

Abstract
Cultured primary neurons have been of extraordinary value for the study of neuronal anatomy, cell biology, and physiology. While use of neuronal cell lines has ease and utility, there are often caveats that arise due to their mitotic nature. This methods article presents detailed methodology for the preparation, purification, and culture of adult murine sensory neurons for the study of herpes simplex virus lytic and latent infections. While virology is the application for our laboratory, these cultures also have broad utility for neurobiologists and cell biologists. While these primary cultures have been highly informative, the methodology is challenging to many investigators. Through publication of this highly detailed protocol, it is our hope that the use of this culture system can spread in the field to allow more rapid progress in furthering our understanding of neurotropic virus infection.
Keywords: Primary neuron culture, Neuron purification, Neurotropic virus infection
1 Introduction
Herpes simplex virus 1 (HSV-1) establishes lifelong infection in the neurons of the trigeminal ganglia (TG), where it may remain latent for extended periods of time. Periodic reactivation events result in the formation of cold sores, herpetic keratitis, or herpetic encephalitis [1]. In vivo models of infection can provide insight into viral pathogenesis, and have demonstrated that neuronal innate immune responses are crucial for control of HSV-1 [2]. However, the involvement of epithelial and immune cells confounds the analysis of specific neuronal contributions to this process.
To study neuronal innate immune responses specifically, it is useful to use cultured primary neurons. For the study of HSV-neuron interactions, there has been significant recent focus on the sensory trigeminal ganglion or TG [3–6]. To achieve this, the TG must be dissected, enzymatically digested to liberate the neurons, and the neurons isolated by gradient separation. This yields a mixed neuronal culture that accurately represents the neuronal subpopulations observed in the intact adult TG. To further isolate specific neuronal subpopulations, it is possible to perform secondary enrichment using flow cytometry.
These methods provide several important advantages: (1) use of adult mice ensures the neuronal innate immune responses are fully mature; (2) it is possible to isolate neurons from transgenic mouse strains; (3) long-term culture (up to 3–4 weeks) facilitates long-term experimental procedures. Thus, this model lends itself to a diverse range of inquiries into neuronal innate immune responses. In this chapter, we will describe our detailed methodology for the isolation, purification, and culture of these murine adult sensory neurons.
2 Materials
2.1 Equipment
CO2 for euthanasia.
Biosafety hood (see Note 1).
Rotator at 37 ºC (in an incubator or a water bath).
Centrifuge suitable for 15 ml and 50 ml conical tubes, achieving 800 μg, with the option of slow acceleration and deceleration.
2.2 Plastic Ware, Glassware, and Tools
Coverslips (12 mm, autoclaved).
100 mm plastic petri dish, sterile.
Parafilm.
Large scissors.
Small scissors.
Micro-scissors.
Large forceps.
Fine forceps.
Large curved hemostat.
Small curved hemostat.
10 ml plastic syringe, with luer-lock tip.
20-gauge needle, bevel removed (see Note 13).
Disposable absorbent bench paper.
Raised perforated stage + tray (optional, see Note 19).
Plastic ziptop bags.
Ice + bucket.
6-well plate, or small plastic dishes, sterile.
50 ml and 15 ml screw cap tube with conical base.
0.22 μm filter, ×2.
Glass Pasteur pipettes, sterile.
Rubber bulb for Pasteur pipette.
Plastic Pasteur pipettes, sterile.
Hemocytometer.
24-well plate.
Pipettes and sterile tips (p10, p200, p1000).
2.3 Chemicals and Reagents
Hank’s Buffered Salt Solution (HBSS): 0.137 M NaCl, 5.4 mM KCl, 0.25 mM Na2HPO4, 0.1 g glucose, 0.44 mM KH2PO4, 1.3 mM CaCl2, 1.0 mM MgSO4, 4.2 mM NaHCO3
20 μg/ml Poly-D-lysine (PDL) in HBSS. Prepare 150 μl per coverslip.
180 μg/ml mouse laminin in HBSS. Prepare 150 μl per coverslip.
Sterile distilled H2O.
Mice: 6–10 weeks old.
Phosphate-buffered saline (PBS).
Papain solution: 120 units papain (see Notes 2 and 3), 1 μg L-cysteine, 1 mM NaHCO3, in 3 ml HBSS. The papain will lower the pH of the solution, and the NaHCO3 will raise it. The solution will appear cloudy, but will clarify over several minutes. For best results, prepare 1–2 h before use and incubate at 37 °C for at least 1 h. Filter-sterilize immediately before use.
Collagenase/Dispase solution (C/D): 210 units collagenase type II, 1.1 units neutral protease, in 3 ml HBSS (see Note 4). Prepare 1–2 h before use, store at 4 °C, and then bring up to 37°C for use. Filter-sterilize immediately before use.
Opti-work solution: 0.66 g/ml OptiPrep in 0.8% w/v sodium chloride. Use this to prepare the Optiprep gradient layers.
Optiprep gradient layers: Opti-work diluted in NBA-W to the following densities: 0.3 g/ml (1st layer), 0.23 g/ml (2nd layer), 0.16 g/ml (3rd layer), 0.1 g/ml (4th layer, see Note 5). Each gradient requires 1 ml of each layer.
Neurobasal A Medium (NBA).
Neurobasal-A Work (NBA-W): 2% B27 in NBA.
Neurobasal-A Complete (NBA-C): 2% B27, 1% GlutaMAX, 50 ng/ml Neurturin, 50 ng/ml Neuronal growth factor, 50 ng/ml Glial-derived neurotrophic factor. Make FRESH for each media change.
NBA-FUDR: NBA-C supplemented with 60 μM 5′-fluoro-2′-deoxyuridine (FUDR).
2.4 Additional Materials and Solutions for Sorting by FACS
Collagenase/Dispase solution (C/D): 420 units collagenase type II, 1.1 units neutral protease, in 3 ml HBSS. Prepare 1–2 h before use, store at 4 °C, then bring up to 37 °C for use. Filter-sterilize immediately before use. This solution replaces the lower dose C/D solution listed in Subheading 2.3.
40 μm cell strainer.
Percoll gradient layers: 28% and 12%, in NBA-W. Prepare 4 ml of each gradient layer per gradient.
Phenol Red-free NBA.
Fluorescent stains for appropriate cell surface markers, or else endogenously expressed fluorescent markers.
Flow cytometry device capable of sorting live cells under sterile conditions, fitted with a 100 mm nozzle and a 2.0 neutral density filter.
2.5 Additional Materials and Solutions for Microfluidics
150 μm microfluidic devices—sonicate them for 10 min in distilled water in a bath sonicator, then submerge them in 70% ethanol for at least 30 min, and then air dry.
Glass coverslips, 24 × 50 mm (1 per microfluidic device)—submerge the coverslips in 70% ethanol for at least 30 min, and then allow them to partially air dry.
100 mm dish, lined with parafilm.
Basal medium (any type) supplemented with 10% FBS.
500 μg/ml PDL in HBSS.
10 μg/ml laminin in HBSS.
3 Methods
This protocol takes approximately 4 h if performed by an experienced scientist (Fig. 1). It includes the following general stages: preparing the reagents and workstation, mouse euthanasia, mouse dissection, transcardial perfusion, TG dissection, enzymatic digestion, counting and plating the neurons (see Note 1). A video demonstration of the perfusion process can be found in the Journal of Visual Experiments [7]; the neuron isolation process is based on descriptions by Malin and Bertke [3, 4].
Fig. 1.

Steps in the neuron isolation process. Coverslips should be coated with PDL and Laminin the day before neuron isolation. This will take a few minutes, followed by >3 h and overnight incubation, respectively. Neuron isolation should take a total of ~4 h. This will include ~1 h of setup, <1 h to isolate the TGs, <1 h of enzymatic digestions (20 min each, with some additional time for spin down), and ~1 h to separate, wash, count, and seed the neurons. This is followed by >1 h incubation, and then 3–4 days culturing, at which point the neuron cultures are ready for use in experiments
3.1 Prepare Coated Coverslips
Start preparing 1–2 days before neuron isolation.
Prepare a 100 mm sterile petri dish lined with parafilm (ethanol wiped, air dried).
Take 12 mm coverslips (autoclaved) and arrange them on the parafilm, with at least 2–3 mm left between slips. This setup encourages liquids to form a bubble on the coverslips, rather than spilling off them.
Apply 150 μl PDL per coverslip. Close the dish and incubate at 37°C for at least 3 h, or as long as overnight (see Note 6).
Remove the PDL, and wash the coverslips three times with sterile distilled H2O. It is convenient to use the vacuum to remove the liquids during this process. However, be sure to use a sterile tip or Pasteur pipette, and make sure there are no residual liquids in the vacuum tube, which might drip onto the coverslips.
Allow the coverslips to air-dry in the hood.
Apply 150 μl laminin per coverslip. Close the dish and incubate at 37 °C overnight.
The next day, wash the coverslips once in NBA-W. They are now ready for use.
Do not allow laminin-coated coverslips to dry, as laminin will crystalize.
Coated coverslips should be used the day after laminin is applied, and never store for later use.
3.2 Prepare Enzymes and NBA-W
3.3 Prepare Your Workstation
Prepare within 1 h of the time you expect to start isolating TGs. For 10 mice, prepare 3 ml of papain, 3 ml of C/D, and 50 ml of NBA-W (see Notes 2–4). Use some NBA-W to prepare your workstation. Incubate the remainder at 37 °C until needed.
It is convenient to have three work areas: a mouse euthanasia area, a perfusion area, and a dissection area. The euthanasia area may be in or out of the hood, as space permits. The perfusion and dissection areas must be in the hood (see Note 1). If hood-space is limited, it is possible to perform perfusion and dissection in the same area.
Mouse euthanasia area: It is most convenient to have the mouse euthanasia area in the hood; however, it can be out of the hood if space is limited. Mice should be euthanized one at a time, never in batches (see Note 9). Therefore, use an empty mouse cage to expose individual mice to CO2. Make sure that this station is in compliance with your institution’s animal welfare protocols.
Perfusion area: lay out disposable absorbent bench paper (see Note 19), and the following instruments: small scissors, large hemostat, small hemostat, large forceps, large scissors, ziptop bag. Take a 10 ml syringe and attach a 20-gauge needle to it. Use the large hemostat to grasp the beveled edge of the needle, then bend it gently back and forth until it breaks off—you now have a blunted needle (see Note 13). Make sure to deposit the broken-off beveled edge in the sharps waste. Fill the syringe with ice-cold sterile PBS, and set it next to the surgery tools.
Dissection area: lay out disposable absorbent bench paper and the following instruments: small scissors, micro-scissors, fine forceps, small ice bucket with ice, small petri dish or 6-well plate with 2–3 ml NBA-W (on ice, for isolated TGs, see Notes 7 and 8), 50 ml conical tube with PBS (on ice, for re-fills of the syringe), a larger bottle with PBS (on ice, for re-fills of the 50 ml conical tube).
3.4 Mouse Euthanasia and Dissection
Expose one mouse to CO2 according to your institution’s animal welfare protocols (see Note 9). When agonal gasping has ceased squeeze the footpad to confirm the mouse is unconscious. DO NOT perform a secondary method of euthanasia, such as cervical dislocation. Transfer the mouse to the dissection area, and proceed to the next step.
Pinch the abdominal skin above the diaphragm, and use the small scissors to make a small incision in the abdominal skin. Pinch the skin above and below the cut and pull firmly toward the head and tail, respectively. This will widen the cut, exposing the abdominal muscles and the lower portion of the ribcage. Pull until the cut has reached the back of the mouse, so you have unobstructed access to the diaphragm (Fig. 2a).
Use the large forceps to pinch the abdominal muscles directly below the ribcage and cut to left and right, along the lowest rib (Fig. 2a).
Use the large forceps to grasp the xiphoid cartilage (at the bottom of the rib cage), and pull slightly up. This will cause the viscera to fall away from the ribcage, exposing the diaphragm.
Using the small scissors, puncture the diaphragm at one side of the mouse, and cut along the ribcage to the other side (Fig. 2b). Take great care not to accidentally puncture or cut the lungs and heart, since this would compromise the perfusion process.
Using the small scissors cut the rib cage at left and right, from the lowest rib toward the head (Fig. 2b). The front of the rib cage is now attached to the carcass only at the top.
Using the large hemostat, grasp the xiphoid cartilage, lock the hemostat, and fold toward the head. The heart and lungs are now fully exposed (Fig. 2c). The heart may be encased in a thin layer of connective tissue and fat. This is normal. Use the large forceps to gently peel away the fat, fully exposing the ventricles and atria.
Fig. 2.

Transcardial perfusion. Lay the mouse on its back, with the tail toward you, and the head away from you. (a) Use small scissors to cut the skin and muscle right below the ribcage. (b) Cut the diaphragm and ribcage, then (c) fold the ribcage up and away, exposing the heart. Use large forceps to grasp the heart while inserting the needle into the left ventricle, taking care not to punch clear through (d–f). Use a small hemostat (locked) to secure the needle in the heart, and then cut a hole in the right atrium (d). Begin perfusing with PBS
3.5 Transcardial Perfusion (See Notes 10–12)
Use the forceps to grasp the heart in the middle, with the atria left free at the top, and the apex of the ventricles extending 2–3 mm out at the bottom (Fig. 2d). The grip should be firm enough to hold the heart while you insert the needle, but not so firm as to damage the tissue or block the needle.
While holding the heart in place, position the blunted needle tip slightly to the right of the apex, so as to enter the left ventricle (Fig. 2d). Push the needle slowly and firmly into the heart (see Notes 13 and 14). You will encounter some initial resistance because the needle is blunted. However, once you punch through the muscle wall and into the ventricle the needle should slide in smoothly. Insert ~5–7 mm of the needle, depending on the size of the heart. Take care to insert the needle toward the aorta, without swiveling it to left and right, or pitching it up or down (Fig. 2e, f).
Keep the needle and syringe steady as you set aside the large forceps, and pick up the small hemostat. Use the hemostat to clamp the heart onto the needle (see Notes 15 and 16). Lock the hemostat, and gently put it down.
Use the small scissors to cut a hole in the right atrium (Fig. 2d). A little blood may flow out.
Slowly press the plunger on the syringe (see Note 17). This will push PBS into the mouse vasculature, pushing blood out of the organs and perfusing them with PBS. Blood, and eventually PBS, will flow out of the right atrium. The liver and paws may turn pale, the lungs may inflate and turn white, and PBS may drip out of the nose and mouth (see Notes 18 and 19). Perfuse the mouse with 5–10 ml PBS over the course of ~60 s.
Once the mouse is perfused, unlock the hemostat and remove the needle.
Use the large scissors to remove the head from the carcass (see Note 20). Discard the carcass (into the ziptop bag), and transfer the head to the dissection area.
3.6 TG Dissection
Using the small scissors, cut the skin on the top of the head, from the base of the spine to between the eyes. Fold the skin flaps under the chin of the mouse and use them to hold the head securely while you work.
The brain should be visible through the thin layer of bone at the top of the skull. Cut the skull along the left and right, with the cuts meeting a point between the eyes. The skull top can now be flipped up and away from you, exposing the brain (Fig. 3a).
Insert the large or small forceps between the frontal lobe and the skull, and flip the brain back toward you, exposing the base of the skull and the TGs (Fig. 3b, c). The optic chiasm will be exposed and ripped during this process. The pituitary gland also rests on the base of the skull, perpendicular to the TGs. Discard it by scraping the gland down and away from the TGs.
The TGs are laid on the top of the skull, to left and right. Each TG has two branches threaded through holes in the base of the skull and extending toward the nose and eyes (see Note 21). Snip the branches at the point where they thread into the skull. Slide the micro-scissors under and along the TGs to cut the connective tissue which anchors the TG to the skull (Fig. 3c).
Use the fine forceps to gently lift the TGs out and place them in the ice cold NBA-W (see Note 22).
Fig. 3.
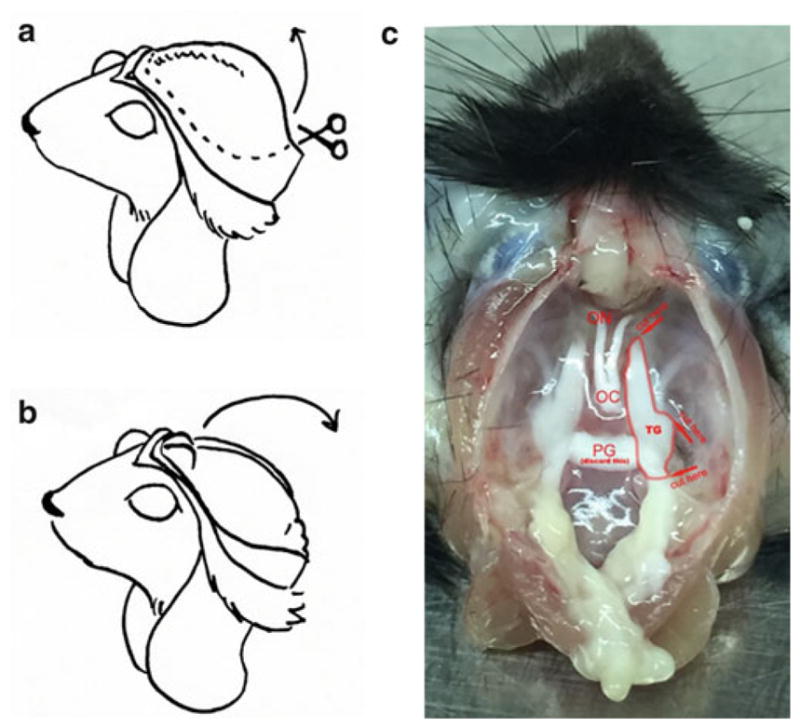
TG isolation. After separating the head from the carcass, cut the skin from neck to between the eyes, and fold the skin flaps under the mouse’s chin (a). Cut the exposed skull along left and right, to between the eyes, then lift the bone up and away (a). Pull the brain out toward you (b), exposing the base of the skull (c). TG dissection: Picture shows a mouse head after brain removal. Trigeminal ganglion is double lined. Arrows indicate cutting sites and the orientation of the cutting. Abbreviations: TG trigeminal ganglion, ON optic nerve, OC optic chiasm, PG pituitary gland. The optic nerves and chiasm should be visible at the top, the TGs along the base to either side, and the pituitary gland along the base between the TGs. Cut the connective tissue anchoring the TGs to the skull, and sever the nerve where it enters the skull—you should now be able to lift out the TGs (C)
3.7 Enzymatic Digestion
Filter-sterilize the papain through a 0.22 μm filter.
Collect the TGs and papain in a 50 ml conical tube, making sure that all the TGs are in the enzyme solution.
Place the 50 ml conical tube, slightly tilted, on a rotator set to ~200 rpm (the gentle agitation improves the enzymatic digestion), and incubate at 37 °C for 20 min.
During this time, warm the C/D solution to 37 °C ten minutes before use.
At the end of the incubation spin down the TGs (200 g, 1 min), and remove the supernatant (see Note 23).
Filter-sterilize the C/D directly onto the TG pellet, and incubate as before (tilted, ~200 rpm, 37 °C, 20 min).
During this time, prepare the gradient (see Subheading 3.8 and Notes 10 and 26).
At the end of the incubation, spin down the TGs (400 g, 4 min), and remove the supernatant.
Add 1 ml of NBA-W to the TGs and triturate gently through a p-1000 pipette tip (~10 times). The TGs should dissociate fully, resulting in a cloudy suspension of cells (see Notes 24 and 25).
Add NBA-W for a final volume of 3 ml per gradient (see Subheading 3.8).
3.8 Gradient Separation
Each gradient can be loaded with up to 10 dissociated TGs (isolated from 5 mice). The volume loaded onto each gradient should always be 3 ml. Therefore, if you have isolated more than 10 TGs, prepare the appropriate number of gradients (up to 4, see Note 10) and bring the volume of tissue homogenate to 6, 9, or 12 ml (3 ml per gradient).
For each gradient you intend to layer, prepare the 4 Optiprep dilutions as described in Subheading 2.3 (see Note 5). Layer the gradient in a 15 ml conical tube (see Notes 26–28), with the densest (1st) layer at the bottom, and the least dense (4th) layer at the top. Handle carefully so as not to disturb the gradient.
Layer 3 ml of cell suspension (containing up to 10 dissociated TGs) on the top of the gradient.
Load the gradient into the centrifuge (see Note 28), and spin the gradient at 800 × g for 20 min, with a slow acceleration and a slow deceleration.
During this time, wash the coverslips with NBA, if not already washed.
Once the gradient spin is complete, remove the 15 ml conical tubes to the hood. As before, handle carefully so as not to disturb the gradient.
Inspect the gradient (Fig. 4). The NBA-W that contained the dissociated TGs should be clear. The lightest (uppermost) layer of the gradient may contain some debris, and there should be a thick band of debris at the junction between this layer and the next. Further down, there should be two distinct bands at the junctures between the gradient layers. Both the bands contain neurons (small neurons and glial cells in the upper band, large neurons in the lower). The bottom layer of the gradient should be clear, with a small pellet at the bottom (see Note 29).
If desired, you may remove some of the NBA-W and some of the first band, though this is not necessary.
Use a p200 or p1000 pipette to collect the lower two bands, as well as the second gradient layer, and most of the third. Avoid collecting the thick band of debris in the third band. Collect this liquid (~2 ml in total) into a new 15 ml conical tube. It is sometimes useful to move the tip gently in circular motions while collecting, so as to collect cells from all the areas of the band.
Wash the OptiPrep away: Add 5 ml of NBA-W, then spin at 670 g for 4 min to pellet. Discard the supernatant, resuspend in 1 ml of NBA-W, then add 5–10 ml NBA-W, spin at 400 g for 2 min, and discard the supernatant.
Resuspend the neurons in NBA-W (100 μl per mouse). At this point, it is possible to combine the output of several gradients, if appropriate.
Fig. 4.
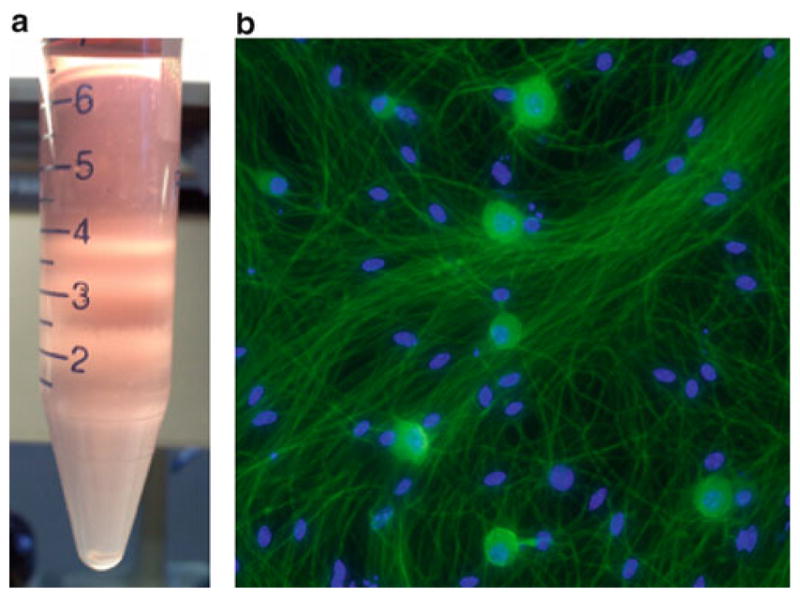
Gradient separation and culture. (a) After the gradient has been spun, there should be four visibly distinct cell suspension layers and a pellet. Collect the liquid and two cell suspension layers from just above the 2.5 ml line to just below the 1 ml line, a total of ~2 ml. Discard the remainder. (b) Fluorescence microscopy of cultured TG neurons for 4 days stained with β-3 tubulin (green) and Dapi (blue)
3.9 Seeding and Culturing Neurons
Count the neurons using a hemocytometer according to the manufacturer’s instructions (see Note 30). Neurons will appear phase-bright and quite large, and their size will also be apparent over a range of focal distances. The sample will also contain debris and supporting cells (which will appear much smaller and flatter than the neurons).
The expected yield is ~15,000–20,000 neurons per mouse (or 4–6 coverslips per mouse). This may vary between mouse strains (e.g., C57B/6 J yields are slightly lower than 129SVEV yields) (see Notes 31–33).
Add NBA-W to dilute the neuron suspension to a concentration of 60 neurons per μl.
Aspirate the NBA from the coverslips, and seed 60 μl per coverslip (a total of 3600 neurons per coverslip, seeded at an approximate density of 32 cells per mm2).
Incubate the coverslips at 37 °C for 1–2 h. During this time the neurons settle and attach. A pad of debris will settle on the top of the cells, and will be visible to the naked eye. This debris will eventually slough off as the cultures are fed. Do not try to tap, scrape, or aspirate the pad off—this is likely to result in significant neuronal loss.
During this time prepare NBA-C with FUDR (400 μl per coverslip) and warm it to 37 °C.
If desired, gently aspirate the seeding media from the coverslips (some of the debris pad might also come off—this is ok).
Using fine forceps, transfer the coverslips to a 24-well plate, and add 400 μl NBA-FUDR to each well (see Note 34). The FUDR will inhibit glial cell division and will prevent them from overrunning the culture.
Incubate at 37 °C for 3–4 days; then replace media with NBA-C (no FUDR). The neurons are now ready for experimentation.
For continued culturing, replace neuron media every 3–4 days with 400 μl freshly prepared NBA-C. Appearance of cultures should resemble Fig. 4b.
3.10 Culturing Neurons (See Notes 35–38)
Once seeded, the neurons will rapidly grow neurites (Fig. 4b). These will be visible within ~4–8 h, and will form a dense mat at the coverslip surface within ~24 h. The neurites often form whorls, and any scratches in the PDL + laminin coating will be evident in the pattern of neurite growth. The pattern of neurite growth will also appear different on precoated coverslips (which can be purchased).
The neurons will appear to be unevenly seeded, with very sparse seeding toward the edges of the coverslip. Nonetheless, over most of the coverslip area neurons should be evenly distributed. It is normal to observe 2–6 neurons which have settled together. If the coverslip is tapped or jostled before the neurons have settle the neurons may become concentrated, almost confluent, in some areas and very sparse in others.
The neurons and glial cells are not stationary on the coverslips, and some migration may be observed over the course of several days, particularly if live imaging is performed.
The neurons do form synapses and signal to each other in culture. This may affect several biological processes, including Ca2+ dynamics and expression of MHC-II.
Cultures should not be kept out of the incubator for extended periods of time, since exposure to prolonged cold, changes in pH, or changes in O2/CO2 tension are harmful to the neurons.
Neurons may be cultured for as long as 3–4 weeks ex-vivo. During this time the neurons will undergo a process of aging, which will result in some loss of viability, although this should not be extensive.
The cultures are mixed, and include multiple neuronal subtypes, as well as supporting satellite glial cells. The glial cells are found in the upper of the two neuronal bands, where many of the smaller neurons are also found. It is therefore not possible to produce a purely neuronal culture unless further purification methods are used (see Subheading 3.12). The FUDR in the media will prevent any initial expansion of this population. After 3–4 days in vitro, the glial cells lose most of their replicative license and some will apoptose. By 3 weeks in vitro there should be significantly fewer glial cells.
3.11 Neuronal Sorting by Flow Cytometry
If desired, newly isolated neurons can be sorted into subpopulations by flow cytometry. To identify neuronal subpopulations, use dyes or antibodies to fluorescently label the appropriate cell surface markers. Alternately, isolate neurons from transgenic mice expressing fluorescently tagged markers. We describe here a modification of the main protocol that facilitates sorting of non-peptidergic neurons, a subpopulation of sensory neurons [8].
A major concern for neuron sorting is the risk of clogging the FACS with tissue debris or large neurons. To avoid this, we have introduced several modifications of the protocol, listed below.
For enzymatic digestion (Subheading 3.8) use 420 units of collagenase II, instead of the usual 210 units.
Immediately after dissociating the tissue and resuspending in 3 ml of NBA-W (Subheading 3.8), pass the suspension through a 40 μm cell strainer. This will remove tissue fragments and the larger neurons. Non-peptidergic neurons have a smaller diameter and will pass through the strainer.
Replace the Optiprep gradient (Subheading 3.9) with a percoll gradient. Layer the percoll gradient in the following order: 4 ml 28% percoll, 4 ml 12% percoll, 3 ml dissociated, strained TG homogenate. This removes unwanted pieces of non-dissociated tissue.
Spin the gradient at 1300 g for 10 min. The neurons will drop into a pellet. To recover the neurons, discard all the layers without disturbing the pellet. Proceed to wash as described above (Subheading 3.4).
After the final wash, resuspend the neurons in Phenol Red-free NBA-C. Phenol Red can interfere with the excitation-emission fluorescence and affect the sorting process.
If using endogenously labeled neurons, proceed directly to step 9. If labeling cell surface markers, incubate the neurons with the appropriate fluorescent dye or antibody. We incubate the neurons for 5 min with fluorescently labeled Isolectin Banderifolia B4 (IB-4), which binds to non-peptidergic neurons. We do not wash the neurons after this step, since the IB-4 signal on neurons is easily detectable above the background.
As soon as possible, sort the neurons at your flow cytometry facility.
The FACS instrument must run under sterile conditions to avoid contaminating the neuron cultures. To avoid clogging, we used a 100 mm nozzle. Finally, we installed a 2.0 neutral density filter. Gate the neurons using logarithmic scale for the FSC and SSC.
3.12 Seeding Neurons in Microfluidics Devices
Compartmentalized cultures, such as Campenot chambers [2, 9] and microfluidic devices ([10], and described herein), separate neuronal cell bodies and proximal neurites from distal projections. This provides an in vitro model of in vivo neuronal architecture, and facilitates specific treatment of the distal projections.
In microfluidic devices, positive pressure from the cell body compartment to the axonal compartment prevents the diffusion of large molecules (>1 kDa) in the opposite direction (e.g., DiI applied to the distal projections, see Fig. 5d). It also encourages neurite growth through the capillaries. Positive pressure is achieved by filling the compartments with unequal volumes of media (see Subheading 3.12, steps 19 and 21).
Assembly and coating of the microfluidic devices involves 3 overnight incubations; therefore, you should begin this process 3 days before you intend to isolate the neurons.
Transfer one partially dried coverslip into a parafilm-lined dish, and allow it to air dry completely. This will prevent the coverslip from adhering too firmly to the parafilm.
Identify the side of the microfluidic device lithographed with channels and microgrooves (see Note 39).
Mount the device onto the coverslip, with the lithographed side facing down. The device will attach immediately. Use forceps to gently press the device to the coverslip, paying special attention to the microfluidic borders. This will ensure the device is properly attached. If correctly mounted, the microgrooves will act as capillaries.
Let it settle for 15 min, then remove the parafilm strip.
Fill compartment 1 with 200 μl of 10% FBS medium, and wait for the central channel to be filled completely (Fig. 5a, see Note 40).
Fill compartment 2 with 200 μl of 10% FBS medium.
Cover the dishes and place in an incubator overnight, allowing the capillaries to slowly fill. On the next day check that capillaries have been filled with medium.
Fill compartment 3 with 200 μl of 10% FBS medium and wait for the central channel to fill.
Fill compartment 4 with 200 μl of 10% FBS medium. Wait for 15 min. The devices are now ready to be coated.
Remove the media and fill each compartment with 200 μl of 500 μg/ml PDL in HBSS, then place in an incubator overnight, allowing the capillaries to slowly fill.
On the next day wash the compartments three times with HBSS.
Apply 200 μl of 10 μg/ml laminin in HBSS to each of the compartments, and then place in an incubator overnight, allowing the capillaries to slowly fill. The devices are now ready for neuron seeding.
Prepare neurons according to the usual protocol described above. After the last wash, resuspend neurons in NBA-C at a concentration of 105 neurons/6 μl (this typically entails the use of ~1 mouse per device).
Remove Laminin from all compartments, and seed 3 μl of cell suspension in compartment 1 as close as possible to the central channel (Fig. 5a). Tilt the plate to a 45° angle, and wait for 10 min (Fig. 5a). This will allow the neurons to flow down the central channel and settle along the entrance to the capillaries.
Seed 3 μl in compartment 2, as close as possible to the central channel, tilt the plate at a 45° angle in the opposite orientation, and wait for 10 min.
Fill compartment 1 and 2 with 200 μl NBA-C + FRDU (each), and compartment 3 and 4 with 100 μl NBA-C (each).
Incubate the devices for 3 days. Neurite growth through the capillaries should be evident on the first day, and should be robust by 3 days, at which point the devices are ready for use in experiments.
When performing experiments, make sure to fill chambers 3 and 4 with 100 μl of media each, and chambers 1 and 2 with 200 μl each.
Fig. 5.

Use of microfluidic chambers to culture TG neurons. (a) Schematic representation of a microfluidic device with compartments enumerated to follow the protocol above. The picture (right) shows a microfluidic device filled with NBA complete medium (all compartments) and NBA complete medium plus the cell tracker DiI (compartments 3 and 4). (b) Representative image of trigeminal neurons cultured for 3 days in a microfluidic device. Neurons were stained with β-3 tubulin (blue). Neuron cell bodies are present only in the upper compartment, with axons projecting through the capillaries to the lower compartment. (c) The same culture as described in b. The cell tracker DiI (orange) was added to the lower compartment. This treatment stains only those neurons that have grown axonal projections through the capillaries
Acknowledgments
Development of the protocols described in this article was supported by PO1 AI098681, and by a pilot grant to JR Cabrera from the Hitchcock Foundation.
Footnotes
Primary neuronal cultures are susceptible to contamination. Work should therefore be carried out in a biosafety hood whenever possible, and particularly when handling neurons. All the reagents and tools should be sterile at the time of use. The tools should be received sterile or autoclaved or wiped with 70% ethanol. The reagents should be received sterile or autoclaved or filter sterilized. Some reagents are unavoidably exposed to non-sterile conditions (e.g., powdered reagents being weighed), and these should be filter sterilized before use.
Most of the solutions needed should be prepared fresh. However, the following stock solutions can be prepared and stored at 4 °C: 0.66 g/ml OptiPrep in 0.8% w/v sodium chloride (Opti-work), 50 μg/ml L-Cystein in HBSS, 9% (w/v) NaHCO3 in double-distilled water, 40 mM FUDR in HBSS. Stock solutions of PDL, Laminin, B27, Neurturin, NGF, and GDNF should be stored at −20 °C. It is possible to prepare the C/D enzyme solution ahead of time and freeze it; however, we have observed that freeze-thawed C/D sometimes do not digest the TG tissue as well as fresh C/D.
The potency of papain varies between batches; therefore, calculate the volume required to achieve 120 units of activity on a per-batch basis. Using Worthington papain, this is usually ~90–120 μl.
The powdered enzymes are very light, and susceptible to static electricity. This can be partially ameliorated by wiping the 50 ml conical tube with a wet paper towel.
An easy guide to preparing the OptiPrep gradient layers: combine Opti-Wand NBA-W in the following ratios: 450 μl +550 μl (1st layer), 350 μl + 650 μl (2nd layer), 250 μl + 750 μl (3rd layer), 150 μl + 850 μl (4th layer).
Handling coverslips: Once you have started preparing the coverslips, they should be handled using fine forceps. Touching the coated side with forceps or a pipette tip will scrape off the PDL/Laminin coating, or else remove cells and damage neurites. Therefore, grasp the coverslip toward the edge and avoid touching the central section as much as possible.
Stack the ice and place the petri dish or 6-well at an angle toward you, so the NBA-W pools and the TGs are easier to deposit.
To avoid confusion when isolating TGs from multiple mouse strains, it is helpful to use a 6-well plate and write the mouse strains on the underside of the wells.
Streamlined process: It is possible to streamline the TG extraction process if two people are available and the hood is large enough. While one person dissects the TGs from the first mouse, the other perfuses the second mouse, and the third mouse is euthanized. In this way, two experienced scientists can isolate the TGs at a rate of ~3 min per mouse. Do not euthanize mice in batches, since neuronal loss increases the longer TGs remain in the euthanized mouse.
Maximal work load: The maximum number of mice per batch is 20. This number is due to two constraints: (1) At ~3 min per mouse, only 20 mice can be processed in <1 h (neuronal loss increases the longer TGs are stored); (2) At ~5 min per gradient, only four gradients can be layered in 20 min (during the C/D incubation). Each gradient separates neurons from up to five mice. An additional constraint is imposed if isolating neurons of different genotypes—these must be isolated on separate gradients.
Practice: many of the steps described require practice. In our experience transcardial perfusion and gradient separation are the most likely to require practice before mastery.
Perfusion is performed in order to remove as much blood as possible from the TGs. This makes dissection easier and lowers neuronal loss due to red blood cell induced toxicity.
Using a beveled tip needle often results in accidentally pushing the tip clear through the mouse’s heart. Moreover, the mouse heart is so small that a beveled edge might not fit completely within the left ventricle (especially if using younger, female mice). The blunted needle has a much shorter tip, and is harder to push through the heart tissue. Thus, it is less likely to pierce clear through the heart.
If you are unable to insert the needle, it may be because the heart is not held firmly enough (and is therefore slipping out of the forceps) or is held too firmly (the forceps are compressing the tissue, making it harder to insert the needle). Adjust the forceps’ grasp of the heart.
If the needle slips out of the heart before you have secured it with a hemostat, gently grasp the heart in the forceps, find the previously made puncture, and gently reinsert the needle.
Take care not to pull too much on the heart and hemostat, as this may damage the tissue or dislodge the needle.
If you are not able to depress the plunger of the syringe, it is probable that the needle is blocked by the surrounding tissue. Without releasing the hemostat lock, pull the syringe slightly toward you. If the needle is still blocked, release the hemostat lock, move the needle inside the heart to reposition, and relock the hemostat. If the needle is still blocked, a tissue fragment may have become lodged in the needle tip, blocking it (this is rare). Discard the blocked needle and install a new, blunted needle.
If there are signs of poor perfusion (liver and paws do not clear, lungs very inflated and white), the rate of perfusion may be too high. Lessen the rate of perfusion. Otherwise, continue—a poorly perfused mouse will still yield some neurons.
During perfusion blood and PBS will spill out of the right atrium. The liquids will be absorbed by the absorbent bench pad. However, if >3 mice are to be perfused the pad may become saturated, making the workstation messy and inconvenient. To avoid this, we have made use of a raised perforated stage, placed above a pad-lined tray. The liquids drip through the perforations and are absorbed by the pad, leaving the work surface mostly clear. It is convenient, though not necessary, to keep some paper towels at hand to periodically wipe this work surface.
It is easier to cut through the spine if you hold the mouse carcass up by the ears, so the shoulders hang down from the neck.
The TG-CNS junction is located toward the back of the skull, near the brainstem. Sometimes, a little CNS matter remains attached here after the brain has been removed, identifiable as a yellowish extension of the TG. It is possible, though not necessary, to remove this CNS tissue.
If you have difficulty in getting the TGs out of the skull, the connective tissue or the TG branches into the skull may not have been fully cut. Use the micro-scissors to cut around the TGs and fully sever the nerve branches.
After the papain digestion, the pellet is not stable, and can be easily disturbed. It is ok to leave a small volume of the supernatant (~100–200 μl) above the pellet.
If the TGs do not fully dissociate on trituration this indicates poor enzymatic digestion. The enzyme was not fully active (too few units, no L-cystein, too cold, not incubated at 37 °C for long enough), or was not properly agitated during incubation. It is not possible to correct this problem at this stage. Avoid forceful or prolonged trituration, as this will not significantly increase the yield, and may be toxic to neurons.
Avoid generating bubbles or foam or triturating forcefully, as this is toxic to the neurons and will reduce your yield.
How to layer a gradient: The key to layering a gradient is to allow a thin stream of the lighter liquid to flow slowly down the conical wall. When it reaches the denser liquid at the bottom it will form a new layer, rather than mix. Pipette the densest (1st) layer into the conical. Use a Pasteur pipette and bulb to mix and collect the 2nd layer (avoid generating bubbles). Hold the conical at a ~ 45° angle, use the tip of the Pasteur pipette to draw a line of liquid up from the 1st layer, and slowly squeeze out the 2nd layer. The 2nd layer should slide down the line of liquid and form a new layer on the top of the 1st, without mixing. Once layered, there should be a visible diffraction line between the two layers. It should take an experienced scientist ~1 min per layer, or ~5 min per gradient. Hence, it is not advisable to attempt >4 gradients during the C/D incubation time. Once layered, the gradient is stable for up to an hour, but for optimal results it should be prepared immediately before use.
If there is no visible diffraction at the expected boundary between the layers, the gradient may have been poorly layered. This might happen if: (1) a layer is applied directly at the surface of the previous layer; (2) the liquid is allowed to slide down a dry surface (it will bead and roll rapidly down the surface); (3) the stream of lighter liquid is applied too fast or in too great a volume. Alternately, the gradient was jostled, and the layers have mixed. It is not possible to salvage this gradient; a new one should be layered. If the 3 ml of dissociated TG have already been layered, remove the cells and media, collecting also a portion of the 4th gradient layer. Add ~5 ml NBA-W and spin down (400 g, 4 min) to wash the Optiprep out. Discard the supernatant, resuspend the cells in 3 ml NBA-W, and re-layer on the new gradient.
Make sure to handle the gradient gently, avoiding any bumps or sudden movements, so as not to disturb the gradient. Make sure the centrifuge is correctly balanced, so as to avoid an emergency brake during the spin.
If there are poorly formed bands after the gradient spin, the gradient may have been jostled or subjected to an emergency stop in the centrifuge, or else the gradient was poorly layered and hence the bands did not form correctly. Collect the entire volume into a 50 ml conical, add an excess of NBA-W, mix, and spin down (~500 g, ~5 min) to wash the Optiprep out. Discard the supernatant, resuspend the cells in 3 ml NBA-W, and re-layer on a new gradient.
It may be difficult to determine which cells are neurons and which are glial cells or debris. This is primarily a matter of experience. Once you have looked at cultured neurons under a microscope you will be able to identify them in a hemocytometer. The neurons will appear phase bright, and will continue to be phase bright over a range of focal distances. Move with the focus dial back-and-forth as you scan the field of view. The debris and glial cells will be out of focus while the neurons remain partially in focus. If you are still not confident, consider staining the neurons for neuronal markers (such as beta-III tubulin or IB4) to train your eye to identify neurons.
The number of neurons may be much lower than expected. This can be due to several reasons, including poor enzymatic digestion, poor gradient separation or collection, loss of neuronal viability during extraction (due to prolonged extraction process, trituration, bubbles or foam), miscalculated volume for resuspension after washes. There is no way to increase the yield at this point. However, care should be taken in future extractions to avoid the possible issues listed.
The number of neurons may be much higher than expected. This may be due to a specific mouse strain, or else the count included some non-neuronal cells. Recount the cells and make sure to count only those that are definitely neurons.
The isolated neuron suspension may include excess tissue debris. This can be due to poor enzymatic digestion, gradient separation, or collection. It is not possible to remove the debris at this stage. Some debris will naturally lift off during media changes. Care should be taken in future extractions to avoid the possible causes listed.
When adding media to neuron cultures, always apply the media gently to the well wall. Applying the media directly onto the neurons may cause them to detach due to shear stress. This will be detectable by a microscope as a “hole” surrounded by ripped axons.
If there is early loss of neuronal viability in culture (over the course of 1–5 days), the neurons may have been exposed to stress during isolation (hypoxia, sheer stress during trituration, bubbles). There is no way to salvage these cultures. Take care during isolation to avoid stressing the neurons. If poor viability persists over several isolation attempts, consider there might be a problem with one of the media ingredients. One of the reagents may have expired or been subjected to freeze-thaw damage. Alternately, you may have received a batch that was compromised (contaminated with chemical or biological reagents, damaged during shipping, etc.). Take note of the lot numbers of your media ingredients, and of the aliquots currently in use. Try systematically replacing each of the ingredients (use a new aliquot, order the same product from the previous lot, or order it from a different vendor). Lastly, one of the reagents may have unanticipated incompatibility with your neuronal cultures. This may be particularly true of FUDR and other antimitotics. Try using different concentrations of FUDR, or else using a different antimitotic.
If the culture is overrun with glial cells, the initial isolation may have been poor in neurons, and rich in glial cells. For purer cultures, avoid the layer closest to the top-most band, which contains debris but also glial cells. This may result in lower yields. You may choose to collect only the lower neuronal band, and this will result in purer cultures of larger neurons, but with fewer of the small neuronal subtypes. Alternately, the FUDR may not inhibit the glial cells. Consider making fresh FUDR, increasing the dose of FUDR, or else adding another mitotic inhibitor, such as aphidicolin. However, this may prove toxic to the neurons, since it inhibits DNA damage repair. Make sure to test the new antimitotic.
If there is a lot of blebbing and cell debris, cells (either neurons or glial cells) are dying. To some extent, this is normal (since glial cells and even neurons may gradually die in culture over the course of 2–3 weeks). Check the cultures regularly and make sure they are not exposed to any additional stress (delayed media change, changes in temperature or pH, etc.).
If many neurons appear grainy, or else the neurites are peeling off the coverslip, this indicates that the neurons are dying. Grainy neurons might yet be salvaged by changing the media, although this may have adverse effects on future assays. However, if the neurites are detaching the cultures cannot be salvaged.
It is possible to reuse the devices, once they have been cleaned and autoclaved.
The 10% FBS medium prepares all the surfaces in the micro-fluidic chamber (including capillaries) for the next steps. The FBS lowers the surface tension of the liquid, allowing the medium to fill the capillaries and central channels. PBS and serum-free medium will not fill the capillaries properly, resulting in the formation of bubbles, which will block neurite growth.
References
- 1.Smith G. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol. 2012;66:153–176. doi: 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosato PC, Leib DA. Neuronal interferon signaling is required for protection against herpes simplex virus replication and pathogenesis. PLoS Pathog. 2015;11(7):e1005028. doi: 10.1371/journal.ppat.1005028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malin SA, Davis BM, Molliver DC. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc. 2007;2(1):152–160. doi: 10.1038/nprot.2006.461. [DOI] [PubMed] [Google Scholar]
- 4.Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, Margolis TP. A5-positive primary sensory neurons are Nonpermissive for productive infection with herpes simplex virus 1 in vitro. J Virol. 2011;85(13):6669–6677. doi: 10.1128/JVI.00204-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosato PC, Leib DA. Intrinsic innate immunity fails to control herpes simplex virus and vesicular stomatitis virus replication in sensory neurons and fibroblasts. J Virol. 2014;88(17):9991–10001. doi: 10.1128/JVI.01462-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katzenell S, Leib DA. Herpes simplex virus and interferon signaling induce novel Autophagic clusters in sensory neurons. J Virol. 2016;90(9):4706–4719. doi: 10.1128/JVI.02908-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gage GJ, Kipke DR, Shain W. Whole animal perfusion fixation for rodents. J Vis Exp. 2012;65:e3564. doi: 10.3791/3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci. 2015;18(1):145–153. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- 9.Pazyra-Murphy MF, Segal RA. Preparation and maintenance of dorsal root ganglia neurons in compartmented cultures. J Vis Exp. 2008;17(20) doi: 10.3791/951. pii: 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cabrera JR, Viejo-Borbolla A, Martinez-Martín N, Blanco S, Wandosell F, Alcamí A. Secreted herpes simplex virus-2 glycoprotein G modifies NGF-TrkA signaling to attract free nerve endings to the site of infection. PLoS Pathog. 2015;11(1):e1004571. doi: 10.1371/journal.ppat.1004571. [DOI] [PMC free article] [PubMed] [Google Scholar]