

Abstract
Background
Disease modifying therapies (DMTs) are urgently needed for neurodegenerative diseases (NDD) such as Alzheimer’s disease (AD) and many other disorders characterized by protein aggregation and neurodegeneration. Despite advances in understanding the neurobiology of NDD, there are no approved DMTs.
Discussion
Defining disease-modification is critical to drug-development programs. A DMT is an intervention that produces an enduring change in the trajectory of clinical decline of an NDD by impacting the disease processes leading to nerve cell death. A DMT is neuroprotective, and neuroprotection will result in disease modification. Disease modification can be demonstrated in clinical trials by a drug-placebo difference in clinical outcomes supported by a drug-placebo difference on biomarkers reflective of the fundamental pathophysiology of the NDD. Alternatively, disease modification can be supported by findings on a staggered start or delayed withdrawal clinical trial design. Collecting multiple biomarkers is necessary to support a comprehensive view of disease modification.
Conclusion
Disease modification is established by demonstrating an enduring change in the clinical trajectory of an NDD based on intervention in the fundamental pathophysiology of the disease leading to nerve cell death. Supporting data are collected in clinical trials. Effectively defining a DMT will assist in NDD drug development programs.
Keywords: Alzheimer’s disease, Frontotemporal dementia, Progressive supranuclear palsy, Corticobasal degeneration, Amyotropic lateral sclerosis, Multiple system atrophy, Disease modification, Disease modifying therapy
Background
Alzheimer’s disease (AD) and many other neurodegenerative disorders (NDD) are becoming more frequent as the world’s population ages; they constitute an impending public health catastrophe. AD doubles in frequency every 5 years after the age of 65 and affects up to 50% of those over age 85 [1]. Similarly, Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS) are age-related conditions and are increasing in frequency with the aging of the population [2, 3]. Progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), frontotemporal dementia (FTD), dementia with Lewy bodies (DLB), Huntington’s disease (HD), multiple system atrophy (MSA), chronic traumatic encephalopathy (CTE), spinocerebellar ataxias and a variety of more rare proteinopathies are NDD. Cumulatively, NDD affect millions of individuals worldwide, costing governments billions of dollars in healthcare expenditures and lost economic productivity [4]. Treatment to ameliorate the social and personal tragedy of NDD is an urgent global priority.
NDD are protein aggregation disorders with neurodegeneration [5]. Neurodegeneration is defined as progressive loss of neurons and their processes with a corresponding progressive impairment in neuronal function [6]. Advances in neuroscience have provided new insights into NDD including their many shared features [7]. Impaired autophagy, protein aggregation, inflammation, oxidative injury, genetic and epigenetic features, mitochondrial impairment, apoptosis, reduced growth factor effects, and loss of synaptic plasticity are involved across NDD. Differing phenotypes of NDD reflect regional central nervous system (CNS) patho-geographies based on the molecular characteristics of the aggregating protein and genetic and environmental influences. PD is associated with substantia nigra and related regional changes; FTD with asymmetric frontal and temporal alterations; HD with striatal impact; and AD with initial medial temporal effects. The progression of NDD is increasingly understood in terms of prion-like transmission of proteins along disease-relevant pathways [8]. Degeneration in NDD occurs in networks of related neurons as identified by functional magnetic resonance imaging (fMRI) studies. These networks also serve as pathways for the propagation of proteins that aggregate in the course of NDD [9, 10].
Despite progress in understanding NDDs, there is a marked translational gap from biology to treatment and no disease-modifying therapy (DMT) has been approved for any NDD [11–15]. Aggressive efforts are required for seeking candidate molecules with possible disease-modification (DM) effects, developing new animal models that more accurately predict human efficacy, and advancing clinical trial methods and conduct [11, 16–18].
Among the clinical trial issues to be addressed in this important enterprise is the definition of DMT. Sponsors cannot plan development program without a clear understanding of how DMTs are defined, and trials cannot be designed and outcome measures chosen without such a definition. Preliminary work on defining DMTs has been accomplished by regulatory agencies that govern drug labeling (U.S. Food and Drug Administration [FDA]; European Medicines Agency [EMA]) and broad consensus building is needed to insure agreement among stakeholders representing trial design, biomarkers, biostatistics, regulation, commercial development, and patients and families. Cummings and Fox [19] recently offered a preliminary definition of DMT for AD. Here, the concept of neurodegeneration, DMT, the relationship of DM to neuroprotection, and the application of the definition to multiple NDDs are described.
Definition of Neurodegeneration and disease-modifying therapy
Neurodeneration is defined as loss of neurons and their processes (axons, dendrites, synapses) with a corresponding progressive impairment in neuronal function [6]. A DMT is defined as an intervention that produces an enduring change in the clinical progression of an NDD by interfering in the underlying pathophysiological mechanisms of the disease process leading to nerve cell death [19]. Two types of data supporting an intervention as a DMT have been identified: 1) the intervention produces a significant drug-placebo difference on accepted clinical outcome(s) and has a consistent effect on biomarkers considered reflective of the fundamental pathophysiology of an NDD, or 2) the intervention produces a positive outcome on a staggered start or delayed withdrawal clinical trial design consistent with an enduring change in clinical course. In both cases, slowing of disease progression on clinical measures is evident; in the former, biological evidence of DM supports the clinical measures.
Disease modification and Neuroprotection
The effect of a DMT is to produce an enduring change in the trajectory of clinical decline of the NDD by impacting the disease processes leading to neuronal death [19]. Neuroprotection is defined as an intervention that favorably influences the disease process or underlying pathogenesis to produce lasting benefits for patients [20, 21]. Effective neuroprotection results in disease modification and efficacious neuroprotective therapies are disease-modifying (Fig. 1).
Fig. 1.
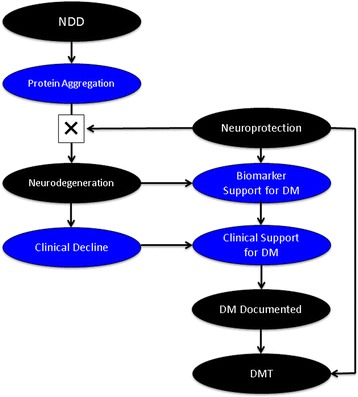
The conceptual framework for disease-modification in neurodegenerative disorders included neuroprotection preventing neurodegeneration with biomarker and clinical support for a disease-modifying therapy (DMT)
Neuroprotection might be achieved by direct effects on neurons (primary neuroprotection) or by interfering with processes that lead secondarily to cell death such as inflammation. The final common pathway to be achieved for DM is neuroprotection.
Biomarker support for disease-modification
Most development programs rely on biomarkers to provide support for DM. The biological change associated with DM is neuroprotection and biomarker support for DM depends on demonstration of neuronal preservation. DM and neuronal preservation cannot be observed directly and must be inferred from other types of evidence including biomarkers. To support DM, the biomarker must be indicative of a change in the processes leading to the loss of neurons.
Evidence regarding which biomarkers reliably reflect processes leading to cell death is evolving (Fig. 2). Cell death is the end product of a cascade of events and which elements of the cascade are critical to measure in support of DM has not been completely ascertained. Given the uncertainties regarding biomarkers as reporters of DM, the FDA has advised collecting multiple biomarkers in the course of trials to support a drug effect on the underlying biology of the NDD. In its guidance on “Alzheimer’s Disease: Developing Drugs for Treatment of Early Stage Disease” [22] the FDA noted that a biomarker effect must reflect a pathophysiological entity that is fundamental to the underlying disease process. They observed that there is currently insufficient evidence on which to base a hierarchical structuring of biomarkers and encouraged trial sponsors to analyze the results of multiple biomarkers independently.
Fig. 2.
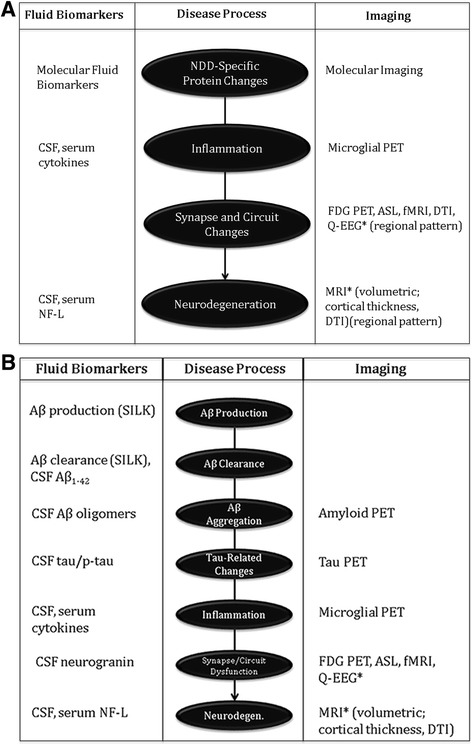
Biomarkers of neurodegenerative disorders (a) and of Alzheimer’s disease (b). The exact order of the cascade of events from Aß production to cell death is not fully determined (Aß – amyloid ß protein; AD – Alzheimer’s disease; ASL – arterial spin labeling; DTI – diffusion tensor imaging; FDG – fluoroeoxyglucose; fMRI functional magnetic resonance imaging; NF-L – neurofilament light; PET – positron emission tomography; p-tau – phosphorylated tau protein; Q-EEG – quantitative electroencephalography; SILK – stable isotope-labeled kinetics). *The regional pattern of these changes may be specific to the neurodegenerative disease
Magnetic resonance imaging (MRI) is a measure of brain volume and shows increasing brain atrophy with progression of AD and other NDD [23–25]. MRI is a candidate biomarker for DM. In AD clinical trials, however, MRI has often shown greater atrophy in active treatment than placebo groups and its ability to function as a biomarker to support DM has not been shown [26, 27]. Diffusion tensor imaging (DTI) assessments are candidate measures for loss of spiny neurons and their projections in HD [28]. Measures of iron and neuromelanin have promise as MRI outcomes assessing neurodegeneration in PD [29], and a combined measure of third ventricle, midbrain, and frontal lobe has been proposed to measure DM effects in clinical trials in PSP [30]. Brain boundary shift intervals and ventricular boundary shift intervals correlate with disease progression in FTD and may perform well in clinical trials to show drug-placebo differences in atrophy with an implied effect on neurodegeneration [24].
In AD, removal of plaque amyloid has been demonstrated with several immunotherapies with no corresponding clinical benefit and reducing fibrillar amyloid demonstrated with amyloid imaging is not by itself support of DM [31, 32]. Removal of plaques was associated with a beneficial impact on cognitive decline in studies of aducanumab, suggesting that fibrillar amyloid removal may correlate with DM in some therapeutic circumstances [33]. Effects on soluable forms of amyloid may be key to establishing DM with anti-amyloid therapies in AD [34].
Tau PET detects neurofibrillary tangles closely associated with cell death and may serve as a marker for neurodegeneration in AD [35, 36]. Some tau-related tracers may be valuable as measures of neurodegeneration in tauopathies such as CTE [37]. High CSF tau levels predict cognitive decline in AD, PD, and Creutzfeldt-Jakob disease [38–40]. These observations demonstrate a relationship between CSF tau and clinical course suggesting that drug-placebo differences in tau elevation may be one means of supporting DM with a biomarker. CSF tau is not abnormal in all NDD; in addition, changes in tau might not be equally supportive of DM across all phases of NDDs.
Emerging biomarkers may assist in supporting DM in DMT trials. Measures of neurofilament light chain protein show the protein to be increased in CSF of patients with AD, FTD, and ALS [41, 42] suggesting that it may be a marker of neurodegeneration relevant to several NDD populations. Drug-placebo differences in elevation of this peptide might offer support for DM. VILIP-1 and neurogranin are additional proteins shown to be abnormal in AD and to predict cognitive decline [43, 44]. These biomarkers may assist in demonstrating DM in AD.
The principal biological means by which DM will be achieved are uncertain and how the intervention will be reflected in serum, CSF, or imaging biomarkers is currently unprooven. Biomarker data of multiple types indicating a consistent, robust effect on fundamental disease pathophysiology in concert with compelling clinical information is most likely to demonstrate that an agent is a DMT. Biomarkers from multiple classes – neurodegeneration, biomarkers specific to the agent’s mechanism of action – are more consistent with DM than measures within a single class of biomarkers [19]. Biomarkers may be specific to a disease or reflective of the general neurodegerative process and present across multiple disease states (Fig. 2). Measures of Aß are specific to AD, alpha-synuclein measures are abnormal in PD and DLB, and measures of Huntington or TDP-43 would be reflective of HD and FTD or ALS, respectively. Changes in brain metabolism (fluorodeoxyglucose) or circuit function (functional MRI) may have regional patterns specific to disease-states [45, 46]. Interventions at any level of the cascade leading to cell death may be neuroprotective and preventive of neurodegenation. Such agents would be DMTS. Biomarkers assist in identifying the presence, type, and magnitude of change induced by therapy.
Trial designs to support disease-modification
The central feature of a DMT is an intervention that produces an enduring change in the course of NDD such that if the treatment is interrupted the patient does not resume the same level of function as one not treated. The concept of enduring change is embedded in the staggered start and delayed withdrawal clinical trial designs [47–50] (Fig. 3). In the staggered start trial, the participants in the delayed therapy group do not catch up with the initial therapy group if the treatment is a DMT. Catching up would be expected with a symptomatic therapy and not with a DMT since the course of the original group has been permanently altered by the DMT. Similarly, in the delayed withdrawal design, one group is withdrawn from therapy to determine if they assume the level of function of a placebo-treated group. Failure to decline to the untreated level is evidence of DM, whereas returning to the same level as an untreated group suggests that the treatment under study has symptomatic effects without DM [22, 48, 49, 51, 52].
Fig. 3.

Delayed start design (a). The delayed treatment group (3) does not catch up with the early treatment group (2). (1) is the untreated, placebo group. Staggered withdrawal design (b). The withdrawn group (3) does not decline to the level of the placebo group (1)
These trials are cumbersome, the duration of periods of observation off drug to draw comparisons with the treatment group, ethical challenges to withdrawing patients from treatment, and statistical issues related to adequate powering of the studies have all contributed to lack of use of these designs to support DM. The ADAGIO study in PD used a staggered start design [53]. A benefit was observed with the 1 mg dose and not the 2 mg dose leading to ambiguity in interpreting the trial in favor of DM by rasagiline [54]. Advances in analytic approaches to two-period designs may improve the utility of these designs and broaden their application [50].
Staggered start analyses have been used at trial termination when patients on placebo for the double-blind portion of the trial are switched to active therapy. This provides an opportunity to see if the placebo-treated patients catch up with those on treatment throughout the trial. This approach was used for analysis of the open label extension data of the Expedition and Expedition II data of solanezumab trials for treatment of AD [55]. The analysis suggested continued benefit, although DM was not confirmed in a subsequent trial [56]. This approach to a staggered start is compromised by the open-label nature of the analysis population (i.e., knowledge by patients and investigators that all patients are on active treatment in the extension period). The observation might help support DM in conjunction with other analyses and biomarker outcomes.
Disease-modification applies across neurodegenerative disorders
NDD are protein aggregation disorders with neurodegeneration [5]. AD, PD, PSP, CBD, FTD, ALS, DLB, HD, MSA, CTE, spinocerebellar ataxias and a variety of more rare proteinopathies are NDD [57, 58]. In all cases the same principles of DM, neurodegeneration, neuroprotection, and defining a DMT apply. Biomarkers, trial designs, many aspects of the underlying neurobiology, regulatory reviews, and even some interventions apply across NDD [7]. To achieve DM in any of these NDD, neuroprotection must be achieved. The enduring change critical to the concept of DM can be demonstrated by clinical measures plus biomarkers or by DM-type of trial designs. Some biomarkers such as neurofilament light chain protein may reflect later stages of cell death applicable to several NDD [41, 42]. Use of other biomarkers will vary depending on the pathobiology of the specific NDD; disease-specific biomarkers typically reflect events earlier in the cascade of processes leading to cell death [59] (Fig. 2). Biomarkers of target engagement are critical in drug development but do not necessarily imply neuroprotection and may not be supportive of DM.
Conclusion
DMTs are urgently needed to address the global public health crisis posed by AD and other NDD. NDD are steadily progressive clinically, reflecting an on-going loss of nerve cells and corresponding brain dysfunction. The biological changes are reflected in biomarkers. DM can be shown by a change in clinical course and corresponding alteration in fluid and imaging biomarkers. The enduring change in disease trajectory can also be demonstrated by staggered start or delayed withdrawal clinical trial designs. DMTs are neuroprotective and neuroprotection is the basis for DM. Neuroprotection may be achieved through interruption of upstream NDD-specific processes leading to cell death or through affecting downstream processes reflecting the intracellular final common pathway of neuronal death. Biomarker changes associated with successful DM may reflect the mechanism of action of the agent, level of interruption, and impact on downstream processes. Target engagement biomarkers may not equate to DM. A repertoire of biomarkers is needed to understand the full impact of a DMT. Implementing the definition and collecting evidence in support of DM will facilitate the development of DMTs.
Acknowledgements
Not applicable.
Funding
JC acknowledges funding from the National Institute of General Medical Sciences (Grant: P20GM109025) and support from Keep Memory Alive.
Availability of data and materials
Not applicable.
Author’s contributions
JC is responsible for all aspects of the manuscript including analysis, manuscript drafting and revisions, and final approval.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
JC has provided consultation to Abbvie, Acadia, Actinogen, Alzheon, Anavex, Avanir, Axovant, Boehinger-Ingelheim, Bracket, Eisai, Forum, GE Healthcare, Genentech, Intracellular Interventions, Lilly, Lundbeck, Medavante, Merck, Neurocog, Novartis, Orion, Otsuka, Pfizer, Piramal, QR, Roche, Suven, Sunovion, Takeda and Toyama pharmaceutical and assessment companies.
References
- 1.Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer's disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci. 2009;11:111–128. doi: 10.31887/DCNS.2009.11.2/cqiu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coupe C, Gordon PH. Amyotrophic lateral sclerosis - clinical features, pathophysiology and management. European Neurol Rev. 2013;8:38–44. doi: 10.17925/ENR.2013.08.01.38. [DOI] [Google Scholar]
- 3.Levy G. The relationship of Parkinson disease with aging. Arch Neurol. 2007;64:1242–1246. doi: 10.1001/archneur.64.9.1242. [DOI] [PubMed] [Google Scholar]
- 4.Shah H, Albanese E, Duggan C, Rudan I, Langa KM, Carrillo MC, et al. Research priorities to reduce the global burden of dementia by 2025. Lancet Neurol. 2016;15:1285–1294. doi: 10.1016/S1474-4422(16)30235-6. [DOI] [PubMed] [Google Scholar]
- 5.Hampel H, O’Bryant SE, Durrleman S, Younesi E, Rojkova K, Escott-Price V, et al. A precision medicine initiative for Alzheimer’s disease: the road ahead to biomarker-guided integrative disease modeling. Climacteric. 2017;20:107–118. doi: 10.1080/13697137.2017.1287866. [DOI] [PubMed] [Google Scholar]
- 6.Jack CR, Jr., Wiste HJ, Weigand SD, Knopman DS, Mielke MM, Vemuri P, et al. Different definitions of neurodegeneration produce similar amyloid/neurodegeneration biomarker group findings. Brain 2015;138:3747–3759. [DOI] [PMC free article] [PubMed]
- 7.Cummings J. Pillai J (Eds.): neurodegenerative diseases: unifying principles. United Kingdom: Oxford University Press; 2016. [Google Scholar]
- 8.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62:42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agosta F, Weiler M, Filippi M. Propagation of pathology through brain networks in neurodegenerative diseases: from molecules to clinical phenotypes. CNS Neurosci Ther. 2015;21:754–767. doi: 10.1111/cns.12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci. 2015;16:109–120. doi: 10.1038/nrn3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cummings J, Morstorf T, Lee G. Alzheimer's disease drug development pipeline: 2016. Alzheimers Dement. 2016;2:222–232. doi: 10.1016/j.trci.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jinawath N, Bunbanjerdsuk S, Chayanupatkul M, Ngamphaiboon N, Asavapanumas N, Svasti J, et al. Bridging the gap between clinicians and systems biologists: from network biology to translational biomedical research. J Transl Med. 2016;14:324. doi: 10.1186/s12967-016-1078-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mott M, Koroshetz W. Bridging the gap in neurotherapeutic discovery and development: the role of the National Institute of Neurological Disorders and Stroke in translational neuroscience. Neurotherapeutics. 2015;12:651–654. doi: 10.1007/s13311-015-0366-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pankevich DE, Altevogt BM, Dunlop J, Gage FH, Hyman SE. Improving and accelerating drug development for nervous system disorders. Neuron. 2014;84:546–553. doi: 10.1016/j.neuron.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sabbagh JJ, Kinney JW, Cummings JL. Alzheimer's disease biomarkers: correspondence between human studies and animal models. Neurobiol Dis. 2013;56:116–130. doi: 10.1016/j.nbd.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Finkbeiner S. Bridging the valley of death of therapeutics for neurodegeneration. Nat Med. 2010;16:1227–1232. doi: 10.1038/nm.2222. [DOI] [PubMed] [Google Scholar]
- 17.Millington C, Sonego S, Karunaweera N, Rangel A, Aldrich-Wright JR, Campbell IL, et al. Chronic neuroinflammation in Alzheimer's disease: new perspectives on animal models and promising candidate drugs. Biomed Res Int. 2014;2014:309129. doi: 10.1155/2014/309129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang R, Liu X, Li S, Li XJ. Transgenic animal models for study of the pathogenesis of Huntington's disease and therapy. Drug Des Devel Ther. 2015;9:2179–2188. doi: 10.2147/DDDT.S58470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cummings J, Fox N. Defining disease modifying therapy for Alzheimer's disease. J Prev Alz Dis. 2017;4:109–115. doi: 10.14283/jpad.2017.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ravina BM, Fagan SC, Hart RG, Hovinga CA, Murphy DD, Dawson TM, et al. Neuroprotective agents for clinical trials in Parkinson's disease: a systematic assessment. Neurology. 2003;60:1234–1240. doi: 10.1212/01.WNL.0000058760.13152.1A. [DOI] [PubMed] [Google Scholar]
- 21.Wiendl H, Elger C, Forstl H, Hartung HP, Oertel W, Reichmann H, et al. Gaps between aims and achievements in therapeutic modification of neuronal damage ("neuroprotection") Neurotherapeutics. 2015;12:449–454. doi: 10.1007/s13311-015-0348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.U.S. Food and Drug Administration. Guidance for Industry Alzheimer's Disease: Developing drugs for the treatment of early stage disease. In: 2013. [http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm338287.pdf].
- 23.Apostolova LG, Thompson PM. Brain mapping as a tool to study neurodegeneration. Neurotherapeutics. 2007;4:387–400. doi: 10.1016/j.nurt.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon E, Rohrer JD, Kim LG, Omar R, Rossor MN, Fox NC, et al. Measuring disease progression in frontotemporal lobar degeneration: a clinical and MRI study. Neurology. 2010;74:666–673. doi: 10.1212/WNL.0b013e3181d1a879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vemuri P, Wiste HJ, Weigand SD, Knopman DS, Trojanowski JQ, Shaw LM, et al. Serial MRI and CSF biomarkers in normal aging, MCI, and AD. Neurology. 2010;75:143–151. doi: 10.1212/WNL.0b013e3181e7ca82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64:1563–1572. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- 27.Novak G, Fox N, Clegg S, Nielsen C, Einstein S, Lu Y, et al. Changes in brain volume with bapineuzumab in mild to moderate Alzheimer's disease. J Alzheimers Dis. 2015;49:1123–1134. doi: 10.3233/JAD-150448. [DOI] [PubMed] [Google Scholar]
- 28.Douaud G, Behrens TE, Poupon C, Cointepas Y, Jbabdi S, Gaura V, et al. Vivo evidence for the selective subcortical degeneration in Huntington's disease. NeuroImage. 2009;46:958–966. doi: 10.1016/j.neuroimage.2009.03.044. [DOI] [PubMed] [Google Scholar]
- 29.Lehericy S, Vaillancourt DE, Seppi K, Monchi O, Rektorova I, Antonini A, et al. The role of high-field magnetic resonance imaging in parkinsonian disorders: pushing the boundaries forward. Mov Disord. 2017;32:510–525. doi: 10.1002/mds.26968. [DOI] [PubMed] [Google Scholar]
- 30.Hoglinger GU, Schope J, Stamelou M, Kassubek J, Del Ser T, Boxer AL, et al. Longitudinal magnetic resonance imaging in progressive supranuclear palsy: a new combined score for clinical trials. Mov Disord. 2017;32:842–852. doi: 10.1002/mds.26973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu E, Schmidt ME, Margolin R, Sperling R, Koeppe R, Mason NS, et al. Amyloid-beta 11C-PiB-PET imaging results from 2 randomized bapineuzumab phase 3 AD trials. Neurology. 2015;85:692–700. doi: 10.1212/WNL.0000000000001877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 33.Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 34.Kayed R, Lasagna-Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis. 2013;33(Suppl 1):S67–S78. doi: 10.3233/JAD-2012-129001. [DOI] [PubMed] [Google Scholar]
- 35.Passamonti L, Vazquez Rodriguez P, Hong YT, Allinson KS, Williamson D, Borchert RJ, et al. 18F-AV-1451 positron emission tomography in Alzheimer's disease and progressive supranuclear palsy. Brain. 2017;140:781–791. doi: 10.1093/brain/aww340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, et al. Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between beta-amyloid and Tauopathy. JAMA Neurol. 2016;73:1070–1077. doi: 10.1001/jamaneurol.2016.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrio JR, Small GW, Wong KP, Huang SC, Liu J, Merrill DA, et al. Vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proc Natl Acad Sci U S A. 2015;112:E2039–E2047. doi: 10.1073/pnas.1409952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen OS, Chapman J, Korczyn AD, Warman-Alaluf N, Nitsan Z, Appel S, Kahana E, Rosenmann H. CSF tau correlates with CJD severity and cognitive decline. Acta Neurol Scand. 2016;133:119–23. [DOI] [PubMed]
- 39.Degerman Gunnarsson M, Lannfelt L, Ingelsson M, Basun H, Kilander L. High tau levels in cerebrospinal fluid predict rapid decline and increased dementia mortality in Alzheimer's disease. Dement Geriatr Cogn Disord. 2014;37:196–206. doi: 10.1159/000355556. [DOI] [PubMed] [Google Scholar]
- 40.Liu C, Cholerton B, Shi M, Ginghina C, Cain KC, Auinger P, et al. CSF tau and tau/Abeta42 predict cognitive decline in Parkinson's disease. Parkinsonism Relat Disord. 2015;21:271–276. doi: 10.1016/j.parkreldis.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaiottino J, Norgren N, Dobson R. Topping J, Nissim a, Malaspina a, et al. increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One. 2013;8:e75091. doi: 10.1371/journal.pone.0075091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Landqvist Waldo M, Frizell Santillo A, Passant U, Zetterberg H, Rosengren L, Nilsson C, et al. Cerebrospinal fluid neurofilament light chain protein levels in subtypes of frontotemporal dementia. BMC Neurol. 2013;13:54. doi: 10.1186/1471-2377-13-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kvartsberg H, Duits FH, Ingelsson M, Andreasen N, Ohrfelt A, Andersson K, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer's disease. Alzheimers Dement. 2015;11:1180–1190. doi: 10.1016/j.jalz.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 44.Tarawneh R, Lee JM, Ladenson JH, Morris JC, Holtzman DMCSF. VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology. 2012;78:709–719. doi: 10.1212/WNL.0b013e318248e568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou J, Greicius MD, Gennatas ED, Growdon ME, Jang JY, Rabinovici GD, et al. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain. 2010;133:1352–1367. doi: 10.1093/brain/awq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cummings J, Zhong K, Cordes D. Drug development in Alzheimer's disease - the role of default mode network assessment in phase II. US Neurol. 2017; In press
- 47.Bodick N, Forette F, Hadler D, Harvey RJ, Leber P, McKeith IG, et al. Protocols to demonstrate slowing of Alzheimer disease progression. Position paper from the international working group on harmonization of dementia drug guidelines. The disease progression sub-group. Alzheimer Dis Assoc Disord. 1997;11(Suppl 3):50–53. [PubMed] [Google Scholar]
- 48.Leber P. Observations and suggestions on antidementia drug development. Alzheimer Dis Assoc Disord. 1996;10(Suppl 1):31–35. doi: 10.1097/00002093-199601031-00009. [DOI] [PubMed] [Google Scholar]
- 49.Leber P. Slowing the progression of Alzheimer disease: methodologic issues. Alzheimer Dis Assoc Disord. 1997;11(Suppl 5):S10–S21. [PubMed] [Google Scholar]
- 50.McDermott MP, Hall WJ, Oakes D, Eberly S. Design and analysis of two-period studies of potentially disease-modifying treatments. Control Clin Trials. 2002;23:635–649. doi: 10.1016/S0197-2456(02)00238-6. [DOI] [PubMed] [Google Scholar]
- 51.European Medicine Agency . Draft guideline on the clinical investigation of medicines for the treatment of Alzheimer's disease and other dementias. 2016. Committee for medicinal products for human use. [Google Scholar]
- 52.Cummings JL. Defining and labeling disease-modifying treatments for Alzheimer's disease. Alzheimers Dement. 2009;5:406–418. doi: 10.1016/j.jalz.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 53.Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A, et al. A double-blind, delayed-start trial of rasagiline in Parkinson's disease. N Engl J Med. 2009;361:1268–1278. doi: 10.1056/NEJMoa0809335. [DOI] [PubMed] [Google Scholar]
- 54.Sampaio C, Ferreira JJ. Parkinson disease: ADAGIO trial hints that rasagiline slows disease progression. Nat Rev Neurol. 2010;6:126–128. doi: 10.1038/nrneurol.2010.2. [DOI] [PubMed] [Google Scholar]
- 55.Liu-Seifert H, Andersen SW, Lipkovich I, Holdridge KC, Siemers EA. Novel approach to delayed-start analyses for demonstrating disease-modifying effects in Alzheimer's disease. PLoS One. 2015;10:e0119632. doi: 10.1371/journal.pone.0119632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abbott A, Dolgin E. Failed Alzheimer's trial does not kill leading theory of disease. Nature. 2016;540:15–16. doi: 10.1038/nature.2016.21045. [DOI] [PubMed] [Google Scholar]
- 57.Dunkel P, Chai CL, Sperlagh B, Huleatt PB, Matyus P. Clinical utility of neuroprotective agents in neurodegenerative diseases: current status of drug development for Alzheimer's, Parkinson's and Huntington's diseases, and amyotrophic lateral sclerosis. Expert Opin Investig Drugs. 2012;21:1267–1308. doi: 10.1517/13543784.2012.703178. [DOI] [PubMed] [Google Scholar]
- 58.Whitcup SM. Clinical trials in neuroprotection. Prog Brain Res. 2008;173:323–335. doi: 10.1016/S0079-6123(08)01123-0. [DOI] [PubMed] [Google Scholar]
- 59.Ewers M, Mattsson N, Minthon L, Molinuevo JL, Antonell A, Popp J, et al. CSF biomarkers for the differential diagnosis of Alzheimer's disease: a large-scale international multicenter study. Alzheimer Dement. 2015;11:1306–1315. doi: 10.1016/j.jalz.2014.12.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.