

Abstract
Cancer subtypes discovery is the first step to deliver personalized medicine to cancer patients. With the accumulation of massive multi-level omics datasets and established biological knowledge databases, omics data integration with incorporation of rich existing biological knowledge is essential for deciphering a biological mechanism behind the complex diseases. In this manuscript, we propose an integrative sparse K-means (is-K means) approach to discover disease subtypes with the guidance of prior biological knowledge via sparse overlapping group lasso. An algorithm using an alternating direction method of multiplier (ADMM) will be applied for fast optimization. Simulation and three real applications in breast cancer and leukemia will be used to compare is-K means with existing methods and demonstrate its superior clustering accuracy, feature selection, functional annotation of detected molecular features and computing efficiency.
Keywords and phrases: Cancer subtype, omics integrative analysis, overlapping group lasso, admm
1. Introduction
While cancer has been thought to be a single type of disease, increasing evidence from modern transcriptomic studies have suggested that each specific cancer may consist of multiple subtypes, with different disease mechanisms, survival rates and treatment responses. Cancer subtypes have been extensively studied, including in leukemia [Golub et al. (1999)], lymphoma [Rosenwald et al. (2002)], glioblastoma [Parsons et al. (2008); Verhaak et al. (2010)], breast cancer [Lehmann et al. (2011); Parker et al. (2009)], colorec-tal cancer [Sadanandam et al. (2013)] and ovarian cancer [Tothill et al. (2008)]. These subtypes usually have strong clinical relevance since they show different outcome, and might be responsive to different treatments [Abramson et al. (2015)]. However, single cohort/single omics (e.g., transcriptome) analysis suffers from sample size limitation and reproducibility issues [Simon et al. (2003); Simon (2005); Domany (2014)]. Over the years, large amount of omics data are accumulated in public databases and depositories, for example, The Cancer Genome Atlas (TCGA) http://cancergenome.nih.gov, Gene Expression Omnibus (GEO) http://www.ncbi.nlm.nih.gov/geo/, Sequence Read Archive (SRA) http://www.ncbi.nlm.nih.gov/sra, just to name a few. These datasets provided unprecedented opportunities to reveal cancer mechanisms via combining multiple cohorts or multiple-level omics data types (a.k.a. horizontal omics meta-analysis and vertical omics integrative analysis; see below) [Tseng, Ghosh and Feingold (2012)]. Omics integrative analysis has been found successful in many applications: (e.g., breast cancer [Koboldt et al. (2012)], stomach cancer [Bass et al. (2014)]). On the other hand, a tremendous amount of biological information has been accumulated in public databases. Proper usage of these prior information (e.g., pathway information and miRNA targeting gene database) can greatly guide the modeling of omics integrative analysis.
In the literature, researchers have applied various types of clustering methods for high-throughput experimental data (e.g., microarray) to identify novel disease subtypes. Popular methods include hierarchical clustering [Eisen et al. (1998)], K-means [Dudoit and Fridlyand (2002)], mixture model-based approaches [Xie, Pan and Shen (2008); McLachlan, Bean and Peel (2002)] and nonparametric approaches [Qin (2006)], for analysis of single transcriptomic study. Resampling and ensemble methods have been used to improve stability of the clustering analysis [Kim et al. (2009); Swift et al. (2004)] or to pursue tight clusters by leaving scattered samples that are different from major clusters [Tseng (2007); Tseng and Wong (2005); Maitra and Ramler (2009)]. Witten and Tibshirani (2010) proposed a sparse K-means algorithm that can effectively select gene features and perform sample clustering simultaneously. To extend single-study techniques towards integration of multiple omics data sets, Tseng, Ghosh and Feingold (2012) categorized omics data integration into two major types: (A) horizontal omics meta-analysis and (B) vertical omics integrative analysis. For horizontal meta-analysis, multiple studies of the same omics data type (e.g., transcriptome) from different cohorts are combined to increase sample size and statistical power, a strategy often used in differential expression analysis [Ramasamy et al. (2008)], pathway analysis [Shen and Tseng (2010)] or subtype discovery [Huo et al. (2016)]. In contrast, vertical integrative analysis aims to integrate multi-level omics data from the same patient cohort (e.g., gene expression data, genome-wide profiling of somatic mutation, DNA copy number, DNA methylation or microRNA expression from the same set of biological samples [Richardson, Tseng and Sun (2016)]). In this paper, we focus on vertical omics integrative analysis for disease subtype discovery. Several methods for this purpose have been proposed in the literature. Lock and Dunson (2013) fitted a finite Dirichlet mixture model to perform Bayesian consensus clustering that allows common clustering across omics types as well as omics-type-specific clustering. The model, however, does not perform proper feature selection, and thus is not suitable for high-dimensional omics data. Shen, Olshen and Ladanyi (2009) proposed a latent variable factor model (namely iCluster) to cluster cancer samples by integrating multi-omics data. The method does not incorporate prior biological knowledge and requires extensive computing due to EM algorithm with large matrix operation. We will use the popular iCluster method as the baseline method to compare in this paper.
The central question we ask in this paper is: “Can we identify cancer subtypes by simultaneously integrating multi-level omics datasets and/or utilizing existing biological knowledge to increase accuracy and interpretation?” Several statistical challenges will arise when we attempt to achieve this goal: (1) If multi-level omics data are available for a given patient cohort, what kind of method is effective to achieve robust and accurate disease subtype detection via integrating multi-omics data? (2) Since only a small subset of intrinsic omics features are relevant to the disease subtype characterization, how can we perform effective feature selection in the high-dimensional integrative analysis? (3) With the rich biological information (e.g., targeted genes of each miRNA or potential cis-acting regulatory mechanism between copy number variation, methylation and gene expression), how can we fully utilize the prior information to guide feature selection and clustering? In this paper, we propose an integrative sparse K-means (IS-K means) approach by extending the sparse K-means algorithm with overlapping group lasso technique to accommodate the three goals described above. The lasso penalty in the sparse K-means method allows effective feature selection for clustering. In the literature, (nonoverlapping) group lasso [Yuan and Lin (2006)] has been developed in a regression setting to encourage features of the same group to be selected or excluded together. The approach, however, has two major drawbacks: (1) it does not allow sparsity within groups (i.e., a group of features are either all selected or all excluded), and (2) the penalty function does not allow overlapping groups. For the first issue, Simon et al. (2013) proposed a sparse group lasso with both an l1 lasso penalty and a group lasso penalty to allow sparsity within groups while the approach does not allow overlapping groups. For the latter issue, overlapping group information from biological knowledge is frequently encountered in many applications. In genomic application, for example, the targeted genes of two miRNAs are often overlapped or two pathways may contain overlapping genes. Jacob, Obozin-ski and Vert (2009) proposed a duplication technique to allow overlapping groups in regression setting while the approach does not allow sparsity within groups. In this paper, we attempt to simultaneously overcome both aforementioned difficulties in a clustering setting, which brings optimization challenges beyond the duplication technique by Jacob, Obozinski and Vert (2009) and the sparse group lasso optimization by Simon et al. (2013). In our proposed IS-K means method, we will develop a novel reformulation of l1 lasso penalty and overlapping group lasso penalty so that a fast optimization technique using alternating direction method of multiplier (ADMM) [Boyd et al. (2011)] can be applied (see Section 3.4.1).
The rest of the paper is structured as following. Section 2 gives a motivating example. Section 3 establishes the method and optimization procedure. Sections 4.1–4.3 comprehensively compares the proposed method with the popular iCluster method using simulation and two breast cancer applications on multilevel omics data. Section 4.4 provides another type of IS-K means application of pathway-guided clustering on single transcriptomic study. Section 5 includes the final conclusion and discussion.
2. Motivating example
Figure 1(A) shows a clustering result using single study sparse K-means (detailed algorithm see Section 3.1) on the mRNA, methylation and copy number variation (CNV) datasets separately from 770 samples in TCGA. As expected, they generate very different disease subtyping without regulatory inference across mRNA, methylation and CNV. In this example, single study sparse K-means fails to consider that different omics features belonging to the same genes are likely to contain cis-acting regulatory mechanisms related to the disease subtypes. Figure 1(B) combines the three datasets to perform IS-Kmeans. The IS-K means generates a single disease subtyping and takes into account of the prior regulatory knowledge between mRNA, methylation and CNV. The prior knowledge can also be a pathway database (e.g., KEGG, BioCarta and Reactome) or knowledge of miRNA targets prediction databases (e.g., PicTar, TargetScan, DIANA-microT, miRanda, rna22 and PITA) [Witkos, Koscianska and Krzyzosiak (2011); Fan and Kurgan (2015)]. Incorporating such prior information of feature grouping increases statistical power and interpretation. Figure 1(C) shows a simple example of such group prior knowledge. Pathway 𝒯1 includes mRNA1, mRNA2, mRNA3 and mRNA6 while pathway 𝒯2 includes mRNA3, mRNA4, mRNA5 and mRNA7. Note that mRNA3 appears in both pathway 𝒯1 and 𝒯2, which requires our algorithm to allow overlapping groups. Our goal is to develop a sparse clustering algorithm integrating multi-level omics datasets and the aforementioned prior regulatory knowledge by overlapping group lasso. The algorithm is also suitable for single omics dataset with incorporating prior overlapping pathway information (see the leukemia examples in Section 4.4).
Fig. 1.
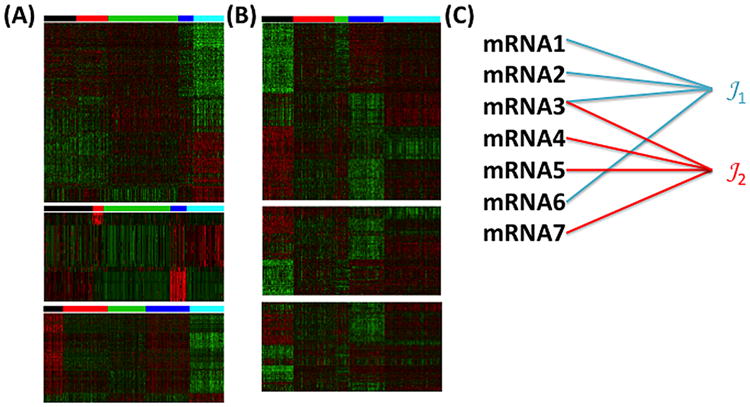
(A) Clustering of mRNA (upper heatmap) CNV (middle heatmap) and methylation (lower heatmap) profiles separately results in different five clusters of breast cancer subtypes (represented by color bars of five colors). (B) IS-Kmeans merges mRNA (upper heatmap) CNV (middle heatmap) and methylation (lower heatmap) and perform sample clustering. Inter-omics biological knowledge is also taken into account by overlapping group lasso. (C) An illustrating example of design of overlapping group lasso penalty term Ω(z) to incorporate prior knowledge of pathway information. Here,
3. Method
3.1. K-Means and sparse K-means
Consider Xjq the gene expression intensity of gene j and sample q. The K -means method [MacQueen (1967)] targets to minimize the within-cluster sum of squares (WCSS):
| (3.1) |
where K is the number of clusters, J is the number of genes (features), C = (C1, C2, …, Ck) denotes the clustering result containing partitions of all samples into K clusters, nk is the number of samples in cluster k and dpq,j = (Xjp – Xjq)2 denotes the squared Euclidean distance of gene j between sample p and q. One drawback of K -means is that it assumes all J features with equal weights in the distance calculation. In genomic applications, J is usually large but biologically only a small subset of genes may contribute to the sample clustering. Witten and Tibshirani (2010) tackled this problem by proposing a sparse K -means approach with lasso regularization on gene-specific weights. They found that direct application of lasso regularization to equation (3.1) will result in a meaningless null solution. Instead, they utilized the fact that minimizing WCSS is equivalent to maximizing between-cluster sum of squares (BCSS) since WCSS and BCSS add up to a constant value of total sum of squares [TSSj = BCSSj(C) + WCSSj(C)]. The optimization in equation (3.1) is equivalent to
| (3.2) |
The lasso regularization on gene-specific weights in equation (3.2) gives the following sparse K-means objective function:
| (3.3) |
where zj denotes weight for gene j, C = (C1,…, Ck) is the clustering result, K is the pre-estimated number of clusters and ‖z‖1 and ‖z‖2 arethe l1 and l2 norm of the weight vector z = (z1,…,zJ). The regularization shrinks most gene weights to zero and μ is a tuning parameter to control the number of nonzero weights (i.e., the number of intrinsic genes for subtype characterization). This objective function can be rewritten in its Lagrangian form:
3.2. Integrative sparse K-means (IS-K means)
We extend the sparse K-means objective function to group structured sparse K-means. Here, we consider J to be the total number of features combing all levels of omics datasets. In order to make features of different omics data types on the same scale and comparable, we normalized BCSSj by TSSj and denote
We put the overlapping group lasso penalty term Ω(z) in the objective function:
| (3.4) |
where γ is the penalty tuning parameter controlling the numbers of nonzero features, α ∈ [0, 1] is a term controlling the balance between individual feature penalty and group feature penalty. If α = 1, there is no group feature penalty term and the objective function is equivalent to sparse K-means objective function after standardizing each feature. If α = 0, there is no individual feature penalty and only group feature penalty exists. The overlapping group lasso penalty term is defined as
where 𝒢0 is the number of (possibly overlapping) feature groups from prior biological knowledge, wg ∈ ℝ is the group weight coefficient for group g, mg = (mg1,…, mgJ) is the design vector of the gth feature group and ○ represents Hadamard product. The design of wg and mg is discussed in Section 3.3. Note that features with no group information are also treated as a group by itself (a group only contains a feature); such a design is to avoid bias towards a feature with no group information by receiving no penalization. The feature groups can either come from existing biological databases (e.g., pathway or miRNA target database), or from basic biological cis-regulatory knowledge (CNV and methylation features in the neighborhood of a nearby gene region). The first term in equation (3.4) encourages large weights for features with strong clustering separability. The second term is an l1 norm lasso penalty to encourage sparsity. Finally, Ω(z) serves as overlapping group lasso to encourage features in the prior knowledge groups to be selected simultaneously (or discarded together). The intuition of group lasso is that if we transform the Lagrange form of Ω(z) to its constraint form, it becomes an elliptic constraint and features of the same group are preferred to be selected together [Yuan and Lin (2006); Jacob, Obozinski and Vert (2009)]. The combination of l1 norm lasso penalty and overlapping group lasso penalty Ω(z) serves to achieve a sparse feature selection and also encourages (but does not force) features of the same group to be selected together.
Remark. Since different types of omics datasets may have different value ranges and distributions, additional normalization may be needed in the preprocessing. For example, the commonly-used beta values from methy-seq (defined as “methylation counts”/“total counts”) represent the proportions of methylation and range between 0 and 1. A logit transformation to so-called M-values is closer to Gaussian distribution and is more suitable to integrate with other omics data. Similarly, log-transformation of expression intensities from microarray, log-transformation of RPKM/TPM (summarized expression values) from RNA-seq and log-ratio values of CNV values from SNP arrays have been shown to be roughly Gaussian distributed and are proper for multi-omics integration. Another possibility is by replacing Euclidean distance to an appropriate distance measurement (e.g., Gower's distance for binary categorical and ordinal data, and Bray– Curtis dissimilarity for count data). Under this scenario, equation (3.4) remains valid under such modification and we only need to incorporate partition around medoids (PAM) [Kaufman and Rousseeuw (1987)] instead of K -means in the optimization procedure in Section 3.4.1. However, heterogeneity of different distance measurement may require extra different sparsity penalties and this is beyond consideration in this paper.
3.3. Design of overlapping group lasso penalty
In this section, we discuss and justify the design of overlapping group lasso penalty for wg and mg. We denote by 𝒯g as the collection of features in group g (1 ≤ g ≤ 𝒢0) and define frequency of feature j appearing in different groups: h(j) = Σ1≤g≤𝒢0𝕀{j ∈ 𝒯g}. We also define the intrinsic feature set ℐ (i.e., features that contribute to the underlying true sample clustering) and the nonintrinsic feature set ℐ̄. We first state an “unbiased feature selection” principle under a simplified situation:
Definition 3.1 (“Unbiased Feature Selection” principle). Suppose equal separation ability in all intrinsic features ℐ = {j : Rj = R > 0} and no separation ability in nonintrinsic features ℐ̄ = {j : Rj = 0} under the true clustering label. The proposed overlapping group lasso design (wg and mg) is said to satisfy the “unbiased feature selection” principle if under equation (3.4), it generates equal weights zj = 1/√|ℐ| for j ∈ ℐ and zj = 0 for j ∈ ℐ̄ given any prior knowledge of feature groups 𝒯g, 1 ≤ g ≤ 𝒢0.
The theorem below states an overlapping group lasso penalty design that satisfies the “unbiased feature selection” principle when all features are intrinsic features (i.e., ℐ̄ = (ϕ).
Theorem 3.1. Consider and mg = (mg1, …,mgj, …, mgJ) in equation (3.4). Suppose equal separation ability for all features R1 = … = RJ = R (ℐ̄ = ϕ) and further assume R > γ. The design of satisfies the “unbiased feature selection” principle such that optimum solution of z from equation (3.4) generates zj = 1/√J, ∀j.
Theorem 3.1 gives a design of overlapping group lasso penalty such that given equal separation ability for all features, the feature selection is not biased by the prior group knowledge. When all the groups are nonoverlapping, h(j) = 1, ∀j, then
where |𝒯g| is number of features in group 𝒯g, which is the nonoverlapping group lasso penalty [Yuan and Lin (2006)]. However, this weight design is not applicable when the underlying intrinsic feature set is sparse (i.e., ℐ̄ ≠ ϕ). If there are many nonintrinsic features inside group g, the intrinsic features in group g is over penalized since wg is inflated by the contribution of nonintrinsic features. Therefore, we propose the following overlapping group lasso penalty and show that the design satisfies the “unbiased feature selection” principle when the intrinsic feature set is sparse:
| (3.5) |
Theorem 3.2. Suppose the intrinsic feature set ℐ = {j : Rj = R > 0} and the nonintrinsic feature set ℐ̄ = {j : Rj = 0}. We further assume R > γ. The overlapping group lasso penalty in equation (3.5) satisfies the “unbiased feature selection” principle such that the optimum solution of z from equation (3.4) is zj = 1/√|ℐ| for j ∈ ℐ and zj = 0 for j ∈ ℐ̄.
Note that we take into account both the nonintrinsic features and the intrinsic features in the penalty design in equation (3.5). Only intrinsic features contribute to the group weight coefficient wg. The design vector mg remains the same as nonoverlapping group lasso. In practice, the intrinsic feature set ℐ is unknown. We follow the coefficient design of adaptive lasso [Zou (2006)] and adaptive group lasso [Huang, Horowitz and Wei (2010)], which have been discussed in the literature and they maintain a consistency property under certain mild conditions. Specifically, we set α = 1 in equation (3.4) where only individual feature penalty is considered and use the solution ẑ to define estimated intrinsic feature set ℐ̂ = {j : ẑj > 0} and nonintrinsic feature set ℐ̄̂ = {j : ẑj = 0} for equation (3.5). In the example of Figure 1(C), suppose all 7 features are intrinsic genes. Pathway 𝒯1 contains mRNA1, mRNA2, mRNA3 and mRNA6, reflecting prior knowledge from pathway databases. Similarly, group for pathway 𝒯2 contains mRNA3, mRNA4, mRNA5 and mRNA7. As a result, m1 = (1, 1,1/2, 0, 0,1,0) and m2 = (0,0, 1/2,1,1, 0, 1) and
Note that in our example mRNA3 is shared by pathway groups 𝒯1 and 𝒯2, representing overlapping group lasso penalty.
3.4. Optimization
In this section, we discuss major issues for optimization of equation (3.4). First, we introduce transformation of equation (3.4) such that l1 norm penalty can be absorbed in l2 norm group penalty. Second, we introduce the optimization procedure for the proposed objective function. Third, we discuss how to use ADMM to optimize the weight term, which is critical and a difficult problem since it involves both the l1 norm penalty and overlapping group lasso penalty. Last, we discuss the stopping rule for the optimization.
3.4.1. Reformulation and iterative optimization
We use the fact that γα‖z‖1 can be rewritten as and zj = (0,…, zj, …., 0)⊤ with only the jth element nonzero. In other words, the l1 norm penalty of a single feature can be deemed as group penalty with only one feature within a group. Therefore, we can rewrite objective function equation (3.4) as
| (3.6) |
s.t. ‖z‖2 ≤ 1, zj ≥ 0, where ϕj = (ϕj1, …, ϕjJ), ϕji = 1 if j = i and ϕji = 0 if j ≠ i. We combine J and 𝒢0 groups and the combined groups are of size 𝒢 = J + 𝒢0. Define
Therefore, we can rewrite objective function equation (3.6) as
| (3.7) |
where R(C) = (R1(C), …, RJ(C))⊤. The optimization procedure are outlined below:
Initialize weight z using the original sparse K-means method without the group lasso term.
Given weight z, use weighted K-means to update cluster labels C [R is the normalized WCSS so minimizing −R(C)⊤z is essentially weighed K-means]. This is a nonconvex problem so multiple random starts are recommended to alleviate local minimum problem.
Given the cluster label C, R is fixed so optimizing the objective function is a convex problem with respect to solving weight z. We use ADMM in the next subsection to update weight z.
Iterate 2 and 3 until converge.
The detailed algorithm for Step 3 is outlined in Section 3.4.2 and the stopping rules of Step 3 and Step 4 are described in Section 3.4.3.
3.4.2. Update weight using ADMM
Alternating direction method of multiplier (ADMM) [Boyd et al. (2011)] is ideal for solving the optimization in equation (3.7). We introduce an auxiliary variable xg and write down the augmented Lagrange:
| (3.8) |
s.t. ‖z‖2 ≤ 1, zj ≥ 0 and xg = βg ○ z. This problem [equation (3.8)] is clearly equivalent to the original objective function [equation (3.7)], since for any feasible z the terms added to the objective is zero. ρ is the augmented Lagrange parameter which will be discussed in more detail in Section 3.4.4. Here, the augmented Lagrange is minimized jointly with respect to the two primal variables xg, z and the dual variable yg. In ADMM, xg, z and yg are updated in an alternating or sequential fashion [Boyd et al. (2011)], and thus the optimization problem can be decomposed into three parts. Given ( , z and ), the new iteration of (x+, z+ and y+g) in equation (3.8) is updated as in the following:
where the updating equation of and z+ are derived from equation (3.8) and the the updating equation of is imbedded in ADMM procedure [Boyd et al. (2011)]. We can derive close form solution for xg part and z part by the Karush– Kuhn–Tucker (KKT) condition. Details are given in the Appendix:
Define we have where (·)+ = max(0, ·).
Define and , where βg = (βg1, βg2, …, βgJ)⊤, xg = (xg1, xg2,…, xgJ)⊤ and yg = (yg1, yg2, …, ygJ)⊤. The solution is given as following: we define If Σj fj(u)2 < 1, Otherwise, and u is selected s.t. ‖z+‖2 = 1.
3.4.3. Stopping rules
We have two algorithms which require stopping rules. For ADMM in the optimization of Step 3, the primal residual of group g in ADMM iteration t is: rt = xt – βg ο zt, and the l2 norm of primal residual is . The l2 norm of dual residual is: . We set our ADMM stopping criteria such that simultaneously rt < 10−10 and νt < 10−10. For convergence of IS-K means, we iterate weighted K-means (Step 2) and updating weight by ADMM (Step 3) until converge. (i.e., ), where represents the zj estimate in the cth iteration of the IS-K means algorithm.
3.4.4. Augmented Lagrangian parameter ρ
Augmented Lagrangian parameter ρ controls the convergence of ADMM. In fact, large value of ρ will lead tosmall primal residual by placing a large penalty on violations of primal feasibility. And conversely, small value of ρ tend to produce small dual residual, but it will result in a large primal residual by reducing the penalty on primal feasibility [Boydet al. (2011)]. An adaptive scheme of varying ρ to balance the primal and dualresidual has been proposed [He, Yang and Wang (2000); Wang and Liao (2001)]
which greatly accelerates ADMM convergence in practice:
We set η = 10 and τincr = τdecr = 2. The intuition behind this scheme is to control both primal and dual residuals for converging to zero simultaneously.
3.5. Select tuning parameters
In the objective function of IS-K means, the number of clusters K is pre-specified. The issue of estimating K has been widely discussed in the literature and has been well recognized as a difficult and data-dependent problem. [Milligan and Cooper (1985); Kaufman and Rousseeuw (1990)]. Here, we suggest the number of clusters to be estimated in each study separately using conventional methods such as prediction strength [Tibshirani and Walther (2005)] or gap statistics [Tibshirani, Walther and Hastie (2001)] and jointly compared across studies (such that the numbers of clusters are roughly the same for all studies) for a final decision before applying integrative sparse K-means. Below we assume that a common K is pre-estimated for all omics datasets.
Another important parameter to be determined is α, which controls the balance between individual feature penalty and overlapping group penalty. According to equation (3.4), α = 1 means we only emphasize on individual feature penalty and ignore overlapping group penalty. In this case, the IS-K means is equivalent to sparse K-means. α = 0 means we only emphasize the overlapping group penalty and ignore the individual feature penalty. Simon et al. (2013) argued that there is no theoretically optimal selection for α because selection of α relates to multiple factors such as accuracy of prior group information and sparsity within groups. In general, a large α (e.g., α = 0.95) is suitable when prior group information may not be accurate or features within selected groups may be sparse. On the other hand, if we expect mild sparsity within groups and high accuracy of prior group information, a small α (e.g., α = 0.05) help select features by groups. In Section 4.1.2, we have performed simulation of different level of prior group information accuracy (θ = 1 and θ = 0.2) and found that α = 0.5 generates robust and high performance results in the sensitivity analysis. As a result, we apply α = 0.5 throughout the paper unless otherwise indicated.
The last tuning parameter is γ, which is the penalty coefficient. When γ is large, we place large penalty on the objective function and end up with less selected features. When γ is small, we put a small penalty and will include more features. We follow and extend the gap statistic procedure [Tibshirani, Walther and Hastie (2001)] to estimate γ:
For each feature in each omics type, randomly permute the gene expression (permute samples). This creates a permuted data set X(1). Repeat for B times to generate X(1), X(2), …, X(B).
-
For each potential tuning parameter γ, compute the gap statistics as below:
(3.9) where is from observed data, where z*, C* are the min-imizer of the objective function in equation (3.4) given γ. Ob(γ) is similar to O(γ) but generated from permuted data X(b).
For a range of selections of γ, select γ* such that the gap statistics in equation (3.9) is minimized.
Figure 2 shows an example of a simulated dataset that will be discussed in Section 4.1. In this example, we used α = 0.5 for IS-K means and the minimum gap statistics corresponded to 1778 genes, which is very close to the underlying truth 1800. The gap statistics for α = 0.05, 0.95,1 are plotted in supplementary materials [Huo and Tseng (2017), Figure S1] and they all provided adequate γ estimation. In practice, calculating gap statistics from a chain of γ can be performed efficiently by adopting a warm start for adjacent γ's. For example, after calculating O(γ1), the resulting weights can be used as an initial value for the next nearby γ2 = γ1 + Δ to calculate O(γ2) in the optimization iteration for fast convergence.
Fig. 2.

Selection oftuning parameter γ. This figure was from the simulated dataset in Section 4.1 with α = 0.5. x-axis represents tuning parameter γ. Red curve and left y-axis denote the corresponding gap statistics. Black curve and right y-axis denote the corresponding number of selected features. The blue dots (γ = 0.21) represent where the gap statistics is minimized, and the corresponding number of selected feature is 1778.
4. Result
We evaluated integrative sparse K-means (IS-K means) on simulation datasets in Section 4.1, multiple-level omics applications using breast cancer TCGA (combining mRNA expression, DNA methylation and copy number variation) and METABRIC (combining mRNA expression and copy number variation) examples in Section 4.2 and 4.3, and a pathway-guided single transcriptomic application in leukemia in Section 4.4. In the simulation, the underlying sample clusters and intrinsic feature set were known and we demonstrated the better performance of IS-K means compared to iCluster and sparse K-means by cluster accuracy, feature selection and computing time. For the TCGA and METABRIC application, the underlying true clustering and intrinsic feature set were not known. We evaluated the performance by clustering similarity using adjusted Rand index (ARI) [Hubert and Arabie (1985)] with subtype definition by PAM50 [Parker et al. (2009)], cis-regulatory groups, survival difference between clusters and computing time. In the leukemia examples, the disease subtypes were defined by observable fusion gene aberration. We evaluated the performance by clustering accuracy (ARI) and pathway enrichment analysis on selected genes.
4.1. Simulation
4.1.1. Simulation setting
To assess the performance of integrative sparse K-means with different choices of α and compare to the original sparse K -means and iCluster, we simulated K = 3 subtypes characterized by several groups of subtype predictive genes in each of S = 2 omics datasets with 1 ≤ s ≤ S as the omics dataset index (e.g., s = 1 represents gene expression and s = 2 represents DNA methylation). The prior group information was imposed between groups of subtype predictive genes across omics datasets. These prior group information represent the possibility that a group of genes and DNA methylations might be co-regulated. To best preserve the data nature of genomic studies, we also simulated confounding variables, correlated gene structure and noninformative genes. Below is the generative process:
-
Subtype predictive genes (intrinsic feature set).
Denote by Nk is the number of subjects in subtype k (1 ≤ k ≤ 3). We simulate N1 ∼ POI(40), N2 ∼ POI(40), N3 ∼ POI(30) and the number of subjects is N = Σk Nk. Simulate S = 2 omics datasets, which share the samples and subtypes. Specifically, we denote s = 1 to be the gene expression dataset and s = 2 to be the DNA methylation dataset.
Simulate M = 30 feature modules (1 ≤ m ≤ M) for each omics dataset. Denote nsm to be the number of features in omics dataset s and module m. For each module in s = 1, sample n1m = 30 genes. For each module in s = 2, sample n2m = 30 methylations. Therefore, there will be of 1800 subtype predictive features among two omics datasets.
Denote by μskm is the template gene expression (on log scale) of omics dataset s (1 ≤ s ≤ S), subtype k (1 ≤ k ≤ 3) and module m (1 ≤ m ≤ M). Simulate the template gene expression μskm ∼ N(9, 22) with constrain maxp,q |μspm – μsqm| ≥ 1, where p, q denote two subtypes. This part defines the subtype mean intensity for each module in all omics datasets. Note that since in equation (3.4) we used for standardization, performance of the algorithm is robust to gene expression distribution (e.g., the Gaussian assumption here).
In order to tune the signal of the template gene expression, we introduce a relative effect size f > 0, such that . If f = 1, we do not tune the signal. If f < 1, we decrease the signal and if f > 1, we amplify the signal.
Add biological variation to the template gene expression and simulate for each module m, subject i (1 ≤ i ≤ Nk) of subtype k and omics dataset s.
Simulate the covariance matrix Σmks for genes in module m, subtype k and omics dataset s, where 1 ≤ m ≤ M, 1 ≤ k ≤ 3 and 1 ≤ s ≤ S. First simulate , where Φ = 0.5Insm×nsm + 0.5Jnsm×nsm, W−1 denotes the inverse Wishart distribution, I is the identity matrix and J is the matrix with all elements equal 1. Then Σmks is calculated by standardizing such that the diagonal elements are all 1 's.
Simulate gene expression levels of genes in cluster m as , where 1 ≤ i ≤ Nks, 1 ≤ m ≤ M, 1 ≤ k ≤ 3 and 1 ≤ s ≤ S.
-
Noninformative genes.
Simulate 5000 noninformative genes denoted by g (1 ≤ g ≤ 5000) in each omics dataset. First, we generate the mean template gene expression μsg ∼ N(9, 22). Then we add biological variance to generate
-
Confounder impacted genes.
Simulate C = 2 confounding variables. In practice, confounding variables can be gender, race, other demographic factors or disease stage etc. These will add heterogeneity to each study to complicate disease subtype discovery. For each confounding variable c (1 ≤ c ≤ C), we simulate R = 10 modules in each omics dataset. For each of these modules rc (1 ≤ rc ≤ R), sample number of genes nrc = 30. Therefore, totally 600 confounder impacted genes are generated in each omics dataset. This procedure is repeated in all S omics datasets.
For each omics dataset s (1 ≤ s ≤ S) and each confounding variable c, sample the number of confounder subclass hsc = k. The N samples in omics dataset s will be randomly divided into hsc subclasses.
Simulate confounding template gene expression μslrc ∼ N(9, 22) for confounder c, gene module r, subclass l (1 ≤ l ≤ hsc) and omics dataset s. Similar to Step a5, we add biological variation to the confounding template gene expression . Similar to Steps a6 and a7, we simulate gene correlation structure within modules of confounder impacted genes.
-
Gene grouping information.
We assume omics dataset s = 1 and s = 2 have prior group information on subtype predictive gene modules. There are M = 30 modules in each omics dataset.
Suppose subtype predictive genes in the mth module of the first omics dataset are grouped with methylation features in the second omics dataset (totally n1m + n2m = 30 + 30 = 60 features are in the same group). With probability 1 – θ (0 ≤ θ ≤ 1), each feature out of the 60 features will be randomly replaced by a confounder impacted gene or noninformative gene. Note that the same replaced feature can appear in multiple subtype predictive gene groups. We set θ = 1 and 0.2 to reflect 100%, 20% accuracy of prior group information.
4.1.2. Simulation result
For IS-K means, the tuning parameter γ was selected by gap statistics introduced in Section 3.5. Table 1 shows the result of gap statistics to select the best γ in the simulation of α = 0.5, θ = 1. The smallest gap statistics was selected at γ = 0.21 that correspond to selecting 1778 features, which was close to the underlying truth. Similarly, gap statistics result for α = 1, 0.95, 0.05 are in the supplementary materials [Huo and Tseng (2017), Figure S1]. For simulation, we generated two scenarios with relative effect size f = 0.6 and f = 0.8. The complete simulation result of f = 0.6 is shown in Table 1 and the result for f = 0.8 is in the supplementary materials [Huo and Tseng (2017), Table S1]. For iCluster and sparse K-means, we allowed them to choose their own optimum tuning parameters. Note that sparse K-means was adopted to each individual omics datatype. We used ARI [Hubert and Arabie (1985)] and Jaccard index [Jaccard (1901)] to evaluate the clustering and feature selection performance. ARI calculated similarity of the clustering result with the underlying true clustering in simulation (range from −1 to 1 and 1 represents exact same partition compared to the underlying truth). Jaccard index compared the similarity and diversity of two feature sets, defined as the size of the intersection of two feature sets divided by the size of the union of two feature sets (range from 0 to 1 and 1 represent identical feature sets compared to the underlying truth). Clearly, IS-K means outperformed iCluster and individual study sparse K-means in terms of ARI and Jaccard index. IS-K means and sparse K-means outperformed iCluster in terms of computing time. Within IS-K means, we compared feature selection in terms of area under the curve (AUC) of ROC curve, which avoids the issue of tuning parameter selection. When θ = 1 (representing the grouping information is accurate), smaller α (representing larger emphasize on grouping information) yielded better feature selection performance in terms of AUC as expected. However, when θ = 0.2 (representing many errors in the grouping information), smaller α yielded worse performance in terms of AUC. Note that α = 0.5 gives robustness and performs well in the two extremes of θ = 1 and θ = 0.2. In all applications below, we will apply α = 0.5 unless otherwise noted.
Table 1.
Comparison table of simulation with relative effect size f = 0.6. We simulated B = 100 times and calculated mean and standard deviation of each quantity. θ denotes the probability grouping information is correct for each feature inside groups. α is the tuning parameter balancing the emphasis between individual penalty and group penalty. For each method, we allow its own tuning parameter selection method to optimize its performance
| θ | Method | α | ARI | Jaccard index | AUC | # features | Time (mins) |
|---|---|---|---|---|---|---|---|
| 1 | IS-K means | 1 | 0.940 (0.239) | 0.781 (0.202) | 0.943 (0.138) | 1465 | 0.44 |
| 0.95 | 0.940 (0.239) | 0.791 (0.204) | 0.945 (0.136) | 1483 | 0.52 | ||
| 0.5 | 0.940 (0.239) | 0.779 (0.202) | 0.971 (0.084) | 1420 | 0.56 | ||
| 0.05 | 0.940 (0.239) | 0.946 (0.214) | 0.997 (0.012) | 1723 | 0.67 | ||
| 0.2 | IS-K means | 1 | 0.940 (0.239) | 0.781 (0.202) | 0.943 (0.138) | 1465 | 0.44 |
| 0.95 | 0.940 (0.239) | 0.783 (0.202) | 0.943 (0.138) | 1469 | 0.57 | ||
| 0.5 | 0.940 (0.239) | 0.602 (0.159) | 0.943 (0.134) | 1105 | 0.57 | ||
| 0.05 | 0.940 (0.239) | 0.467 (0.096) | 0.888 (0.111) | 2824 | 1.2 | ||
| iCluster | 0.374 (0.323) | 0.383 (0.274) | 1239 | 26 | |||
| Sparse K means 1 | 0.312 (0.370) | 0.105 (0.101) | 896 | 0.12 | |||
| Sparse K means 2 | 0.361 (0.424) | 0.204 (0.124) | 2137 | 0.13 |
4.1.3. Data perturbation
We also evaluated the stability of the algorithm against data perturbation. Instead of Gaussian distribution in the data generative process, we utilized heavy tailed t-distribution to generate the expression. In the simulation setting Step a3, the template gene expression is simulated from a t-distribution with degree of freedom 3, location parameter 9 and scale parameter 2. In Step a4, we set relative effect size f = 0.6 and f = 0.8, respectively. In Step a5, is simulated from a t-distribution with degree of freedom 3, location parameter s and scale parameter . The result for data perturbation is in supplementary materials [Huo and Tseng (2017), Tables S5 and S6]. The resulting message remains almost the same as the conclusion in Section 4.1.2. Therefore, our proposed algorithm is robust against non-Gaussian or heavy tail distributions.
4.2. Integrating TCGA breast cancer mRNA, CNV and methylation
We downloaded TCGA breast cancer (BRCA) multi-level omics datasets from TCGA NIH official website. TCGA BRCA gene expression (IlluminaHiSeq RNAseqV2) was downloaded on 04/03/2015 with 20,531 genes and 1095 subjects. TCGA BRCA DNA methylation (Methylation450) was downloaded on 09/12/2015 with 485,577 probes and 894 subjects. TCGA BRCA copy number variation (BI gis-tic2) was downloaded on 09/12/2015 with 24,776 genes and 1079 subjects. There were 770 subjects with all these three omics data types. Features (probes/genes) with any missing value were removed. For gene expression, we transformed the FPKM value by log2(· + 1), where 1 is a pseudo-count to avoid undefined log2(0), such that the transformed value was on continuous scale. For methylation, the Methylation450 platform provided beta value with range 0 < β < 1, where 0 represents un-methylated and 1 represents methylated. We transformed the beta value to M value, which is defined by a logit transformation . Therefore, methylation characterized by M value is on a continuous scale, similar to mRNA and CNV. If multiple methylation probes matched to the same gene symbol, we selected one methylation probe as a representative, which had the largest average correlation with other methylation probes of the same gene symbol. We ended up with 20,147 methylation probes with unique gene symbols.
We filtered out 50% low expression genes (unexpressed genes) and then 50% low variance genes (noninformative genes). 50% low expression genes are genes with the lowest 50% mean of gene expression across samples and 10,250 genes remained after this filtering step. 50% low variance genes are genes with the lowest 50% variance of gene expression across samples and 5125 genes remained after this filtering step. We obtained 4815 CNV features and 5035 methylation features by matching to the 5125 gene symbols. The features from three different omics datasets that shared the same cis-regulatory annotation (same gene symbol) were grouped together to form 5125 feature groups. In this case, each group had one mRNA gene expression, one CNV gene and/or one methylation probe. Each group contained candidate multi-omics regulatory information because CNV and methylation could potentially regulate mRNA expression. We applied IS-K means with α = 0.5, sparse K-means by directly merging three omics datasets together as well as iCluster. Number of clusters K was set to be 5 since it was well established that breast cancer has 5 subtypes by PAM50 definition [Parker et al. (2009)]. For a fair comparison, we selected the tuning parameter for each method such that number of selected features are close to 2000.
For evaluation purposes, we investigated three categories of groups among selected features: G1, G2 and G3. G3 represents feature groups (gene symbol) where all three types (mRNA, CNV and methylation) of features are selected. Similarly, G2 represents feature groups (gene symbol) where only two types of features are selected; G1 represents feature groups (gene symbol) where only one type of feature is selected. We also compared the clustering result with PAM50 subtype definition in terms of ARI. The result is shown in Table 2. Clearly, IS-K means obtained more G2 and G3 features than sparse K-means and iCluster. This is biologically more interpretable but not surprising since IS-K means incorporated the multi-omics regulatory information and we expected feature of the same group were encouraged to come out together. Besides, IS-K means has higher ARI compared to sparse K-means and iCluster, indicating the clustering result of IS-K means is closer to PAM50 definition than sparse K-means and iCluster. The 5-by-5 confusion table of IS-K means clustering result and PAM50 subtypes is shown in supplementary materials [Huo and Tseng (2017), Table S3]. One should note the the ARI for all these three methods are not very high. This could be because PAM50 was defined by gene expression only and in our scenario we integrated multi-omics information. The heatmaps of IS-K means result is shown in Figure 1(B). In terms of computing time, IS-K means is nearly 20 times faster than iCluster.
Table 2.
Comparison of different methods using TCGA breast cancer (K = 5). G3 represents feature groups (gene symbol) where all three types of features are selected. Similarly, G2 represents feature groups (gene symbol) where only two types of features are selected; G1 represents feature groups (gene symbol) where only one type of feature is selected. We also compared the clustering result with PA M 50 subtype definition in terms of ARI
| Method | ARI | nfeature | G1 | G2 | G3 | Time |
|---|---|---|---|---|---|---|
| ISKmeans | 0.379 | 2066 | 843 | 538 | 49 | 12.1 mins |
| SparseKmeans | 0.332 | 2034 | 1466 | 284 | 0 | 6.85 mins |
| iCluster | 0.272 | 2475 | 1725 | 375 | 0 | 3.91 hours |
4.3. Integrating METABRIC breast cancer mRNA and CNV
We tested the performance of IS-K means in another large breast cancer multi-omics (sample size n = 1981) dataset METABRIC [Curtis et al. (2012)] with mRNA expression (llu-mina HumanHT12v3) and CNV (Affymetrix SNP 6.0 chip) and survival information. The datasets are available at https://www.synapse.org/#Synapse:syn1688369/wiki/27311. There were originally 49,576 probes in gene expression. If multiple probes matched to the same gene symbol, we selected the probe with the largest IQR (interquartile range) to represent the gene. After mapping the probes to gene symbols, we obtained 19,489 mRNA expression features and 18,538 CNV features, which shared 1981 samples. After filtering out 30% low expression mRNA based on mean gene expression across samples and then 30% low variance mRNA based on variance of gene expression across samples, we ended up with 9504 mRNA features. We obtained 8696 CNV feature symbols by matching with mRNA feature symbols. Therefore, we had totally 18,200 features and 9504 feature groups (share the same gene symbol) among 1981 samples.
We applied IS-K means with α = 0.5, sparse K-means by directly merging three omics dataset together as well as iCluster. The number of clusters K was set to be 5 (same reason in TCGA). For a fair comparison, we selected the tuning parameter for each method such that number of selected features are close to 2000. For evaluation purposes, we similarly defined two categories of groups among selected features. G2 represents feature groups (gene symbol) where both types of features are selected and G1 represents feature groups (gene symbol) where only one type of feature is selected. We also compared the clustering result with PAM50 subtype definition in terms of ARI. The result is shown in Table 3. Similar to the TCGA example in Section 4.2, IS-K means obtained more G2 features than sparse K-means and iCluster. The log-rank test of survival difference for the clustering result defined by IS-K means is more significant than sparse K-means and iClus-ter. Furthermore, IS-K means has higher ARI compared to sparse K-means and iCluster, indicating the clustering result of IS-K means is closer to PAM50 definition than sparse K-means and iCluster. The 5-by-5 confusion table of IS-K means clustering result and PAM50 subtypes are in the supplementary materials [Huo and Tseng (2017), Table S4]. In terms of computing time, IS-K means and sparse K-means are much faster than iCluster.
Table 3.
Comparison of different methods using metabric breast cancer (K = 5). G2 represents feature groups (gene symbol) where all two types of features are selected; G1 represents feature groups (gene symbol) where only one type of feature is selected. The clustering result is compared with PA M 50 subtype definition in terms of ARI. Survival p-value obtained from the log rank test is given for the clustering assignment for each method
| Method | ARI | nfeature | G1 | G2 | p-value | Time |
|---|---|---|---|---|---|---|
| ISKmeans | 0.233 | 1882 | 1494 | 194 | 8.29 × 10−17 | 38.4 mins |
| SparseKmeans | 0.22 | 2004 | 2004 | 0 | 3.04 × 10−13 | 34.3 mins |
| iCluster | 0.0572 | 2471 | 2471 | 0 | 0.143 | 11.8 hours |
4.4. Three leukemia transcriptomic datasets using pathway database as prior knowledge
In the simulations and applications so far (Sections 4.1–4.3), we have focused on using the cis-regulatory mechanism as grouping information for integrating multi-level omics data for sample clustering. In this subsection, we present a different but commonly encountered application of pathway-guided clustering in single transcriptomic study. Specifically, we use pathway information from databases to provide prior overlapping group information (i.e., a pathway is a group containing tens to hundreds of genes and two pathways may contain overlapping genes). A transcriptomic study is used for sample clustering with the overlapping group information. We apply IS-K means to three leukemia transcrip-tomic datasets [Verhaak et al. (2009); Balgobind et al. (2010) and Kohlmann et al. (2008)] separately and using three pathway databases (KEGG, BioCarta and Reac-tome) independently, generating nine IS-K means clustering results (see Table 4). The supplementary materials [Huo and Tseng (2017), Table S2] show a summary description of the three leukemia transcriptomic studies.
Table 4. Comparison of different methods by ARI.
| Verhaak | Kohlmann | Balgobind | |||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|||||
| Method | Pathway | # features | ARI | # features | ARI | # features | ARI |
| Biocarta | 1009 | 0.932 | 1000 | 0.948 | 999 | 0.792 | |
| IS-K means | KEGG | 1002 | 0.901 | 1013 | 0.948 | 990 | 0.792 |
| Reactome | 993 | 0.932 | 994 | 0.948 | 1008 | 0.792 | |
| iCluster | 982 | 0.733 | 1233 | 0.504 | 1020 | 0.214 | |
| sparse K-means | 992 | 0.932 | 998 | 0.948 | 1014 | 0.792 | |
We only considered samples from acute myeloid leukemia (AML) with three fusion gene subtypes: inv(16) (inversions in chromosome 16), t(15; 17) (translo-cations between chromosome 15 and 17), t(8;21) (translocations between chromosomes 8 and 21). These three gene-translocation AML subtypes have been well studied with different survival, treatment response and prognosis outcomes. Since the three subtypes are observable under the microscope, we treated these class labels as the underlying truth to evaluate the clustering performance. The expression data for Verhaak, Balgobind ranged from around [3.169, 15.132] while Kohlmann ranged in [0, 1]. All the datasets were downloaded directly from the NCBI GEO website. Originally, there were 54,613 probe sets in each study. For each study, we removed genes with any missing value in it. If multiple microarray probes matched to the same gene symbol, we selected the probe with the largest interquartile range (IQR) to represent the gene. We ended up with 20,154 unique genes in Verhaak and 20,155 unique genes in Balgobind and Kohlmann. We further filtered out 30% low expression genes in each study, which were defined as 30% of genes with the lowest mean expression. We ended up with 14,108 unique genes in each study.
We obtained the three pathway databases (BioCarta, KEGG and Reactome) from MSigDB (http://www.broadinstitute.org/gsea/msigdb/collections.jsp#C2) as the prior group information to guide feature selection in IS-K means. The original pathway sizes were 217, 186 and 674 for BioCarta, KEGG and Reactome. We only kept pathways with size (number of genes inside pathway) greater or equal to 15 and less or equal to 200 after intersecting with 14,108 unique genes. After gene size restriction, we ended up with 114, 160 and 428 pathways for BioCarta, KEGG and Reactome. Note that these pathway groups have large overlaps (i.e., many genes appear in multiple pathways).
For each of the three studies, we applied IS-K means (with BioCarta, KEGG and Reactome as prior group information, respectively), sparse K-means and iCluster. Note that in this example, IS-K means dealt with single omics dataset with prior knowledge. For a fair comparison, we tuned the parameters so that the number of selected features are close to 1000. The result is shown in Table 4. For Verhaak and Kohlmann, IS-K means and sparse K-means almost recovered the underlying true clustering labels (ARI = 0.901–0.932), while iCluster had relatively smaller ARI (ARI = 0.733). We investigated the heatmap of the clustering result of Verhaak using iCluster (supplementary materials [Huo and Tseng (2017), Figure S2]) to understand reasons of its worse performance (lower ARI) and found that its solution converged to a stable clustering configuration with clear clustering separation. Thus, the worse clustering performance in iCluster likely comes from a local optimum solution. For Balgobind, the clustering results from IS-K means and sparse K-means had smaller ARI (ARI = 0.792) but iCluster performed even worse (ARI = 0.214).
To further evaluate functional annotation of the selected intrinsic genes via each method, we explored pathway enrichment analysis (Figure 3) using BioCarta database via Fisher exact test. Five methods [iCluster, IS-K means (BioCarta), IS-K means (KEGG), IS-K means (Reactome), sparse K-means] were compared. The jittered plot of −log10 p-values is shown in Figure 3. IS-K means (BioCarta) show the most significant pathways consistently across three studies; this is somewhat expected since we used the BioCarta pathway as prior knowledge to guide our feature selection. IS-K means (KEGG) and IS-K means (Reactome) also showed more significant pathways than sparse K-means and iCluster, indicating incorporating prior knowledge indeed improved feature selection (in the sense that the selected feature are more biological meaningful). Note that IS-K means (KEGG) and IS-K means (Reactome) did not have an overfitting issue since the test pathway database (BioCarta) was different from the prior knowledge we utilized. Similarly, the results using KEGG and Reactome as a testing pathway are in supplementary materials [Huo and Tseng (2017), Figure S3].
Fig. 3. Pathway enrichment analysis result for Leukemia BioCarta.

5. Conclusion and discussion
Cancer subtype discovery is a critical step for personalized treatment of the disease. In the era of massive omics datasets and biological knowledge, how to effectively integrate omics datasets and/or incorporate existing biological evidence brings new statistical and computational challenges. In this paper, we proposed an integrative sparse K-means (IS-K means) approach for this purpose. The existing biological information is incorporated in the model and the resulting sparse features can be further used to characterize the cancer subtype properties in clinical application.
Our proposed IS-K means has the following advantages. First, integrative analysis increases clustering accuracy, statistical power and explainable regulatory flow between different omics types of data. The existing biological information is taken into account by using the overlapping group lasso. Fully utilizing the inter-omics regulatory information and external biological information will increase the accuracy and interpretation of the cancer subtype findings. Second, we reformulated the complex objective function into a simplified form where weighted K-means and ADMM can be iteratively applied to optimize the convex sub-problems with closed-form solutions. Due to the nature of classification EM algorithm in K-means and closed-form iteration updates of ADMM, implementation of the IS-K means framework is computationally efficient. IS-K means only takes 10-15 minutes for 15,000 omics features and more than 700 subjects on a standard desktop with single computing thread while iCluster takes almost 4 hours. Third, the resulting sparse features from IS-K means have better interpretation than features selected from iCluster.
IS-K means potentially has the following limitations. The existing biological information is prone to errors and can be updated frequently. Incorporating false biological information may dilute information contained in the data and even lead to biased finding. Therefore, we suggest not to over-weigh the overlapping group lasso term and choose α = 0.5 to adjust for the balance between information from existing biological knowledge and information from the omics datasets. The users can, however, tune this parameter depending on the strength of their prior belief of the biological knowledge. Another limitation is that IS-K means can only deal with one cohort with multiple types of omics data. How to effectively combine multiple cohorts with multi-level omics data will be a future work. R package “ISKmeans” incorporates C++ for fast computing and it is publicly available on GitHub https://github.com/Caleb-Huo/IS-Kmeans as well as the authors’ websites. All the data and code presented in this paper are also available on the authors’ websites.
Supplementary Material
Supplement to “Integrative Sparse K-means with overlapping group lasso in genomic applications for disease subtype discovery” (DOI: 10.1214/17-AOAS1033SUPP;.pdf). This supplementary materials contain 3 figures and 6 tables, regarding results for tuning parameter selection, simulation, leukemia dataset description, comparison of IS-K means and PAM50 clustering results on TCGA multi-omics dataset, heatmaps of Verhaak dataset (leukemia), pathway enrichment analysis result for leukemia using KEGG and Reactome as the testing database.
Acknowledgments
The authors appreciate the many insightful critiques from the reviewers and Associate Editor.
Appendix.
A.1. Proof for Theorem 3.1 and Theorem 3.2
Proof of Theorem 3.1. Given equal separation ability for each feature R1 = … = Rj = … = RJ = R and the proposed design of overlapping group lasso penalty, equation (3.4) becomes
First, we can take away the constraint zj ≥ 0, ∀j. It is easy to see that if any zj < 0, we can always use −zj to replace the solution and the objective function will decrease. We can write down the Lagrange function of equation (3.4) after dropping the constraint zj ≥ 0, ∀j:
Partial derivative of the Lagrange is
It is easy to verify that will make . Since the object function is a convex function, according to sufficiency of the KKT condition, the proposed penalty design will lead to the solution of the “unbiased feature selection” principle.
Proof of Theorem 3.2. For the intrinsic gene set ℐ, we have Rj = R > 0 for j ∈ ℐ. For the nonintrinsic gene set ℐ̄, we have Rj = 0 for j ∈ ℐ̄. Given the proposed design of overlapping group lasso penalty, equation (3.4) becomes
First, we can similarly take away the constraint zj ≥ 0, ∀j. We can write down the Lagrange function of equation (3.4) after dropping the constraint zj ≥ 0, ∀j:
The partial derivative of the Lagrange is
It is easy to verify that if for j ∈ ℐ, zj = 1/√J, j ∈ ℐ̄, zj = 0 and is a zero solution to the partial derivative of the Lagrange function. Note here we set the sub-gradient at zj = 0. Since the object function is a convex function, according to sufficiency of KKT condition, the proposed penalty design leads to the “unbiased feature selection” principle.
A.2. Optimization by KKT condition
There are two optimization problems:
It is a convex optimization problem for with no constraint. The stationarity condition states that the sub-gradient of the objective function will be 0 at the optimum solution. Therefore, we have
where S(v) is the sub-gradient of ‖v‖2 and
If we define , it can be derived that where (·)+ = max(0, ·).
The optimization problem for z+ is a convex optimization problem with two constraints. We first write down the Lagrange function and convert the constrained optimization problem into an un-constrained optimization problem:
such that u ∈ ℝ, u ≥ 0, νj ∈ ℝ and νj ≥ 0 ∀j. Taking gradient of the Lagrange function with respect to z and use the constraints, we can derive the solution to this problem. Define and , where βg = (βg1, βg2, …, βgJ)⊤, xg = (xg1, xg2,…, xgJ)⊤, yg = (yg1, yg2, …, ygJ)⊤, and mg = (mg1, mg2, …, mgJ)⊤. The solution is given as following: we define . If Σj fj(u)2 < 1, . Otherwise, and u is selected s.t. ‖z+‖2 = 1.
References
- Abramson VG, Lehmann BD, Ballinger TJ, Pietenpol JA. Subtyping of triple-negative breast cancer: Implications for therapy. Cancer. 2015;121:8–16. doi: 10.1002/cncr.28914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balgobind BV, Van den Heuvel-Eibrink MM, De Menezes RX, Reinhardt D, Hollink IHIM, Arentsen-Peters STJCM, van Wering ER, Kaspers GJL, Cloos J, de Bont ESJM, Cayuela JM, Baruchel A, Meyer C, Marschalek R, Trka J, Stary J, Beverloo HB, Pieters R, Zwaan CM, den Boer ML. Evaluation of gene expression signatures predictive of cytogenetic and molecular subtypes of pediatric acute myeloid leukemia. Haematologica. 2010;96:221–230. doi: 10.3324/haematol.2010.029660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass A, Thorsson V, Shmulevich I, Reynolds S, Miller M, Bernard B, Hinoue T, Laird P, Curtis C, Shen H, et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd S, Parikh N, Chu E, Peleato B, Eckstein J. Distributed optimization and statistical learning via the alternating direction method of multipliers. Found Trends Mach Learn. 2011;3:1–122. [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domany E. Using high-throughput transcriptomic data for prognosis: A critical overview and perspectives. Cancer Res. 2014;74:4612–4621. doi: 10.1158/0008-5472.CAN-13-3338. [DOI] [PubMed] [Google Scholar]
- Dudoit S, Fridlyand J. A prediction-based resampling method for estimating the number of clusters in a dataset. Genome Biol. 2002;3:1–21. doi: 10.1186/gb-2002-3-7-research0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Kurgan L. Comprehensive overview and assessment of computational prediction of microRNA targets in animals. Brief Bioinform. 2015;16:780–794. doi: 10.1093/bib/bbu044. [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- He BS, Yang H, Wang SL. Alternating direction method with self-adaptive penalty parameters for monotone variational inequalities. J Optim Theory Appl. 2000;106:337–356. MR1788928. [Google Scholar]
- Huang J, Horowitz JL, Wei F. Variable selection in nonparametric additive models. Ann Statist. 2010;38:2282–2313. doi: 10.1214/09-AOS781. MR2676890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert L, Arabie P. Comparing partitions. J Classification. 1985;2:193–218. [Google Scholar]
- Huo Z, Tseng G. Supplement to “Integrative sparse K-means with overlapping group lasso in genomic applications for disease subtype discovery”. 2017 doi: 10.1214/17-AOAS1033SUPP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Z, Ding Y, Liu S, Oesterreich S, Tseng G. Meta-analytic framework for sparse K-means to identify disease subtypes in multiple transcriptomic studies. J Amer Statist Assoc. 2016;111:27–42. doi: 10.1080/01621459.2015.1086354. MR3494636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaccard P. Étude comparative de la distribution florale dans une portion des Alpes et des Jura. Bull Soc Vaud Sci Nat. 1901;37:547–579. [Google Scholar]
- Jacob L, Obozinski G, Vert JP. ICML '09: Proceedings of the 26th Annual International Conference on Machine Learning. ACM; New York: 2009. Group lasso with overlap and graph lasso; pp. 433–440. [Google Scholar]
- Kaufman L, Rousseeuw P. Clustering by Means of Medoids. North-Holland; Amsterdam: 1987. [Google Scholar]
- Kaufman L, Rousseeuw PJ. Finding Groups in Data: An Introduction to Cluster Analysis. Wiley; New York: 1990. MR1044997. [Google Scholar]
- Kim EY, Kim SY, Ashlock D, Nam D. MULTI-K: Accurate classification of microarray subtypes using ensemble k-means clustering. BMC Bioinform. 2009;10:260. doi: 10.1186/1471-2105-10-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmann A, Kipps TJ, Rassenti LZ, Downing JR, Shurtleff SA, Mills KI, Gilkes AF, Hofmann WK, Basso G, Dell’Orto MC, Foà R, Chiaretti S, Vos JD, Rauhut S, Papenhausen PR, Hernández JM, Lumbreras E, Yeoh AE, Koay ES, Li R, Liu W, Williams PM, Wieczorek L, Haferlach T. An international standardization programme towards the application of gene expression profiling in routine leukaemia diagnostics: The microarray innovations in LEukemia study prephase. Br J Haematol. 2008;142:802–807. doi: 10.1111/j.1365-2141.2008.07261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock EF, Dunson DB. Bayesian consensus clustering. Bioinformatics. 2013;29:2610–2616. doi: 10.1093/bioinformatics/btt425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen J. Proc Fifth Berkeley Sympos Math Statist and Probability (Berkeley, Calif, 1965/66) Univ California Press; Berkeley, CA: 1967. Some methods for classification and analysis of multivariate observations; pp. 281–297. MR0214227. [Google Scholar]
- Maitra R, Ramler IP. Clustering in the presence of scatter. Biometrics. 2009;65:341–352. doi: 10.1111/j.1541-0420.2008.01064.x. MR2751457. [DOI] [PubMed] [Google Scholar]
- McLachlan GJ, Bean R, Peel D. A mixture model-based approach to the clustering of microarray expression data. Bioinformatics. 2002;18:413–422. doi: 10.1093/bioinformatics/18.3.413. [DOI] [PubMed] [Google Scholar]
- Milligan GW, Cooper MC. An examination of procedures for determining the number of clusters in a data set. Psychometrika. 1985;50:159–179. [Google Scholar]
- Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JCH, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin ZS. Clustering microarray gene expression data using weighted Chinese restaurant process. Bioinformatics. 2006;22:1988–1997. doi: 10.1093/bioinformatics/btl284. [DOI] [PubMed] [Google Scholar]
- Ramasamy A, Mondry A, Holmes CC, Altman DG. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 2008;5:1320–1333. doi: 10.1371/journal.pmed.0050184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson S, Tseng GC, Sun W. Statistical methods in integrative genomics. Annu Rev Statist Appl. 2016;3:181–209. doi: 10.1146/annurev-statistics-041715-033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–1947. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LCG, Lannon WA, Grotzinger C, Del Rio M, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25:2906–2912. doi: 10.1093/bioinformatics/btp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Tseng GC. Meta-analysis for pathway enrichment analysis when combining multiple genomic studies. Bioinformatics. 2010;26:1316–1323. doi: 10.1093/bioinformatics/btq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R. Development and validation of therapeutically relevant multi-gene biomarker classifiers. J Natl Cancer Inst. 2005;97:866–867. doi: 10.1093/jnci/dji168. [DOI] [PubMed] [Google Scholar]
- Simon R, Radmacher MD, Dobbin K, McShane LM. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J Natl Cancer Inst. 2003;95:14–18. doi: 10.1093/jnci/95.1.14. [DOI] [PubMed] [Google Scholar]
- Simon N, Friedman J, Hastie T, Tibshirani R. A sparse-group lasso. J Comput Graph Statist. 2013;22:231–245. MR3173712. [Google Scholar]
- Swift S, Tucker A, Vinciotti V, Martin N, Orengo C, Liu X, Kellam P. Consensus clustering and functional interpretation of gene-expression data. Genome Biol. 2004;5:R94. doi: 10.1186/gb-2004-5-11-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R, Walther G. Cluster validation by prediction strength. J Comput Graph Statist. 2005;14:511–528. MR2170199. [Google Scholar]
- Tibshirani R, Walther G, Hastie T. Estimating the number of clusters in a data set via the gap statistic. J R Stat Soc Ser B Stat Methodol. 2001;63:411–423. MR1841503. [Google Scholar]
- Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro B, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14:5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- Tseng GC. Penalized and weighted K-means for clustering with scattered objects and prior information in high-throughput biological data. Bioinformatics. 2007;23:2247–2255. doi: 10.1093/bioinformatics/btm320. [DOI] [PubMed] [Google Scholar]
- Tseng G, Ghosh D, Feingold E. Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Res. 2012;40:3785–3799. doi: 10.1093/nar/gkr1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GC, Wong WH. Tight clustering: A resampling-based approach for identifying stable and tight patterns in data. Biometrics. 2005;61:10–16. doi: 10.1111/j.0006-341X.2005.031032.x. MR2129196. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Wouters BJ, Erpelinck CA, Abbas S, Beverloo HB, Lugthart S, Löwenberg B, Delwel R, Valk PJ. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009;94:131–134. doi: 10.3324/haematol.13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SL, Liao LZ. Decomposition method with a variable parameter for a class of monotone variational inequality problems. J Optim Theory Appl. 2001;109:415–429. MR1834183. [Google Scholar]
- Witkos TM, Koscianska E, Krzyzosiak WJ. Practical aspects of microRNA target prediction. Curr Mol Med. 2011;11:93–109. doi: 10.2174/156652411794859250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten DM, Tibshirani R. A framework for feature selection in clustering. J Amer Statist Assoc. 2010;105:713–726. doi: 10.1198/jasa.2010.tm09415. MR2724855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B, Pan W, Shen X. Penalized model-based clustering with cluster-specific diagonal covariance matrices and grouped variables. Electron J Stat. 2008;2:168–212. doi: 10.1214/08-EJS194. MR2386092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Lin Y. Model selection and estimation in regression with grouped variables. J R Stat Soc Ser B Stat Methodol. 2006;68:49–67. MR2212574. [Google Scholar]
- Zou H. The adaptive lasso and its oracle properties. J Amer Statist Assoc. 2006;101:1418–1429. MR2279469. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement to “Integrative Sparse K-means with overlapping group lasso in genomic applications for disease subtype discovery” (DOI: 10.1214/17-AOAS1033SUPP;.pdf). This supplementary materials contain 3 figures and 6 tables, regarding results for tuning parameter selection, simulation, leukemia dataset description, comparison of IS-K means and PAM50 clustering results on TCGA multi-omics dataset, heatmaps of Verhaak dataset (leukemia), pathway enrichment analysis result for leukemia using KEGG and Reactome as the testing database.