγR binding capacities in post-exposure prophylaxis and therapy models of HIV-1 infection in humanized mice revealed a requirement for activating FcγR engagement for optimal therapeutic activity of anti-Env nAbs (33, 129). These findings are consistent with previous observations using a non-human primate pre-exposure prophylaxis model, in which Fc domain variants with minimal capacity for FcγR engagement provided minimal protection against SHIV challenge (125). Importantly, these studies demonstrated that IgGs can mediate suppression of a chronic viral pathogen, HIV-1, and that activating Type I FcγRs are required for this activity in vivo. Given the contribution of FcγR-mediated pathways in the in vivo antiviral activity of anti-Env nAbs, Fc domain engineering for selective engagement of activating FcγRs is expected to lead to the development of anti-Env nAbs with improved therapeutic efficacy.
Other infectious diseases
Studies support an important role for FcγR signaling in protection against a number of pathogens in addition to influenza viruses and HIV. For example, passive transfer of an anti-RSV mAb, palivizumab, with enhanced affinity for the activating FcγRIIIa by virtue of modification with afucosylated Fc glycoforms, reduced RSV lung titers in vivo compared with the fucosylated version; abrogation of palivizumab-FcγR binding further reduced in vivo protection, demonstrating that FcγR-independent neutralization plays little role in protection mediated by palivizumab in vivo (130). Protection against some bacterial and fungal pathogens has also been shown to depend on FcγRs. FcγRIIIa was shown to be required for mAb-mediated protection against S. pneumoniae and numerous experiments have shown that subclass determines protective activity of IgGs against S. aureus and C. neoformans, with affinity for activating FcγRs correlating with improved protection (reviewed in (131)). Based on these studies, mAbs engineered for selectively enhanced binding to activating FcγRs have been developed as passive transfer therapeutics; the in vivo activity of a mAb against the lethal toxin of B. anthracis, for example, was improved through Fc optimization that enhanced binding to activating human FcγRs (34) and, as described above, similarly engineered mAbs have shown increased potency in suppression of HIV rebound (33, 129). In addition to activating FcγRs, signaling through the inhibitory FcγRIIb can also affect susceptibility to infections since it offsets activating FcγR effects. For example, Fcgr2b-/- mice exhibit improved bacterial clearance in models of pneumococcal peritonitis through improved macrophage effector function (132, 133), whereas overexpression of FcγRIIb is associated with increased mortality upon challenge with Streptococcus pneumoniae (132, 133). Collectively, these findings demonstrate the importance of IgG-FcγR-mediated effector functions in protection against a diverse set of infectious organisms in vivo.
Antibody dependent enhancement of infection and/or disease
A long-appreciated, yet poorly understood, property of Type I FcγRs is their occasional ability to mediate ADE of infectivity and disease. An example of ADE can be observed in secondary infection with dengue virus; while primary infection is often asymptomatic or mild in presentation, a second infection with a heterologous dengue serotype can be accompanied by increased viral replication and severe disease. This enhancement is also seen, on occasion, in infants with primary dengue infection who have maternal anti-dengue antibodies that bind, but do not neutralize the infecting virus strain. (134, 135). ADE in secondary dengue is thought to be mediated by cross-reactive, non-neutralizing IgGs generated during the primary virus exposure that increase infectivity of Type I FcγR-bearing cells, including monocytes, macrophages and dendritic cells. ADE of dengue virus infectivity and/or disease has been demonstrated in a variety of in vitro and in vivo infection models; studies have shown that dengue virus infection in the presence of dengue-binding, but non-neutralizing, IgGs can result in more infected cells (136-138), enhanced virus fusion activity (139) and/or suppression of innate immunity (140, 141), but specific FcγR pathways required for ADE in dengue infection have not been identified (142-145). Interestingly, selectivity of patient IgG responses may play a role in susceptibility to severe dengue disease and explain why only a small fraction of patients with pre-existing, non-neutralizing anti-dengue IgGs develop severe disease (146). Analysis of IgGs from patients with mild or severe dengue disease showed that patients with severe disease have Fc structures on anti-dengue IgGs that are selectively enhanced for FcγRIIIa binding (TTW and JVR, unpublished observation). This suggests that production of IgGs with higher affinity for FcγRIIIa may play a role in ADE of disease in dengue infection.
ADE of disease severity not due to increased microbial replication may also occur in some settings due to formation of pathological immune complexes that trigger enhanced inflammation. This mechanism is not well understood, but some speculate that pathological immune complexes were responsible for failure of a RSV vaccine trial in 1968 in which children who received the experimental vaccine were >30-times more likely to be hospitalized with severe RSV infection compared with control subjects and two toddlers died as a result of what was thought to be vaccine-enhanced disease (147). The experimental vaccine was a formalin-killed RSV virus that elicited a large proportion of non-neutralizing antibodies that are thought to have formed insoluble immune complexes with viral proteins which may have mediated cytotoxicity and/or triggered cytokine production leading to enhanced disease (148-150). Disease exacerbation through immune complex deposition was also proposed for patients with fatal 1957 or 2009 pandemic influenza infections who were found to have large quantities of immune complexes deposited in pulmonary tissue (148).
How some microbes cause increased infectivity and/or clinical disease through antibody-mediated mechanisms is not well understood, but the phenomenon likely relies on both host and pathogen determinants. Adaptation to productive replication in activating FcγR-bearing cells such as macrophages and monocytes may be one determinant of ADE. A second determinant could be the antigenic variability of a pathogen because repeated exposures can lead to the presence of cross-reactive, non-neutralizing IgGs which may mediate pathogen uptake by FcγR-expressing cells or, in rare circumstances, may form insoluble immune complexes that enhance inflammation and disease due to complement-mediated inflammation, Type I FcγR-mediated inflammation or direct cytotoxicity.
Conclusions
IgG antibodies represent a major component of the adaptive immune system and have the capacity to mediate pleiotropic effector activities through specific interactions with Type I and Type II FcγRs. In addition to the direct role that IgG antibodies play in protection against malignancies and infections, IgG-mediated signaling also has a key role in shaping adaptive immune responses. This is because antigen is often ‘seen’ in vivo as IgG immune complexes since IgG antibodies that were pre-existing or that are produced early during an adaptive response bind to the antigen during infection or vaccination. For example, one mechanism by which these immune complexes modulate adaptive immunity is by enhancing T-cell responses, as described previously in this chapter. Crosslinking of activating Type I FcγRs on dendritic cells triggers their maturation and leads to enhanced antigen presentation and activation of T-cell responses. This phenomenon is exemplified by in vivo work showing that formation of a protective memory T-cell response (vaccinal response) after rituximab administration is dependent on rituximab/CD20 immune complex formation and expression of FcγRIIa on dendritic cells (12). IgG immune complexes can also act in an autocrine signaling manner to modulate selection of B cells during maturation of an antibody response. This was recently demonstrated in vivo in studies showing that sialylated immune complexes signal through the Type II FcγR, CD23, on B cells to trigger upregulation of the B cell inhibitory FcγRIIb. Increased FcγRIIb expression elevates the threshold for selection of B cells based on affinity of the BCR, ultimately resulting in selection for affinity matured cells (7). Interestingly, it was also shown that the initial step in this autocrine signaling cascade, engagement of CD23, is regulated during maturation of antibody responses in humans; Fc sialylation on antigen-specific IgGs was shown to increase in the days following influenza virus vaccination, with the increased change in sialylation correlating with final affinity of vaccine response. Immunization with sialylated immune complexes was shown, in an influenza system, to result in higher affinity IgG responses with enhanced in vivo potency and breadth of protective activity (7). These findings highlight the interplay of different types of FcγRs in mediating effector activities and modulating adaptive immune responses. Fc domain engineering of therapeutic antibodies to engage particular classes of FcγRs substantially improved their clinical efficacy. Given the contribution of Fc-FcγR interactions to the modulation of adaptive immune responses, specific activation of FcγR-mediated pathways during vaccination could also lead to the generation of robust and sustained adaptive immune responses, with broad and potent protective activity.
Acknowledgments
We wish to thank Meghan K. DiLillo for help preparing the manuscript. We also acknowledge support from the Rockefeller University. Research reported in this publications was supported in part by the Rockefeller University Center for Clinical and Translational Science (UL1TR000043), the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (P01AI100148, U19AI111825, U19AI109946), the National Cancer Institute (R35CA196620, P01CA190174), the Bill & Melinda Gates Foundation (OPP1124068), and DARPA (W31P4Q-14-1-0010). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. SB is an amfAR Mathilde Krim Fellow in Basic Biomedical Research (108977-57-RKVA). RD is a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute.
Footnotes
The authors have no conflicting financial interests.
Literature Cited
- 1.Pincetic A, Bournazos S, DiLillo DJ, Maamary J, Wang TT, Dahan R, Fiebiger BM, Ravetch JV. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol. 2014;15:707–16. doi: 10.1038/ni.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310:1510–2. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 3.Sondermann P, Pincetic A, Maamary J, Lammens K, Ravetch JV. General mechanism for modulating immunoglobulin effector function. Proc Natl Acad Sci U S A. 2013;110:9868–72. doi: 10.1073/pnas.1307864110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrara C, Grau S, Jäger C, Sondermann P, Brünker P, Waldhauer I, Hennig M, Ruf A, Rufer AC, Stihle M, Umaña P, Benz J. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci U S A. 2011;108:12669–74. doi: 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Man YA, Dolhain RJ, Hazes JM. Disease activity or remission of rheumatoid arthritis before, during and following pregnancy. Curr Opin Rheumatol. 2014;26:329–33. doi: 10.1097/BOR.0000000000000045. [DOI] [PubMed] [Google Scholar]
- 6.Theodoratou E, Thaçi K, Agakov F, Timofeeva MN, Štambuk J, Pučić-Baković M, Vučković F, Orchard P, Agakova A, Din FV, Brown E, Rudd PM, Farrington SM, Dunlop MG, Campbell H, Lauc G. Glycosylation of plasma IgG in colorectal cancer prognosis. Sci Rep. 2016;6:28098. doi: 10.1038/srep28098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang TT, Maamary J, Tan GS, Bournazos S, Davis CW, Krammer F, Schlesinger SJ, Palese P, Ahmed R, Ravetch JV. Anti-HA Glycoforms Drive B Cell Affinity Selection and Determine Influenza Vaccine Efficacy. Cell. 2015;162:160–9. doi: 10.1016/j.cell.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van de Geijn FE, Wuhrer M, Selman MH, Willemsen SP, de Man YA, Deelder AM, Hazes JM, Dolhain RJ. Immunoglobulin G galactosylation and sialylation are associated with pregnancy-induced improvement of rheumatoid arthritis and the postpartum flare: results from a large prospective cohort study. Arthritis Res Ther. 2009;11:R193. doi: 10.1186/ar2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Espy C, Morelle W, Kavian N, Grange P, Goulvestre C, Viallon V, Chéreau C, Pagnoux C, Michalski JC, Guillevin L, Weill B, Batteux F, Guilpain P. Sialylation levels of anti-proteinase 3 antibodies are associated with the activity of granulomatosis with polyangiitis (Wegener's) Arthritis Rheum. 2011;63:2105–15. doi: 10.1002/art.30362. [DOI] [PubMed] [Google Scholar]
- 10.Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci U S A. 2008;105:19571–8. doi: 10.1073/pnas.0810163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature. 2011;475:110–3. doi: 10.1038/nature10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiLillo DJ, Ravetch JV. Differential Fc-Receptor Engagement Drives an Anti-tumor Vaccinal Effect. Cell. 2015;161:1035–45. doi: 10.1016/j.cell.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc Natl Acad Sci U S A. 2012;109:6181–6. doi: 10.1073/pnas.1203954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.te Velde AA, de Waal Malefijt R, Huijbens RJ, de Vries JE, Figdor CG. IL-10 stimulates monocyte Fc gamma R surface expression and cytotoxic activity. Distinct regulation of antibody-dependent cellular cytotoxicity by IFN-gamma, IL-4, and IL-10. J Immunol. 1992;149:4048–52. [PubMed] [Google Scholar]
- 15.Uciechowski P, Schwarz M, Gessner JE, Schmidt RE, Resch K, Radeke HH. IFN-gamma induces the high-affinity Fc receptor I for IgG (CD64) on human glomerular mesangial cells. Eur J Immunol. 1998;28:2928–35. doi: 10.1002/(SICI)1521-4141(199809)28:09<2928::AID-IMMU2928>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 16.Bournazos S, Ravetch JV. Fcγ receptor pathways during active and passive immunization. Immunol Rev. 2015;268:88–103. doi: 10.1111/imr.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanakaraj P, Duckworth B, Azzoni L, Kamoun M, Cantley LC, Perussia B. Phosphatidylinositol-3 kinase activation induced upon Fc gamma RIIIA-ligand interaction. J Exp Med. 1994;179:551–8. doi: 10.1084/jem.179.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sánchez-Mejorada G, Rosales C. Fcgamma receptor-mediated mitogen-activated protein kinase activation in monocytes is independent of Ras. J Biol Chem. 1998;273:27610–9. doi: 10.1074/jbc.273.42.27610. [DOI] [PubMed] [Google Scholar]
- 19.Aramburu J, Azzoni L, Rao A, Perussia B. Activation and expression of the nuclear factors of activated T cells, NFATp and NFATc, in human natural killer cells: regulation upon CD16 ligand binding. J Exp Med. 1995;182:801–10. doi: 10.1084/jem.182.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–82. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 21.Martyn KD, Kim MJ, Quinn MT, Dinauer MC, Knaus UG. p21-activated kinase (Pak) regulates NADPH oxidase activation in human neutrophils. Blood. 2005;106:3962–9. doi: 10.1182/blood-2005-03-0859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suh CI, Stull ND, Li XJ, Tian W, Price MO, Grinstein S, Yaffe MB, Atkinson S, Dinauer MC. The phosphoinositide-binding protein p40phox activates the NADPH oxidase during FcgammaIIA receptor-induced phagocytosis. J Exp Med. 2006;203:1915–25. doi: 10.1084/jem.20052085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egesten A, Breton-Gorius J, Guichard J, Gullberg U, Olsson I. The heterogeneity of azurophil granules in neutrophil promyelocytes: immunogold localization of myeloperoxidase, cathepsin G, elastase, proteinase 3, and bactericidal/permeability increasing protein. Blood. 1994;83:2985–94. [PubMed] [Google Scholar]
- 24.Gabay JE, Almeida RP. Antibiotic peptides and serine protease homologs in human polymorphonuclear leukocytes: defensins and azurocidin. Curr Opin Immunol. 1993;5:97–102. doi: 10.1016/0952-7915(93)90087-9. [DOI] [PubMed] [Google Scholar]
- 25.Owen CA, Campbell MA, Boukedes SS, Campbell EJ. Inducible binding of bioactive cathepsin G to the cell surface of neutrophils. A novel mechanism for mediating extracellular catalytic activity of cathepsin G. J Immunol. 1995;155:5803–10. [PubMed] [Google Scholar]
- 26.Odin JA, Edberg JC, Painter CJ, Kimberly RP, Unkeless JC. Regulation of phagocytosis and [Ca2+]i flux by distinct regions of an Fc receptor. Science. 1991;254:1785–8. doi: 10.1126/science.1837175. [DOI] [PubMed] [Google Scholar]
- 27.Sobota A, Strzelecka-Kiliszek A, Gładkowska E, Yoshida K, Mrozińska K, Kwiatkowska K. Binding of IgG-opsonized particles to Fc gamma R is an active stage of phagocytosis that involves receptor clustering and phosphorylation. J Immunol. 2005;175:4450–7. doi: 10.4049/jimmunol.175.7.4450. [DOI] [PubMed] [Google Scholar]
- 28.Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood. 2008;112:935–45. doi: 10.1182/blood-2007-12-077917. [DOI] [PubMed] [Google Scholar]
- 29.Selvaraj P, Carpén O, Hibbs ML, Springer TA. Natural killer cell and granulocyte Fc gamma receptor III (CD16) differ in membrane anchor and signal transduction. J Immunol. 1989;143:3283–8. [PubMed] [Google Scholar]
- 30.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–9. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clynes R, Ravetch JV. Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity. 1995;3:21–6. doi: 10.1016/1074-7613(95)90155-8. [DOI] [PubMed] [Google Scholar]
- 32.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–29. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 33.Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV. Broadly Neutralizing Anti-HIV-1 Antibodies Require Fc Effector Functions for In Vivo Activity. Cell. 2014;158:1243–53. doi: 10.1016/j.cell.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bournazos S, Chow SK, Abboud N, Casadevall A, Ravetch JV. Human IgG Fc domain engineering enhances antitoxin neutralizing antibody activity. J Clin Invest. 2014;124:725–9. doi: 10.1172/JCI72676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DiLillo DJ, Tan GS, Palese P, Ravetch JV. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat Med. 2014;20:143–51. doi: 10.1038/nm.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating Fcgamma receptors on dendritic cells. J Exp Med. 2002;195:1653–9. doi: 10.1084/jem.20020338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bournazos S, Woof JM, Hart SP, Dransfield I. Functional and clinical consequences of Fc receptor polymorphic and copy number variants. Clin Exp Immunol. 2009;157:244–54. doi: 10.1111/j.1365-2249.2009.03980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karassa FB, Trikalinos TA, Ioannidis JP. The role of FcgammaRIIA and IIIA polymorphisms in autoimmune diseases. Biomed Pharmacother. 2004;58:286–91. doi: 10.1016/j.biopha.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, Heward JM, Gough SC, de Smith A, Blakemore AI, Froguel P, Owen CJ, Pearce SH, Teixeira L, Guillevin L, Graham DS, Pusey CD, Cook HT, Vyse TJ, Aitman TJ. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007;39:721–3. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mueller M, Barros P, Witherden AS, Roberts AL, Zhang Z, Schaschl H, Yu CY, Hurles ME, Schaffner C, Floto RA, Game L, Steinberg KM, Wilson RK, Graves TA, Eichler EE, Cook HT, Vyse TJ, Aitman TJ. Genomic pathology of SLE-associated copy-number variation at the FCGR2C/FCGR3B/FCGR2B locus. Am J Hum Genet. 2013;92:28–40. doi: 10.1016/j.ajhg.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Latour S, Fridman WH, Daëron M. Identification, molecular cloning, biologic properties, and tissue distribution of a novel isoform of murine low-affinity IgG receptor homologous to human Fc gamma RIIB1. J Immunol. 1996;157:189–97. [PubMed] [Google Scholar]
- 42.Amigorena S, Salamero J, Davoust J, Fridman WH, Bonnerot C. Tyrosine-containing motif that transduces cell activation signals also determines internalization and antigen presentation via type III receptors for IgG. Nature. 1992;358:337–41. doi: 10.1038/358337a0. [DOI] [PubMed] [Google Scholar]
- 43.Bonnerot C, Briken V, Brachet V, Lankar D, Cassard S, Jabri B, Amigorena S. syk protein tyrosine kinase regulates Fc receptor gamma-chain-mediated transport to lysosomes. EMBO J. 1998;17:4606–16. doi: 10.1093/emboj/17.16.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bergtold A, Desai DD, Gavhane A, Clynes R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity. 2005;23:503–14. doi: 10.1016/j.immuni.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 45.Swanson JA, Hoppe AD. The coordination of signaling during Fc receptor-mediated phagocytosis. J Leukoc Biol. 2004;76:1093–103. doi: 10.1189/jlb.0804439. [DOI] [PubMed] [Google Scholar]
- 46.Desai DD, Harbers SO, Flores M, Colonna L, Downie MP, Bergtold A, Jung S, Clynes R. Fc gamma receptor IIB on dendritic cells enforces peripheral tolerance by inhibiting effector T cell responses. J Immunol. 2007;178:6217–26. doi: 10.4049/jimmunol.178.10.6217. [DOI] [PubMed] [Google Scholar]
- 47.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–6. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 48.Dhodapkar KM, Kaufman JL, Ehlers M, Banerjee DK, Bonvini E, Koenig S, Steinman RM, Ravetch JV, Dhodapkar MV. Selective blockade of inhibitory Fcgamma receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. Proc Natl Acad Sci U S A. 2005;102:2910–5. doi: 10.1073/pnas.0500014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. Genetic modifiers of systemic lupus erythematosus in FcgammaRIIB(-/-) mice. J Exp Med. 2002;195:1167–74. doi: 10.1084/jem.20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest. 2005;115:2914–23. doi: 10.1172/JCI24772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gordon S, Plüddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. 2014;262:36–55. doi: 10.1111/imr.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clynes R, Maizes JS, Guinamard R, Ono M, Takai T, Ravetch JV. Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J Exp Med. 1999;189:179–85. doi: 10.1084/jem.189.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dhodapkar KM, Banerjee D, Connolly J, Kukreja A, Matayeva E, Veri MC, Ravetch JV, Steinman RM, Dhodapkar MV. Selective blockade of the inhibitory Fcgamma receptor (FcgammaRIIB) in human dendritic cells and monocytes induces a type I interferon response program. J Exp Med. 2007;204:1359–69. doi: 10.1084/jem.20062545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yuasa T, Kubo S, Yoshino T, Ujike A, Matsumura K, Ono M, Ravetch JV, Takai T. Deletion of fcgamma receptor IIB renders H-2(b) mice susceptible to collagen-induced arthritis. J Exp Med. 1999;189:187–94. doi: 10.1084/jem.189.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. 1997;90:293–301. doi: 10.1016/s0092-8674(00)80337-2. [DOI] [PubMed] [Google Scholar]
- 56.Pearse RN, Kawabe T, Bolland S, Guinamard R, Kurosaki T, Ravetch JV. SHIP recruitment attenuates Fc gamma RIIB-induced B cell apoptosis. Immunity. 1999;10:753–60. doi: 10.1016/s1074-7613(00)80074-6. [DOI] [PubMed] [Google Scholar]
- 57.Xiang Z, Cutler AJ, Brownlie RJ, Fairfax K, Lawlor KE, Severinson E, Walker EU, Manz RA, Tarlinton DM, Smith KG. FcgammaRIIb controls bone marrow plasma cell persistence and apoptosis. Nat Immunol. 2007;8:419–29. doi: 10.1038/ni1440. [DOI] [PubMed] [Google Scholar]
- 58.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature. 1996;383:263–6. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 59.Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13:277–85. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 60.Blank MC, Stefanescu RN, Masuda E, Marti F, King PD, Redecha PB, Wurzburger RJ, Peterson MG, Tanaka S, Pricop L. Decreased transcription of the human FCGR2B gene mediated by the -343 G/C promoter polymorphism and association with systemic lupus erythematosus. Hum Genet. 2005;117:220–7. doi: 10.1007/s00439-005-1302-3. [DOI] [PubMed] [Google Scholar]
- 61.Su K, Wu J, Edberg JC, Li X, Ferguson P, Cooper GS, Langefeld CD, Kimberly RP. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcgammaRIIb alters receptor expression and associates with autoimmunity. I. Regulatory FCGR2B polymorphisms and their association with systemic lupus erythematosus. J Immunol. 2004;172:7186–91. doi: 10.4049/jimmunol.172.11.7186. [DOI] [PubMed] [Google Scholar]
- 62.Floto RA, Clatworthy MR, Heilbronn KR, Rosner DR, MacAry PA, Rankin A, Lehner PJ, Ouwehand WH, Allen JM, Watkins NA, Smith KG. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat Med. 2005;11:1056–8. doi: 10.1038/nm1288. [DOI] [PubMed] [Google Scholar]
- 63.Henningsson F, Ding Z, Dahlin JS, Linkevicius M, Carlsson F, Grönvik KO, Hallgren J, Heyman B. IgE-mediated enhancement of CD4+ T cell responses in mice requires antigen presentation by CD11c+ cells and not by B cells. PLoS One. 2011;6:e21760. doi: 10.1371/journal.pone.0021760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clynes R, Takechi Y, Moroi Y, Houghton A, Ravetch JV. Fc receptors are required in passive and active immunity to melanoma. Proc Natl Acad Sci U S A. 1998;95:652–6. doi: 10.1073/pnas.95.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, Tedder TF. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199:1659–69. doi: 10.1084/jem.20040119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hamaguchi Y, Xiu Y, Komura K, Nimmerjahn F, Tedder TF. Antibody isotype-specific engagement of Fcgamma receptors regulates B lymphocyte depletion during CD20 immunotherapy. J Exp Med. 2006;203:743–53. doi: 10.1084/jem.20052283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Minard-Colin V, Xiu Y, Poe JC, Horikawa M, Magro CM, Hamaguchi Y, Haas KM, Tedder TF. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood. 2008;112:1205–13. doi: 10.1182/blood-2008-01-135160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Biburger M, Aschermann S, Schwab I, Lux A, Albert H, Danzer H, Woigk M, Dudziak D, Nimmerjahn F. Monocyte subsets responsible for immunoglobulin G-dependent effector functions in vivo. Immunity. 2011;35:932–44. doi: 10.1016/j.immuni.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 69.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–8. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 70.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–7. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 71.Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, Neri TM, Ardizzoni A. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26:1789–96. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 72.Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, Lamy A, Penault-Llorca F, Frébourg T, Michel P, Sabourin JC, Boissière-Michot F. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27:1122–9. doi: 10.1200/JCO.2008.18.0463. [DOI] [PubMed] [Google Scholar]
- 73.Liu SD, Chalouni C, Young JC, Junttila TT, Sliwkowski MX, Lowe JB. Afucosylated antibodies increase activation of FcgammaRIIIa-dependent signaling components to intensify processes promoting ADCC. Cancer Immunol Res. 2015;3:173–83. doi: 10.1158/2326-6066.CIR-14-0125. [DOI] [PubMed] [Google Scholar]
- 74.Horton HM, Bernett MJ, Pong E, Peipp M, Karki S, Chu SY, Richards JO, Vostiar I, Joyce PF, Repp R, Desjarlais JR, Zhukovsky EA. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res. 2008;68:8049–57. doi: 10.1158/0008-5472.CAN-08-2268. [DOI] [PubMed] [Google Scholar]
- 75.Gerdes CA, Nicolini VG, Herter S, van Puijenbroek E, Lang S, Roemmele M, Moessner E, Freytag O, Friess T, Ries CH, Bossenmaier B, Mueller HJ, Umana P. GA201 (RG7160): a novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin Cancer Res. 2013;19:1126–38. doi: 10.1158/1078-0432.CCR-12-0989. [DOI] [PubMed] [Google Scholar]
- 76.Abes R, Gelize E, Fridman WH, Teillaud JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood. 2010;116:926–34. doi: 10.1182/blood-2009-10-248609. [DOI] [PubMed] [Google Scholar]
- 77.Hilchey SP, Hyrien O, Mosmann TR, Livingstone AM, Friedberg JW, Young F, Fisher RI, Kelleher RJ, Jr, Bankert RB, Bernstein SH. Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: support for a “vaccinal effect” of rituximab. Blood. 2009;113:3809–12. doi: 10.1182/blood-2008-10-185280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kwak LW, Campbell MJ, Czerwinski DK, Hart S, Miller RA, Levy R. Induction of immune responses in patients with B-cell lymphoma against the surface-immunoglobulin idiotype expressed by their tumors. N Engl J Med. 1992;327:1209–15. doi: 10.1056/NEJM199210223271705. [DOI] [PubMed] [Google Scholar]
- 79.de Bono JS, Rha SY, Stephenson J, Schultes BC, Monroe P, Eckhardt GS, Hammond LA, Whiteside TL, Nicodemus CF, Cermak JM, Rowinsky EK, Tolcher AW. Phase I trial of a murine antibody to MUC1 in patients with metastatic cancer: evidence for the activation of humoral and cellular antitumor immunity. Ann Oncol. 2004;15:1825–33. doi: 10.1093/annonc/mdh472. [DOI] [PubMed] [Google Scholar]
- 80.Taylor C, Hershman D, Shah N, Suciu-Foca N, Petrylak DP, Taub R, Vahdat L, Cheng B, Pegram M, Knutson KL, Clynes R. Augmented HER-2 specific immunity during treatment with trastuzumab and chemotherapy. Clin Cancer Res. 2007;13:5133–43. doi: 10.1158/1078-0432.CCR-07-0507. [DOI] [PubMed] [Google Scholar]
- 81.Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med. 2014;65:185–202. doi: 10.1146/annurev-med-092012-112807. [DOI] [PubMed] [Google Scholar]
- 82.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, Leming PD, Lipson EJ, Puzanov I, Smith DC, Taube JM, Wigginton JM, Kollia GD, Gupta A, Pardoll DM, Sosman JA, Hodi FS. Survival, Durable Tumor Remission, and Long-Term Safety in Patients With Advanced Melanoma Receiving Nivolumab. Journal of Clinical Oncology. 2014;32:1020–30. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A. Safety and Tumor Responses with Lambrolizumab (Anti–PD-1) in Melanoma. New England Journal of Medicine. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab plus Ipilimumab in Advanced Melanoma. New England Journal of Medicine. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity through Reduction of Intratumoral Regulatory T Cells. Cancer Immunology Research. 2013;1:32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 87.Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, Brogdon JL. Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. The Journal of Experimental Medicine. 2013;210:1685–93. doi: 10.1084/jem.20130573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating Fc[gamma]Rs, leading to antitumor efficacy. Immunol Cell Biol. 2014;92:475–80. doi: 10.1038/icb.2014.26. [DOI] [PubMed] [Google Scholar]
- 89.Dahan R, Sega E, Engelhardt J, Selby M, Korman Alan J, Ravetch Jeffrey V. FcγRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell. 2015;28:285–95. doi: 10.1016/j.ccell.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 90.Furness AJ, Vargas FA, Peggs KS, Quezada SA. Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol. 2014;35:290–8. doi: 10.1016/j.it.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 91.Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015;112:6140–5. doi: 10.1073/pnas.1417320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Comin-Anduix B, Escuin-Ordinas H, Ibarrondo FJ. Tremelimumab: research and clinical development. Onco Targets Ther. 2016;9:1767–76. doi: 10.2147/OTT.S65802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li F, Ravetch JV. Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science. 2011;333:1030–4. doi: 10.1126/science.1206954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li F, Ravetch JV. A general requirement for FcgammaRIIB co-engagement of agonistic anti-TNFR antibodies. Cell Cycle. 2012;11:3343–4. doi: 10.4161/cc.21842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li F, Ravetch JV. Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcgamma receptor engagement. Proc Natl Acad Sci U S A. 2012;109:10966–71. doi: 10.1073/pnas.1208698109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li F, Ravetch JV. Antitumor activities of agonistic anti-TNFR antibodies require differential FcgammaRIIB coengagement in vivo. Proc Natl Acad Sci U S A. 2013;110:19501–6. doi: 10.1073/pnas.1319502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Ostling J, Dahan R, Harris RA, Rantalainen M, Klevebring D, Sund M, Brage SE, Fuxe J, Rolny C, Li F, Ravetch JV, Karlsson MC. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016;15:2000–11. doi: 10.1016/j.celrep.2016.04.084. [DOI] [PubMed] [Google Scholar]
- 98.French RR, Chan HT, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med. 1999;5:548–53. doi: 10.1038/8426. [DOI] [PubMed] [Google Scholar]
- 99.Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, Sullivan P, Mahany JJ, Gallagher M, Kramer A, Green SJ, O'Dwyer PJ, Running KL, Huhn RD, Antonia SJ. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25:876–83. doi: 10.1200/JCO.2006.08.3311. [DOI] [PubMed] [Google Scholar]
- 100.Ruter J, Antonia SJ, Burris HA, Huhn RD, Vonderheide RH. Immune modulation with weekly dosing of an agonist CD40 antibody in a phase I study of patients with advanced solid tumors. Cancer Biol Ther. 2010;10:983–93. doi: 10.4161/cbt.10.10.13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vonderheide RH, Burg JM, Mick R, Trosko JA, Li D, Shaik MN, Tolcher AW, Hamid O. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology. 2013;2:e23033. doi: 10.4161/onci.23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dahan R, Barnhart BC, Li F, Yamniuk AP, Korman AJ, Ravetch JV. Therapeutic Activity of Agonistic, Human Anti-CD40 Monoclonal Antibodies Requires Selective FcgammaR Engagement. Cancer Cell. 2016;29:820–31. doi: 10.1016/j.ccell.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Beppler J, Koehler-Santos P, Pasqualim G, Matte U, Alho CS, Dias FS, Kowalski TW, Velasco IT, Monteiro RC, Pinheiro da Silva F. Fc Gamma Receptor IIA (CD32A) R131 Polymorphism as a Marker of Genetic Susceptibility to Sepsis. Inflammation. 2016;39:518–25. doi: 10.1007/s10753-015-0275-1. [DOI] [PubMed] [Google Scholar]
- 104.Endeman H, Cornips MC, Grutters JC, van den Bosch JM, Ruven HJ, van Velzen-Blad H, Rijkers GT, Biesma DH. The Fcgamma receptor IIA-R/R131 genotype is associated with severe sepsis in community-acquired pneumonia. Clin Vaccine Immunol. 2009;16:1087–90. doi: 10.1128/CVI.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mohsin SN, Mahmood S, Amar A, Ghafoor F, Raza SM, Saleem M. Association of FcgammaRIIa Polymorphism with Clinical Outcome of Dengue Infection: First Insight from Pakistan. Am J Trop Med Hyg. 2015;93:691–6. doi: 10.4269/ajtmh.15-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Garcia G, Sierra B, Perez AB, Aguirre E, Rosado I, Gonzalez N, Izquierdo A, Pupo M, Danay Diaz DR, Sanchez L, Marcheco B, Hirayama K, Guzman MG. Asymptomatic dengue infection in a Cuban population confirms the protective role of the RR variant of the FcgammaRIIa polymorphism. Am J Trop Med Hyg. 2010;82:1153–6. doi: 10.4269/ajtmh.2010.09-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Okuno Y, Isegawa Y, Sasao F, Ueda S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J Virol. 1993;67:2552–8. doi: 10.1128/jvi.67.5.2552-2558.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hilleman MR, Werner JH, Gauld RL. Influenza antibodies in the population of the USA; an epidemiological investigation. Bull World Health Organ. 1953;8:613–31. [PMC free article] [PubMed] [Google Scholar]
- 109.Baltimore RS, Kasper DL, Baker CJ, Goroff DK. Antigenic specificity of opsonophagocytic antibodies in rabbit anti-sera to group B streptococci. J Immunol. 1977;118:673–8. [PubMed] [Google Scholar]
- 110.Townsend AR, Skehel JJ, Taylor PM, Palese P. Recognition of influenza A virus nucleoprotein by an H-2-restricted cytotoxic T-cell clone. Virology. 1984;133:456–9. doi: 10.1016/0042-6822(84)90413-6. [DOI] [PubMed] [Google Scholar]
- 111.Knossow M, Gaudier M, Douglas A, Barrère B, Bizebard T, Barbey C, Gigant B, Skehel JJ. Mechanism of neutralization of influenza virus infectivity by antibodies. Virology. 2002;302:294–8. doi: 10.1006/viro.2002.1625. [DOI] [PubMed] [Google Scholar]
- 112.Barbey-Martin C, Gigant B, Bizebard T, Calder LJ, Wharton SA, Skehel JJ, Knossow M. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology. 2002;294:70–4. doi: 10.1006/viro.2001.1320. [DOI] [PubMed] [Google Scholar]
- 113.Wang TT, Palese P. Textbook of influenza; Chapter 14: Emergence and Evolution of the, 1918, 1957, 1968, and 2009 pandemic virus strains. Chichester, West Sussex, UK: Wiley-Blackwell; 2013. pp. 218–230. [Google Scholar]
- 114.DiLillo DJ, Palese P, Wilson PC, Ravetch JV. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J Clin Invest. 2016;126:605–10. doi: 10.1172/JCI84428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.DiLillo DJ, Tan GS, Palese P, Ravetch JV. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat Med. 2014;20:143–51. doi: 10.1038/nm.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Memoli MJ, Shaw PA, Han A, Czajkowski L, Reed S, Athota R, Bristol T, Fargis S, Risos K, Powers JH, Davey RT, Jr, Taubenberger JK. Evaluation of Antihemagglutinin and Antineuraminidase Antibodies as Correlates of Protection in an Influenza A/H1N1 Virus Healthy Human Challenge Model. MBio. 2016;7:e00417–16. doi: 10.1128/mBio.00417-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mazor Y, Yang C, Borrok MJ, Ayriss J, Aherne K, Wu H, Dall'Acqua WF. Enhancement of Immune Effector Functions by Modulating IgG's Intrinsic Affinity for Target Antigen. PLoS One. 2016;11:e0157788. doi: 10.1371/journal.pone.0157788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Leon PE, He W, Mullarkey CE, Bailey MJ, Miller MS, Krammer F, Palese P, Tan GS. Optimal activation of Fc-mediated effector functions by influenza virus hemagglutinin antibodies requires two points of contact Proc Natl Acad Sci U S A In Press. 2016 doi: 10.1073/pnas.1613225113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schlessinger J, Steinberg IZ, Givol D, Hochman J, Pecht I. Antigen-induced conformational changes in antibodies and their Fab fragments studied by circular polarization of fluorescence. Proc Natl Acad Sci U S A. 1975;72:2775–9. doi: 10.1073/pnas.72.7.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sagawa T, Oda M, Morii H, Takizawa H, Kozono H, Azuma T. Conformational changes in the antibody constant domains upon hapten-binding. Mol Immunol. 2005;42:9–18. doi: 10.1016/j.molimm.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 121.Eryilmaz E, Janda A, Kim J, Cordero RJ, Cowburn D, Casadevall A. Global structures of IgG isotypes expressing identical variable regions. Mol Immunol. 2013;56:588–98. doi: 10.1016/j.molimm.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Torres M, Fernandez-Fuentes N, Fiser A, Casadevall A. The immunoglobulin heavy chain constant region affects kinetic and thermodynamic parameters of antibody variable region interactions with antigen. J Biol Chem. 2007;282:13917–27. doi: 10.1074/jbc.M700661200. [DOI] [PubMed] [Google Scholar]
- 123.Klein F, Mouquet H, Dosenovic P, Scheid JF, Scharf L, Nussenzweig MC. Antibodies in HIV-1 vaccine development and therapy. Science. 2013;341:1199–204. doi: 10.1126/science.1241144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mascola JR, Stiegler G, VanCott TC, Katinger H, Carpenter CB, Hanson CE, Beary H, Hayes D, Frankel SS, Birx DL, Lewis MG. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat Med. 2000;6:207–10. doi: 10.1038/72318. [DOI] [PubMed] [Google Scholar]
- 125.Hessell AJ, Hangartner L, Hunter M, Havenith CE, Beurskens FJ, Bakker JM, Lanigan CM, Landucci G, Forthal DN, Parren PW, Marx PA, Burton DR. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449:101–4. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 126.Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature. 2012;481:81–4. doi: 10.1038/nature10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Caskey M, Klein F, Lorenzi JC, Seaman MS, West AP, Buckley N, Kremer G, Nogueira L, Braunschweig M, Scheid JF, Horwitz JA, Shimeliovich I, Ben-Avraham S, Witmer-Pack M, Platten M, Lehmann C, Burke LA, Hawthorne T, Gorelick RJ, Walker BD, Keler T, Gulick RM, Fätkenheuer G, Schlesinger SJ, Nussenzweig MC. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature. 2015;522:487–91. doi: 10.1038/nature14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lu CL, Murakowski DK, Bournazos S, Schoofs T, Sarkar D, Halper-Stromberg A, Horwitz JA, Nogueira L, Golijanin J, Gazumyan A, Ravetch JV, Caskey M, Chakraborty AK, Nussenzweig MC. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science. 2016;352:1001–4. doi: 10.1126/science.aaf1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Halper-Stromberg A, Lu CL, Klein F, Horwitz JA, Bournazos S, Nogueira L, Eisenreich TR, Liu C, Gazumyan A, Schaefer U, Furze RC, Seaman MS, Prinjha R, Tarakhovsky A, Ravetch JV, Nussenzweig MC. Broadly Neutralizing Antibodies and Viral Inducers Decrease Rebound from HIV-1 Latent Reservoirs in Humanized Mice. Cell. 2014;158:989–99. doi: 10.1016/j.cell.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hiatt A, Bohorova N, Bohorov O, Goodman C, Kim D, Pauly MH, Velasco J, Whaley KJ, Piedra PA, Gilbert BE, Zeitlin L. Glycan variants of a respiratory syncytial virus antibody with enhanced effector function and in vivo efficacy. Proc Natl Acad Sci U S A. 2014;111:5992–7. doi: 10.1073/pnas.1402458111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bournazos S, DiLillo DJ, Ravetch JV. The role of Fc-FcγR interactions in IgG-mediated microbial neutralization. J Exp Med. 2015;212:1361–9. doi: 10.1084/jem.20151267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brownlie RJ, Lawlor KE, Niederer HA, Cutler AJ, Xiang Z, Clatworthy MR, Floto RA, Greaves DR, Lyons PA, Smith KG. Distinct cell-specific control of autoimmunity and infection by FcgammaRIIb. J Exp Med. 2008;205:883–95. doi: 10.1084/jem.20072565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Clatworthy MR, Smith KG. FcgammaRIIb balances efficient pathogen clearance and the cytokine-mediated consequences of sepsis. J Exp Med. 2004;199:717–23. doi: 10.1084/jem.20032197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gonzalez D, Castro OE, Kouri G, Perez J, Martinez E, Vazquez S, Rosario D, Cancio R, Guzman MG. Classical dengue hemorrhagic fever resulting from two dengue infections spaced 20 years or more apart: Havana, Dengue 3 epidemic, 2001-2002. Int J Infect Dis. 2005;9:280–5. doi: 10.1016/j.ijid.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 135.Kliks SC, Nimmanitya S, Nisalak A, Burke DS. Evidence that maternal dengue antibodies are important in the development of dengue hemorrhagic fever in infants. Am J Trop Med Hyg. 1988;38:411–9. doi: 10.4269/ajtmh.1988.38.411. [DOI] [PubMed] [Google Scholar]
- 136.Blackley S, Kou Z, Chen H, Quinn M, Rose RC, Schlesinger JJ, Coppage M, Jin X. Primary human splenic macrophages, but not T or B cells, are the principal target cells for dengue virus infection in vitro. J Virol. 2007;81:13325–34. doi: 10.1128/JVI.01568-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kou Z, Lim JY, Beltramello M, Quinn M, Chen H, Liu S, Martinez-Sobrido L, Diamond MS, Schlesinger JJ, de Silva A, Sallusto F, Jin X. Human antibodies against dengue enhance dengue viral infectivity without suppressing type I interferon secretion in primary human monocytes. Virology. 2011;410:240–7. doi: 10.1016/j.virol.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 138.Dejnirattisai W, Supasa P, Wongwiwat W, Rouvinski A, Barba-Spaeth G, Duangchinda T, Sakuntabhai A, Cao-Lormeau VM, Malasit P, Rey FA, Mongkolsapaya J, Screaton GR. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat Immunol. 2016;17:1102–8. doi: 10.1038/ni.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Flipse J, Diosa-Toro MA, Hoornweg TE, van de Pol DP, Urcuqui-Inchima S, Smit JM. Antibody-Dependent Enhancement of Dengue Virus Infection in Primary Human Macrophages; Balancing Higher Fusion against Antiviral Responses. Sci Rep. 2016;6:29201. doi: 10.1038/srep29201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Modhiran N, Kalayanarooj S, Ubol S. Subversion of innate defenses by the interplay between DENV and pre-existing enhancing antibodies: TLRs signaling collapse. PLoS Negl Trop Dis. 2010;4:e924. doi: 10.1371/journal.pntd.0000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ubol S, Phuklia W, Kalayanarooj S, Modhiran N. Mechanisms of immune evasion induced by a complex of dengue virus and preexisting enhancing antibodies. J Infect Dis. 2010;201:923–35. doi: 10.1086/651018. [DOI] [PubMed] [Google Scholar]
- 142.Beltramello M, Williams KL, Simmons CP, Macagno A, Simonelli L, Quyen NT, Sukupolvi-Petty S, Navarro-Sanchez E, Young PR, de Silva AM, Rey FA, Varani L, Whitehead SS, Diamond MS, Harris E, Lanzavecchia A, Sallusto F. The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe. 2010;8:271–83. doi: 10.1016/j.chom.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, Endy TP, Raengsakulrach B, Rothman AL, Ennis FA, Nisalak A. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis. 2000;181:2–9. doi: 10.1086/315215. [DOI] [PubMed] [Google Scholar]
- 144.Halstead SB, O'Rourke EJ. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J Exp Med. 1977;146:201–17. doi: 10.1084/jem.146.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Moi ML, Takasaki T, Saijo M, Kurane I. Dengue virus infection-enhancing activity of undiluted sera obtained from patients with secondary dengue virus infection. Trans R Soc Trop Med Hyg. 2013;107:51–8. doi: 10.1093/trstmh/trs007. [DOI] [PubMed] [Google Scholar]
- 146.Guzman MG, Alvarez M, Halstead SB. Secondary infection as a risk factor for dengue hemorrhagic fever/dengue shock syndrome: an historical perspective and role of antibody-dependent enhancement of infection. Arch Virol. 2013;158:1445–59. doi: 10.1007/s00705-013-1645-3. [DOI] [PubMed] [Google Scholar]
- 147.Graham BS. Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol Rev. 2011;239:149–66. doi: 10.1111/j.1600-065X.2010.00972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Monsalvo AC, Batalle JP, Lopez MF, Krause JC, Klemenc J, Hernandez JZ, Maskin B, Bugna J, Rubinstein C, Aguilar L, Dalurzo L, Libster R, Savy V, Baumeister E, Aguilar L, Cabral G, Font J, Solari L, Weller KP, Johnson J, Echavarria M, Edwards KM, Chappell JD, Crowe JE, Jr, Williams JV, Melendi GA, Polack FP. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat Med. 2011;17:195–9. doi: 10.1038/nm.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Murphy BR, Prince GA, Walsh EE, Kim HW, Parrott RH, Hemming VG, Rodriguez WJ, Chanock RM. Dissociation between serum neutralizing and glycoprotein antibody responses of infants and children who received inactivated respiratory syncytial virus vaccine. J Clin Microbiol. 1986;24:197–202. doi: 10.1128/jcm.24.2.197-202.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Guihot A, Luyt CE, Parrot A, Rousset D, Cavaillon JM, Boutolleau D, Fitting C, Pajanirassa P, Mallet A, Fartoukh M, Agut H, Musset L, Zoorob R, Kirilovksy A, Combadiere B, van der Werf S, Autran B, Carcelain G Flu BALSG. Low titers of serum antibodies inhibiting hemagglutination predict fatal fulminant influenza A(H1N1) 2009 infection. Am J Respir Crit Care Med. 2014;189:1240–9. doi: 10.1164/rccm.201311-2071OC. [DOI] [PubMed] [Google Scholar]