

Abstract
Background
Inflammatory bowel diseases (IBD) are believed to be driven by dysregulated interactions between the host and the gut microbiota. Our goal is to characterize and infer relationships between mucosal T cells, the host tissue environment and microbial communities in IBD patients that will serve as basis for mechanistic studies on human IBD.
Methods
We characterized mucosal CD4+ T cells using flow cytometry, along with matching mucosal global gene expression and microbial communities data from 35 pinch biopsy samples from IBD patients. We analyzed these data sets using an integrated framework to identify predictors of inflammatory states and then reproduced some of the putative relationships formed among these predictors by analyzing data from the pediatric RISK cohort.
Results
We identified 26 predictors from our combined data set that were effective in distinguishing between regions of the intestine undergoing active inflammation and regions that were normal. Network analysis on these 26 predictors revealed SAA1 as the most connected node linking the abundance of the genus Bacteroides with the production of IL17 and IL22 by CD4+ T cells. These SAA1-linked microbial and transcriptome interactions were further reproduced with data from the pediatric IBD RISK cohort.
Conclusion
This study identifies expression of SAA1 as an important link between mucosal T cells, microbial communities and their tissue environment in IBD patients. A combination of FACS, gene expression and microbial profiling can distinguish between intestinal inflammatory states in IBD regardless of disease types.
Keywords: IBD, mucosal healing, SAA1, systems biology, supervised learning
INTRODUCTION
Inflammatory bowel diseases (IBD) are immune-mediated diseases characterized by chronic intestinal inflammation and are typically categorized as either ulcerative colitis (UC) or Crohn’s Disease (CD). Mucosal healing is recognized both as a measure of disease activity and as part of the treatment goals for IBD patients (1–3). For UC patients, disease location is limited to the large intestinal mucosa and achieving early mucosal healing has been associated with improved clinical outcomes, including reduced incidence of colectomy (4). While disease involvement in CD can occur anywhere along the gastrointestinal tract, mucosal healing also has important prognostic purposes and may prevent the development of complications, such as strictures. Clinical improvement with infliximab is associated with significant healing of mucosal lesions and marked histological improvement of mucosal infiltrates (5). In a Norwegian population-based cohort study, mucosal healing after one year of treatment was predictive of reduced disease activity and subsequent need for active treatment (6). A better understanding of mucosal inflammation and its resolution could facilitate the identification of better biomarkers for prediction of mucosal healing.
While there are significant overlaps in the genetic susceptibility profiles of UC and CD patients, the manifestation of these two types of IBD are considered to be largely distinct (7, 8). IBD pathogenesis is thus not only determined by host genetics, but driven by environmental factors and host-microbial interactions within the intestinal microenvironment (9). As such, intestinal inflammation should be further investigated using multi-parameter data sets characterizing host-microbial relationships. Several studies have already utilized systems biology approaches to investigate IBD (10–12), with the Pediatric RISK Stratification Study (RISK) being the largest one to interrogate host-microbial interactions using microbial and gene expression profiles generated from ileal biopsies obtained from patients with early onset (pediatric) IBD during their initial diagnostic endoscopy, prior to any form of treatment (13).
However, T cell effector function has not been incorporated within any of these multi-parameter studies, despite the importance of T cells in the initiation and resolution of intestinal inflammation during IBD (14). The role of different T cell populations in intestinal pathogenesis has mostly been characterized in mouse models of colitis, which are not completely representative of human IBD. Most human studies characterizing immune cell function have focused on peripheral blood mononuclear cells (PBMCs), although the main target organ in IBD is the intestine (15). Where intestinal lamina propria mononuclear cells (LPMCs) have been studied, it has not been paired with other types of data to allow for systems biology approaches (16). This is mostly due to the challenge of obtaining sufficient material for the generation of different types of data from pinch biopsies. While surgically resected tissue can be an alternative to intestinal pinch biopsies, IBD patients undergoing surgical resection are more likely to have responded poorly to treatment and thus may poorly reflect the broader IBD population.
We have optimized a protocol that allows us to isolate sufficient numbers of LPMCs for: (1) CD4+ T cell cytokine production by multi-color fluorescence-activated cell sorting (FACS), which can be coupled with (2) mucosal gene expression by microarrays and (3) bacterial 16S ribosomal RNA (rRNA) sequencing of mucosa-adherent bacterial communities (17, 18). Using this protocol, we previously reported increased number of T helper 17 (TH17) cells in CD patients and decreased number of T helper 22 (TH22) cells in UC patients (17). In this study, we have focused on the identification of features from these different data types that would best predict inflammation state in IBD and identified expression of serum amyloid A isoform 1 (SAA1), an acute phase protein, as the key node linking microbial communities and TH17/TH22 cells in this network of inflammatory state predictors.
METHODS
All patient samples described here were collected as part of studies approved by the New York University School of Medicine and Mount Sinai Medical Center Institutional Review Boards.
Biopsies acquisition and generation of FACS, 16S microbial and microarray data
Data used in this study were generated from three different sets of biopsies and included both published and unpublished data, with all corresponding references listed in Table 1. IBD biopsies were obtained from adult IBD patients undergoing surveillance colonoscopy at Mount Sinai Medical Center (17). Biopsies from the Mucosal Immunity of Ulcerative Colitis Patients Undergoing Therapy With Trichuris Suis Ova (MUCUS) (Trial ID: NCT01433471, https://clinicaltrials.gov) were collected from two trial subjects (ENR1 and ENR3) at three different time points (baseline, week 12 and week 24 post-treatment). Non-IBD control biopsies of the ileum and the sigmoid colon were obtained from patients referred to the Gastroenterology clinic at the Manhattan campus of the Veteran Affairs Hospital for average risk screening colonoscopy (19, 20). All FACS and 16S rRNA sequencing data were generated as previously described in (17). Microarray data was generated as described in (19). Mucosal inflammatory state of biopsies from adult IBD patients and non-IBD control subjects was determined by pathologists who were reviewing the biopsy samples for standard clinical care. Biopsies from the MUCUS trial were scored based on an agreement between two pathologists using a custom scoring system (Supplemental Table 1).
Table 1.
References for all data set used in this study.
| Data | Cohort | Reference |
|---|---|---|
| FACS | Adult IBD | Leung et al[17] |
| 16S microbial | Adult IBD | Leung et al[17] |
| Microarrays | Adult IBD | Unpublished |
| FACS | MUCUS trial | Unpublished |
| Microarrays | MUCUS trial | Unpublished |
| FACS | Non-IBD control | Unpublished |
| 16S microbial | Non-IBD control | Tang et al[20] |
| Microarrays | Non-IBD control | Unpublished |
Microarray data processing
Microarrays for adult IBD patients and non-IBD control subjects were performed on the Agilent Human SurePrint GE 8 × 60k platform and raw data from Agilent’s Feature Extraction software was processed using limma. Background correction was done using the normal plus exponential convolutional (“normexp”) method, followed by between-array quantile normalization (21). Probes with low expression were removed from the normalized matrix. Only probes with intensity values at least 10% greater than the 95th percentile of the negative control probe intensities across all 41 arrays were kept. Finally, probes that did not have an annotated gene symbol were removed.
Feature selection methods for microarray, 16S microbial and FACS data
Features selection was performed for each data set using the following strategies before fitting the classifier:
Feature selection for gene expression data
Principal Component Analysis (PCA) was done on unique genes. Where splice variant and duplicate probes were present for a gene, the intensity value of the gene was summarized as the median intensity values of its duplicate and/or splice variant probes. To select for highly variable genes, the gene-wise inter-quartile range was computed and only genes with inter-quartile range greater than 0.5 were retained. This resulted in 9822 highly variable unique genes, on which PCA was performed using non-scaled log2 intensity values. The resulting PC scores were plotted on a two-dimensional plot. To select for features that would be used for supervised learning, we first determined the number of PCs that contributed to 80% of the total variance within the gene expression data set (N=12 PCs). We then took the top 10 positive and negative loadings for each of these 12 PCs and concatenated them into a single matrix. This resulted in 164 unique genes. Finally, we used only microarray samples with matching FACS and 16S microbial data for the supervised learning algorithm.
Feature selection for 16S bacterial rRNA sequencing data
The 16S ribosomal RNA (rRNA) sequencing data was generated and processed as described in Leung et al and the resulting Operational Taxonomic Unit (OTU) table was used for this study (17). To exclude low abundance OTUs from the analysis, we removed OTUs that were present in less than 10% of all the biopsy samples. A single unit pseudocount was added to each OTU across all samples, in order to correct for zero counts, and centered log-ratio (CLR) transformation was performed (22). PCA was performed on scaled, CLR-transformed values and we determined the number of PCs contributing to 80% of the total variance within the 16S microbial data set (N=15 PCs). We then took the top 10 positive and negative loadings for each of these 15 PCs and combined them into a single matrix. This resulted in 227 unique OTUs.
Feature selection for CD4+ T cell cytokine data
To help with interpretability, we only selected for FACS gates of double-cytokine combinations for supervised classification. Since each gate was defined as a percentage of the total CD4+ cells, this gave the data a compositional property. As such, we performed CLR transformation for each of the double-cytokine combination. For instance, for the double-cytokine combination of IL4 and IL17, CLR transformation was performed on each of the 4 possible populations of IL4+IL17−, IL4−IL17+, IL4+IL17+ and IL4−IL17−. This was reiterated through each of the 10 double cytokine combinations, resulting in a final matrix of 40 FACS gates that were CLR transformed.
Classification of inflammatory states with sparse generalized canonical correlation analysis (sGCCA)
We used a statistical learning framework known as “sparse generalized canonical correlation analysis” (sGCCA) to discover interactions between the tissue microenvironment (genes expressed on intestinal mucosa, mucosa-adherent bacterial communities) and T cell effector function based on inflammatory states. This framework was chosen as it identified features that were highly correlated within and between the data block(s). This not only allowed for identification of features that highly correlate with a phenotype of interest, but also identified “co-expressed” factors measured using different assays, thereby facilitating discovery of multivariable interactions resulting in a phenotype of interest (for example, inflammatory states) (23, 24).
Briefly, sGCCA seeks to identify a narrow (“sparse”) set of features from each data block to maximize the covariance between each data block in a smaller dimension subspace (spanned by “latent components”), according to a design matrix, where the entries are binary of either 0 (no relationship between the data sets) or 1 (there exists a relationship between the data sets) (23, 24). Sparsity is determined using the l1-penalty, where, in the mixDIABLO implementation, is specified as a user-defined number of non-zero coefficients for each data block.
Although there are three parameters for tuning in mixDIABLO, i.e. (1) the design matrix, (2) the number of latent components and (3) the number of non-zero coefficients for each data block (i.e. sparsity term), we have kept the design matrix to a full design throughout our analysis (i.e. each data block was connected to each other, with the assumption that all three factors in our data sets contributed to inflammation state differences). We also used the suggestion by Lê Cao et al, where the number of components in the model is typically K-1, where K is the total number of different class labels for the classification task (24, 25). Since there were three different class labels (Active, Inactive and Normal) in the adult IBD dataset, we fitted the model with 2 components.
As such, the only parameter we tuned for in the model was the number of features with non-zero coefficients from each data block. A search grid of all possible combinations of number of non-zero coefficients for each data block was generated by imposing the following constraints for each data block: 1–20 non-zero coefficients for FACS, 5–50 for genes, 5–50 for 16S microbial data, each in increment steps of five (24). This helped kept the tuning procedure computationally manageable. Model selection was done by leave-one-out cross validation and performance was evaluated using the overall balanced error rate (BER) based on the average prediction scheme. All data were scaled for model fitting, and the final model was fit with the combination of parameters that was chosen based on the minimum overall BER of component 1, since this would be the more important component.
We ran sGCCA using its mixDIABLO implementation directly through the R package mixOmics and set up model selection using custom R scripts (24). Only IBD biopsies with matching FACS, 16S microbial and microarray data were used for sGCCA (N=35 biopsies). Descriptions of these biopsies are included in Table 2. Since the sGCCA model was trained using data from these 35 biopsies, we will hereby refer to this set of biopsies as the “training data set”. We have also termed the selected variables from the sGCCA model as “predictors” of inflammatory states. However, it should be noted that this does not imply that these features are causative of inflammatory states, but instead are statistically defined to be independent variables of a supervised learning model (26).
Table 2.
Description of pinch biopsies used for sGCCA.
| Classification | Number of samples |
|---|---|
| Inflammation state | Active, N = 12 Inactive, N = 8 Normal, N = 15 |
| Disease type | UC, N = 19 CD, N = 16 |
| Anatomical location | Ascending colon, N = 6 Cecum, N = 2 Descending colon, N = 3 Ileum, N = 3 Rectum, N = 2 Sigmoid, N = 13 Transverse colon, N = 6 |
Unsupervised visualization of inflammation predictors
To objectively assess the effectiveness of the identified predictors in discriminating between the different class labels, we performed independent unsupervised bi-clustering and PCA on the predictors. The log2 intensity values of the predictor genes, as well as the CLR transformed values of the predictor OTUs and FACS gates, were combined into a single data matrix. The values of each predictor were scaled to have a mean of 0 and standard deviation of 1 for bi-clustering and PCA. Results of the bi-clustering were visualized as heatmaps. To further infer the relationships between the predictors, we performed pairwise correlations using Spearman correlation on the predictors. Spearman correlation was based on un-scaled predictor values (i.e. absolute log2 intensity values for gene expression, as well as CLR-transformed values for 16S microbial and FACS data).
Network analysis of inflammation predictors
We took a network approach to facilitate observations of the interactions between these predictors. Each predictor was a node in the network and edges were the inferred relationships between nodes. We first constructed the co-adjacency matrix using the absolute Spearman correlation values generated above with a hard-threshold of 0.5. Self-correlations were removed and only edges with absolute Spearman correlation value greater than 0.5 were included in the final network. A positive correlation value was inferred as a positive relationship, while a negative correlation value was inferred as a negative relationship. Edge directions were not inferred. Each predictor was then ranked by degree of connectedness (i.e. total number of edges originating from a node) to prioritize for predictors that could potentially be of interest for future experimental validation.
Analyses of MUCUS trial biopsies
Microarrays for the MUCUS trial biopsies were generated on the Agilent Human SurePrint GE v2 8 × 60k platform and raw data from the Feature Extraction software was background corrected and quantile-normalized using limma as described above. Since these are repeated measures of the same subjects, we incorporated an additional step to model the per-gene expression value from each biopsy sample using a multivariate approach that took into account the mean effect of a perturbation (i.e. inflammation state in our study) and the individual differential response to the same perturbation (27). The “within-subject variation” can be extracted by subtracting off the between-subject variation from the mean perturbation effect, resulting in a data matrix where the gene expression values were directly proportional to the perturbation effect on each individual (27). Finally, we extracted expression values of the 10 inflammation predictor genes from this within-subject variation matrix for downstream analysis. FACS data from the biopsy samples were CLR-transformed as described above for the training data set and the CLR-transformed values of the 11 inflammation predictor FACS gates were extracted. Values from the combination of these 21 predictors genes and FACS gates were concatenated into a single matrix, which were then scaled and used for PCA and bi-clustering.
Data from non-IBD control subjects
Microarray and FACS analyses of biopsies from non-IBD control subjects were performed as described above for biopsies from adult IBD patients. 16S data was generated as described in (20).
Classification of inflammatory states and regression analysis of SAA1 expression levels in RISK cohort
Data matrix containing reads per kilobase of exon model per million reads (RPKM) values of the RNA-Seq data from the RISK cohort (N=254 samples) was downloaded from NCBI’s Gene Expression Omnibus (GEO) (supplementary file GSE57945_all_samples_RPKM.txt.gz, GEO study GSE57945). Operational Taxonomic Unit (OTU) table for the 16S microbial data was downloaded from Qiita (https://qiita.ucsd.edu/, study 1939, Nov 3, 2016).
We kept the RNA-Seq data from RISK cohort at transcript level and kept only transcripts with at least 5 reads per kilobase of exon model per million reads (RPKM) in at least 5 samples (13). We then added a unit pseudocount to the RPKM values before log2-transformation. The maximum log2 RPKM values for each transcript was computed and the 25th percentile of the per-transcript maximum log2 RPKM value was determined. We kept only transcripts that had a maximum log2 RPKM value greater than the 25th percentile of the per-transcript maximum values. Finally, highly variable transcripts were defined using the inter-quartile range method described above for the adult IBD microarray dataset. This resulted in 4487 transcripts for N=254 samples. Only N=158 RNA-Seq samples with matching 16S data were used for the inflammatory state classification analysis, while all 254 RNA-Seq samples were used for SAA1 regression analysis. For 16S data, we took only samples with matching RNA-Seq data (N=158). We kept only OTUs with at least a total of 10 counts across all samples and were present in at least 10% of all samples. This resulted in 642 OTUs. A single unit pseudocount was added to the OTU counts followed by CLR-transformation.
The sGCCA model was re-trained to classify biopsies with active inflammation and normal biopsies. Normal biopsies included non-inflamed biopsies from Crohn’s disease patients, as well as ileal biopsies from patients with well-controlled UC and non-IBD subjects, for which histopathology reports of the biopsies were not available, consistent with the control samples used in (13). The sGCCA model was fit using one component since there were only two class labels in the RISK cohort (inflamed vs. normal). All other parameters for model selection and fitting were done as described above for the adult IBD data. RNA-Seq log2 RPKM data were not scaled while CLR-transformed 16S data was scaled for sGCCA. We chose these parameters, as this was the only setting that resulted in the validated inflammation marker genes DUOX2 and APOA1 being selected as inflammation predictors (13). Log2 RPKM values of inflammation gene predictors and CLR-transformed values of inflammation OTU predictors were then concatenated into a single matrix and visualized as a heatmap after bi-clustering.
To identify gene and OTU predictors of SAA1 expression levels from the RISK cohort, we used SAA1 expression levels as a response variable and fit a sparse partial least square regression (sPLS-regression) model to the RNA-Seq and 16S microbial data separately. We chose this model as the sGCCA framework cannot be used for a regression problem. Model selection for sPLS-regression was done using 10-fold cross-validation, repeated 10 times. Transcript expression values were not scaled for model fitting, while clr-transformed OTU count from the 16S data was scaled for model fitting, consistent with all other analyses performed in this study. The optimal model was chosen based on the smallest root mean squared error and to ensure stability of the selected variables, the coefficients of all selected predictors were bootstrapped 1000 times and coefficients with zero-containing 90% confidence intervals were removed. Interactions between gene and OTU predictors of SAA1 levels were then inferred using pairwise Spearman correlation value between the predictors. A positive Spearman value was inferred as a positive interaction and a negative Spearman value was inferred as a negative interaction. No threshold was applied, in order to visualize the weaker interactions between the ileal microbiota and SAA1.
RESULTS
Identification of a narrow set of features predictive of inflammatory states using a supervised learning algorithm combined with dimension reduction
Given the limited number of samples (N=35) and the large number of parameters in this study, it would have been a challenge to perform model selection for supervised learning. As such, we first performed feature selection using general dimension reduction technique (by first performing PCA and selecting top loadings from each of the PCs that retain up to 80% of variance) to identify features that would maximize the variance within the total data set and used these selected features for supervised classification by sGCCA. We fit the final sGCCA model using 10 genes, 5 OTUs and 11 FACS gates in 2 components. Using this strategy, we identified a narrow set of features predictive of inflammatory states across UC and CD biopsies (overall balanced error rate, BER=0.42). To independently assess the effectiveness of these predictors in distinguishing between inflammatory states, we subjected predictors from the first component to unsupervised PCA and bi-clustering analysis. We found that most of the biopsies from regions with active inflammation and normal regions were distinguishable using only these 26 predictors. With PCA, the variance in the data set was primarily explained by the differences between biopsies with active inflammation and biopsies from normal regions (53% variance explained by PC1), in a manner that is independent of disease type (Fig. 1A). Visualization of the bi-clustering results showed samples from actively inflamed and normal intestinal regions in two separate clusters, while samples from intestinal regions with inactive inflammation were equally distributed between these two clusters of actively inflamed and normal biopsies (Fig. 1B). This indicates that the predictors were more effective in distinguishing between biopsies from intestinal regions with and without active inflammation, but less effective in predicting an inactive state of inflammation. We further separated these predictors into two different subgroups – predictors with higher abundance in regions with active inflammation and predictors with higher abundance in normal regions. Predictors from within the same subgroup were highly correlated among each other, while inversely correlated with predictors from the other subgroup (Fig. 1C).
Figure 1.
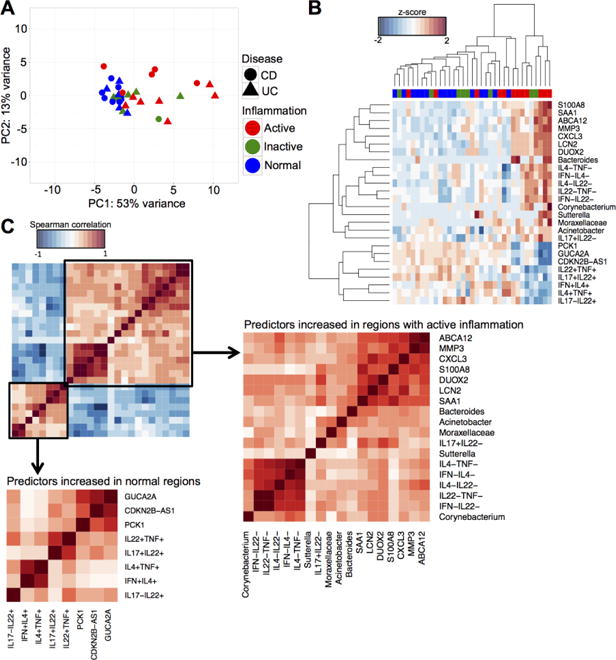
Integrated analysis from N=35 biopsies reveals a set of inflammation-specific features from the FACS, 16S microbial and microarray data, regardless of disease type. (A) PCA of the 26 predictors of inflammatory states. (B) Bi-clustering results of the 26 inflammation predictors visualized as heatmap. Rows are predictors and columns are samples. Red = Active, green = Inactive, blue = Normal. (C) Correlation matrices of predictors of inflammatory states using the Spearman correlation metric. Subsets of predictors were identified that were increased in regions with active inflammation and in normal regions, respectively. Predictors from these two different subsets were highly correlated within subset and inversely correlated between subsets.
We repeated the same unsupervised PCA and bi-clustering analyses on predictors from the second component. With the exception of the CD4+IL17+IL22− and CD4+IL4−IL22− populations, the remaining 24 predictors from component 2 were different from predictors from component 1. We found that predictors from the second component were predominantly increased in ileal CD samples, independent of inflammatory states (Supplemental Fig. 1). This suggests that despite the differences between ileal and colonic tissue microenvironment, there is a set of host and microbial features that could explain inflammatory states independent of anatomical location, as evident by predictors from component 1, which is the more important variable in the model.
In order to demonstrate the effectiveness of the model to classify inflammatory states, we would ideally use an independent test data set to determine precision and recall rates. Unfortunately, none of the published multi-parameter studies (10–13) have incorporated flow cytometry data on T cell effector functions. However, we had established the MUCUS clinical trial to investigate the effect of Trichuris suis ova (TSO) on UC patients (https://clinicaltrials.gov: NCT01433471). As part of this study, we generated unpublished FACS and microarray data from colon biopsies (N=14), collected from two trial subjects over three different time points. We used this small data set to assess how well the combination of gene and FACS predictors could distinguish between disease states. We extracted the values of these predictors from the microarray and FACS data generated using the MUCUS trial biopsies for unsupervised PCA and bi-clustering. Visualization of PCA showed separation of biopsies with active and inactive inflammatory states along PC1, which explained 38% of the total variance in the data set (Fig. 2A). Visualization of the bi-clustering results showed all but two of the inflamed samples clustering separately from the inactive samples and all of the inactive samples clustering together (Fig. 2B).
Figure 2.
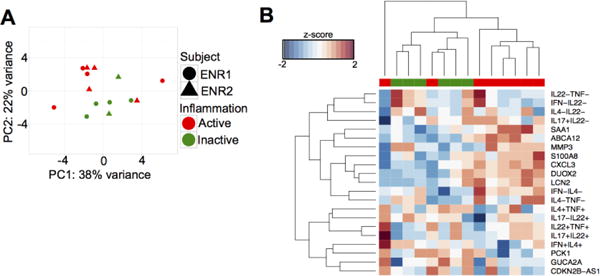
Inflammation predictors can effectively distinguish between inflammatory states in an independent set of biopsies. (A) PCA and (B) bi-clustering using values of the inflammation predictors measured from the MUCUS trial biopsies (N=14 biopsies). Bi-clustering results were visualized as a heatmap, with rows as predictors and columns as samples. Red = Active, green = Inactive.
Network analysis of identified inflammation predictors experimentally validated as well as new interactions
To efficiently understand how the different features interact with each other, we took a network approach with the identified 26 predictors. We computed pairwise Spearman correlation values between these 26 predictors and inferred interactions using these Spearman correlation values, where an interaction was defined using a hard-threshold of absolute Spearman correlation greater than 0.5 and a positive value was inferred as a positive interaction, while a negative value was inferred as a negative interaction (Fig. 3A). We found the SAA1 gene to have the highest degree of connectedness in this network (Fig. 3B). We then focused on the edges connected to the SAA1 gene and found a direct positive relationship between SAA1 and CD4+IL17+IL22− T cells (TH17 cells), as well as between SAA1 and an OTU from the Bacteroides genus (Fig. 3C). We also observed an inverse relationship between SAA1 and the abundance of CD4+IL22+IL17− T cells (TH22 cells), which is a population of CD4+ cells that may be protective to the intestinal mucosa (17, 28).
Figure 3.
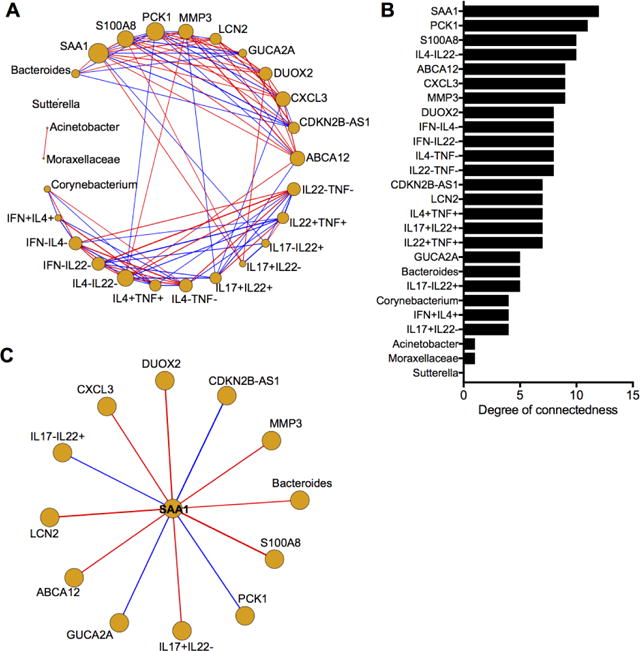
Network analysis of highly correlated inflammation-state predictors. (A) Network inferred for the 26 predictors of inflammatory states using pairwise Spearman correlation with a hard-threshold of |Spearman correlation| > 0.5. Each predictor is represented as a node and edges are the Spearman correlation value between two different predictors. Red edges represent positive correlations, while blue edges represent negative correlations. (B) Predictors ranked by degree of connectedness. (C) All interactions involving SAA1.
Since the inter-individual variation of immune response has been associated with the gut microbiome (29), we asked if these host-microbial interactions during active intestinal inflammation were also present in a state of homeostasis, contributing to individual variation in immune response. To answer this question, we took advantage of our data set collected from non-IBD control subjects, which also included gene expression, 16S microbial and CD4+ T cell FACS data (N=27 biopsies). We extracted the abundance values of the 26 inflammation predictors from this control data set and computed pairwise Spearman correlation values (Supplemental Fig. 2). We found that only 70 of the 650 predictor pairs had absolute correlation values greater than 0.5. In contrast, of the 650 predictor pairs in the adult IBD data set, 174 predictor pairs had absolute correlation values greater than 0.5. This suggests that most of these interactions were uniquely occurring during active inflammation and not during normal intestinal homeostasis.
Analysis of data from the pediatric IBD RISK cohort reproduces similar SAA1-linked interactions
To gain insights on the generalizability of the inflammatory predictors network and SAA1-linked interactions identified from our small data set, we re-trained the sGCCA model using publicly available data from the RISK cohort. We fit the re-trained sGCCA model using 5 genes and 10 OTUs (overall BER = 0.18). We also verified model performance by demonstrating the identification of the experimentally validated DUOX2 and APOA1 genes as two of the inflammatory state predictors in this cohort (Supplemental Fig. 3A). However, we did not identify SAA1 as an inflammatory state predictor from this data set, despite SAA1 being significantly upregulated in inflamed biopsies (Supplemental Fig. 3B). This was likely due to the lack of FACS data from this cohort, since SAA1 was linked to TH17 cells.
To counter this problem, we performed additional regression analysis on samples from the RISK cohort by sparse partial least square regression (sPLS-regression), using the SAA1 expression level as a response variable. The RNA-Seq (N=254 samples) and 16S microbial data (N=158 samples) from the RISK cohort were subjected to separate regression analysis and selected variables from both regression models (N=7 predictor genes and N=19 predictor OTUs, respectively), along with SAA1, were concatenated into a single matrix for pairwise Spearman correlation (Fig. 4A). We then visualized SAA1 and predictors of its expression level as nodes of a network and the corresponding correlation values as edges (Fig. 4B). Six of the predictor genes (SAA2, LCN2, DUOX2, DUOXA2, CYR61 and UBD) were highly and positively correlated with SAA1 expression (range of Spearman correlation = 0.61 – 0.97). Of these six predictor genes, LCN2 and DUOX2 have already been identified to be positively associated with SAA1 in our adult IBD data set. SAA2 is highly homologous with SAA1 and was highly expressed along with SAA1 to modulate IL-17 production in the mouse intestine (30). CYR61 was previously identified as a SAA1-responsive gene from in vitro-polarized mouse TH17 cells cultured with murine recombinant SAA1 (statistically significant with log2 fold change = 0.53, when compared to in vitro-polarized mouse TH17 cells cultured in the absence of murine recombinant SAA1, as reported in the supplementary file GSE71281_G53_veh_vs_SAA_Th17_CuffDiff_Gene_Differential.txt from GEO Series GSE71281) (30). In contrast, the correlations between the predictor OTUs and SAA1 in this data set were weaker (range of Spearman correlation = −0.22 – 0.27, indicated by dotted lines. Nonetheless, an OTU from the Bacteroides genus was identified to be positively associated to SAA1, similar to what we found from our adult IBD data set. On the other hand, most of the other predictor OTUs from the Lachnospiraceae family that were inversely correlated with SAA1 were of the order Clostridiales, which has been demonstrated to have an inverse relationship with Bacteroides (31).
Figure 4.
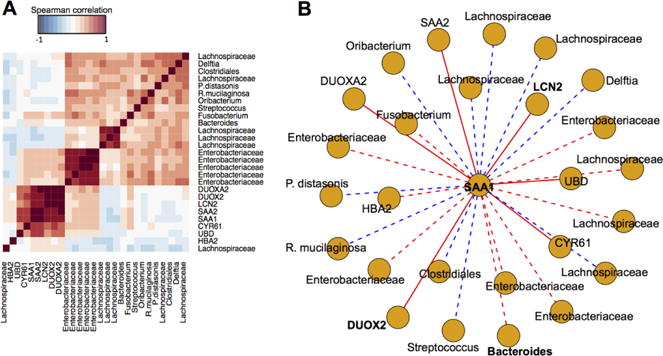
SAAl-linked interactions identified from the RISK cohort are similar to those identified from the adult IBD training data set. (A) Pairwise Spearman correlation values of SAA1 and predictors of SAA1 expression level identified through regression analyses using SAA1 as a response variable. (B) All associations involving SAA1 visualized as a network. Each predictor is represented as a node and edges are the Spearman correlation value between two different predictors. Red edges represent positive correlations, while blue edges represent negative correlations. Dotted edges have |Spearman correlation| less than 0.5 and solid edges have |Spearman correlation| greater than 0.5.
DISCUSSION
In this study, we report a multi-parameter study of intestinal inflammation in IBD and the identification of a narrow set of predictors associated with intestinal inflammatory states. The interaction between SAA1 and CD4+IL17+IL22− T cells served as a proxy marker of model performance, as this interaction has been extensively validated using mouse models (30). SAA1 is an acute-phase reactant during inflammation and is highly conserved in vertebrates (32). SAA1 expression on ileal epithelial cells in the mouse intestine is induced by the attachment of the mouse commensal segmented filamentous bacteria (SFB) and leads to IL-17A production by TH17 cells, through an IL23R/IL22 and Stat3-dependent mechanism (30, 33). We also observed several undescribed interactions involving SAA1 in this study, including a direct positive interaction between SAA1 with an OTU of the Bacteroides genus and negative interaction between SAA1 and CD4+IL22+IL17− T cells. In mouse models, certain Bacteroides spp. have been demonstrated to induce colitis in a host-genotype dependent manner (34, 35). Evidence on how the abundance of Bacteroides changes during intestinal inflammation in IBD patients has been varied, likely due to diversity within the Bacteroides genus (36, 37). We also attempted to verify the reproducibility of these SAA1-linked interactions in the RISK cohort. While the strength of interactions between SAA1 and the OTUs from the RISK cohort were weaker as compared to that from the adult IBD patients, this was likely due to the terminal ileal microbiota being less abundant and less diverse (38–40).
Apart from SAA1, several other predictors identified from our study have also been demonstrated to be associated with intestinal inflammation. For example, the LCN2 gene, another predictor positively associated with SAA1, is increased in serum and feces of IBD patients (41, 42). In the IL10-deficient mouse colitis model, the absence of LCN2 resulted in more severe colitis along with spontaneous intestinal tumor development, secondary to mucosal barrier damage and intestinal microbial dysbiosis (43). This suggests that the expression of LCN2 could be a protective response to intestinal inflammation. LCN2 had also been identified as an IL-17 responsive gene in preosteoblast cell lines (44), in line with its positive association with SAA1, which induces IL-17A production by TH17 cells. On the other hand, the PCK1 gene, a predictor negatively associated with active inflammation, has been reported to be downregulated in inflamed biopsies from IBD patients (both UC and CD) (45).
Disease location is an important consideration in IBD, particularly for CD, which can affect any part of the gastrointestinal tract and as such, disease presentation and management are dependent on the affected anatomical location (46). For example, it was found that microbiota changes in colonic CD were a lot more similar to microbiota changes in UC than microbiota changes in ileocolonic or ileal CD, although it is unclear if these microbial differences are secondary or causative to the inflammation location (11). While our training data consists mostly of colonic biopsies, we only saw ileal-specific trend in predictors identified from component 2 of our inflammatory state classification model (Supplemental Fig. 1). Moreover, several of the predictor genes identified from our training data set (DUOX2, MMP3, CXCL3, S100A8, SAA1, LCN2) were upregulated in the RISK cohort, which was generated from ileal biopsies obtained from treatment-naïve pediatric IBD patients. This suggests that some of the 26 predictors that we have identified, particularly genes such as SAA1, DUOX2 and LCN2, could be a core set of IBD intestinal inflammation features, independent of disease location and disease type. Since a challenge in diagnosing IBD patients is the heterogeneity in its disease manifestation, findings from our study could be clinically relevant, as it points towards the feasibility of being able to predict mucosal inflammation and its resolution in IBD patients using a common set of markers.
Due to the limited number of samples in our study, we sought to manage the challenge of model selection using a combined approach of dimension reduction with supervised classification, similar to a previously described strategy when dealing with sparse set of available samples in the setting of high-dimensional biological data (47). The high overall BER in this model was mostly contributed by classification of samples with inactive inflammation (Supplemental Table 2). When we removed the biopsies with inactive inflammation and repeated the classification task for Active vs. Normal, we saw a great improvement in model performance (overall BER=0.08, data not shown). In addition to poor statistical power, we also could not model inter-individual variability in this small set of samples, since discarding any unpaired samples would have led to further reduction in power. To identify features specific to IBD patients, it would also be more meaningful to classify between biopsies obtained from IBD patients to non-IBD control subjects. We were not able to make this direct comparison as the biopsies from the non-IBD control subjects were collected and processed in a different institution, leading to a confounding batch effect. Future efforts will focus on collecting more biopsies from larger patient cohorts in a longitudinal manner. This will not only address the flaws we have stated above, but will also allow us to test the effectiveness of these predictors in predicting disease progression using finer resolution of classification tasks (such as comparing between different subgroups of patients).
Given the generalizability of our framework and the development of more sensitive genome-wide methods, we also foresee the possibility of incorporating additional data sets, such as chromatin accessibility assay and microbial data from stool (to complement biopsy samples), into the predictive model. Genome-wide chromatin accessibility measurement in clinical samples is now possible using Assay for Transposase-Accessible Chromatin with Sequencing (ATAC-Seq) (48, 49). Functions of specific mucosal immune cell populations can then be inferred using active regulatory elements under physiological conditions, rather than by cytokine stimulation studies. Microbial data from stool samples will facilitate discovery of biomarkers that can be measured using non-invasive methods, which is clinically important in the management of IBD patients given procedural costs and risks.
In conclusion, our study demonstrates the possibility of discovering host-microbial interactions that occur within the inflamed intestinal microenvironment of IBD patients using heterogeneous multi-parameter data sets. These computational predictions, such as the inferred SAA1-associated interactions, can also serve as basis for validation studies using human intestinal CD4+ T cells directly FACS-sorted from gut biopsies to be cultured ex vivo with human recombinant SAA1 and for subsequent genome-wide assays, such as transcriptional profiling and chromatin accessibility assays, to elucidate the mechanisms regulating their differentiation process.
Supplementary Material
Acknowledgments
Parts of the computational workflow were performed on the High Performance Computing facility at New York University. We thank Dr. Kajari Mondal (RISK Program Manager) for assistance with RISK RNA-Seq and 16S data.
Funding: P.L was supported by the National Institutes of Health (AI093811 and DK103788), Broad Medical Research Program Foundation, the Kevin & Marsha Keating Foundation and NY Crohn’s Foundation. I.C was supported by the Doris Duke Charitable Foundation, Grant No. 2014109. Z.P was supported by the National Institutes of Health (U01CA182370, R01AI110372, and R21DE025352). M.A.P and Z.P. are Staff Physicians at the Department of Veterans Affairs New York Harbor Healthcare System. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the U.S. Department of Veterans Affairs or the United States Government.
Footnotes
Conflict of interest: The authors declared no conflict of interest.
Authorship: M.S.T designed and implemented all computational workflows, with input from R.A.B and P.L. R.B, J.M.L and U.M.G performed flow cytometry and generated microarray data from biopsies. R.B and M.J.W coordinated MUCUS trial. Z.P and A.G.N performed histopathology scoring for MUCUS trial biopsies. I.C, D.H, L.B.M, M.A.P, L.E.C, W.M.A, T.U and L.M were GI physicians who collected biopsies. M.J.W and L.B.M obtained IRB protocols. M.S.T wrote the manuscript with input from all authors. P.L and I.C. obtained funding. P.L. provided overall supervision. All authors read and approved of the final manuscript.
Data availability: All microarray data (raw and normalized intensity values) have been deposited on National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) with the accession numbers GSE96665 (Adult IBD biopsies), GSE97011 (non-IBD control biopsies) and GSE96698 (MUCUS trial biopsies). Demultiplexed 16S reads have been deposited on NCBI Sequence Read Archive with the accession numbers SRR5401050 and SRR5401051. FACS data and corresponding metadata files, as well as computational codes, have been included in a supplementary folder for reproducibility purpose.
References
- 1.Rutgeerts P, Vermeire S, Van Assche G. Mucosal healing in inflammatory bowel disease: impossible ideal or therapeutic target? Gut. 2007;56:453–455. doi: 10.1136/gut.2005.088732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dave M, Loftus EV. Mucosal Healing in Inflammatory Bowel Disease—A True Paradigm of Success? Gastroenterology & Hepatology. 2012;8:29–38. [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh A, Palmer R, Travis S. Mucosal Healing As a Target of Therapy for Colonic Inflammatory Bowel Disease and Methods to Score Disease Activity. Gastrointestinal Endoscopy Clinics of North America. 2014;24:367–378. doi: 10.1016/j.giec.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Colombel JF, Rutgeerts P, Reinisch W, et al. Early Mucosal Healing With Infliximab Is Associated With Improved Long-term Clinical Outcomes in Ulcerative Colitis. Gastroenterology. 2011;141:1194–1201. doi: 10.1053/j.gastro.2011.06.054. [DOI] [PubMed] [Google Scholar]
- 5.D’Haens G, Van Deventer S, Van Hogezand R, et al. Endoscopic and histological healing with infliximab anti-tumor necrosis factor antibodies in Crohn’s disease: A European multicenter trial. Gastroenterology. 1999;116:1029–1034. doi: 10.1016/s0016-5085(99)70005-3. [DOI] [PubMed] [Google Scholar]
- 6.Frøslie KF, Jahnsen J, Moum BA, et al. Mucosal Healing in Inflammatory Bowel Disease: Results From a Norwegian Population-Based Cohort. Gastroenterology. 2007;133:412–422. doi: 10.1053/j.gastro.2007.05.051. [DOI] [PubMed] [Google Scholar]
- 7.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peloquin JM, Goel G, Villablanca EJ, et al. Mechanisms of Pediatric Inflammatory Bowel Disease. Annual Review of Immunology. 2016;34:31–64. doi: 10.1146/annurev-immunol-032414-112151. [DOI] [PubMed] [Google Scholar]
- 9.Huttenhower C, Kostic Aleksandar D, Xavier Ramnik J. Inflammatory Bowel Disease as a Model for Translating the Microbiome. Immunity. 40:843–854. doi: 10.1016/j.immuni.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Häsler R, Sheibani-Tezerji R, Sinha A, et al. Uncoupling of mucosal gene regulation, mRNA splicing and adherent microbiota signatures in inflammatory bowel disease. Gut. 2016 doi: 10.1136/gutjnl-2016-311651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imhann F, Vich Vila A, Bonder MJ, et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. 2016 doi: 10.1136/gutjnl-2016-312135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peloquin JM, Goel G, Kong L, et al. Characterization of candidate genes in inflammatory bowel disease–associated risk loci. JCI Insight. :1. doi: 10.1172/jci.insight.87899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haberman Y, Tickle TL, Dexheimer PJ, et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. The Journal of clinical investigation. 2014;124:3617–3633. doi: 10.1172/JCI75436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 140:1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eastaff-Leung N, Mabarrack N, Barbour A, et al. Foxp3+ Regulatory T Cells, Th17 Effector Cells, and Cytokine Environment in Inflammatory Bowel Disease. Journal of Clinical Immunology. 2010;30:80–89. doi: 10.1007/s10875-009-9345-1. [DOI] [PubMed] [Google Scholar]
- 16.Maul J, Loddenkemper C, Mundt P, et al. Peripheral and Intestinal Regulatory CD4+CD25high T Cells in Inflammatory Bowel Disease. Gastroenterology. 2005;128:1868–1878. doi: 10.1053/j.gastro.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 17.Leung J, Davenport M, Wolff M, et al. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal immunology. 2014;7:124–133. doi: 10.1038/mi.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowcutt R, Malter LB, Chen LA, et al. Isolation and cytokine analysis of lamina propria lymphocytes from mucosal biopsies of the human colon. Journal of immunological methods. 2015;421:27–35. doi: 10.1016/j.jim.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolff MJ, Leung JM, Davenport M, et al. TH17, TH22 and Treg cells are enriched in the healthy human cecum. PLoS One. 2012;7:e41373. doi: 10.1371/journal.pone.0041373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang MS, Poles J, Leung JM, et al. Inferred metagenomic comparison of mucosal and fecal microbiota from individuals undergoing routine screening colonoscopy reveals similar differences observed during active inflammation. Gut Microbes. 2015;6:48–56. doi: 10.1080/19490976.2014.1000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurtz ZD, Muller CL, Miraldi ER, et al. Sparse and compositionally robust inference of microbial ecological networks. PLoS computational biology. 2015;11:e1004226. doi: 10.1371/journal.pcbi.1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tenenhaus A, Philippe C, Guillemot V, et al. Variable selection for generalized canonical correlation analysis. Biostatistics (Oxford, England) 2014;15:569–583. doi: 10.1093/biostatistics/kxu001. [DOI] [PubMed] [Google Scholar]
- 24.Singh A, Gautier B, Shannon CP, et al. DIABLO – an integrative, multi-omics, multivariate method for multi-group classification. bioRxiv. 2016 [Google Scholar]
- 25.Lê Cao K, Boitard S, Besse P. Sparse PLS Discriminant Analysis: biologically relevant feature selection and graphical displays for multiclass problems. BMC bioinformatics. 2011:12. doi: 10.1186/1471-2105-12-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hastie T, Tibshirani R, Friedman J. Elements of Statistical Learning. 2nd. Springer; 2006. Overview of Supervised Learning. [Google Scholar]
- 27.Liquet B, Cao K-AL, Hocini H, et al. A novel approach for biomarker selection and the integration of repeated measures experiments from two assays. BMC bioinformatics. 2012;13:325. doi: 10.1186/1471-2105-13-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broadhurst MJ, Leung JM, Kashyap V, et al. IL-22+ CD4+ T cells are associated with therapeutic trichuris trichiura infection in an ulcerative colitis patient. Sci Transl Med. 2010;2:60ra88. doi: 10.1126/scitranslmed.3001500. [DOI] [PubMed] [Google Scholar]
- 29.Schirmer M, Smeekens SP, Vlamakis H, et al. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell. 2016;167:1125–1136.e1128. doi: 10.1016/j.cell.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sano T, Huang W, Hall JA, et al. An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell. 2015;163:381–393. doi: 10.1016/j.cell.2015.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramanan D, Bowcutt R, Lee SC, et al. Helminth infection promotes colonization resistance via type 2 immunity. Science. 2016;352:608–612. doi: 10.1126/science.aaf3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. European Journal of Biochemistry. 1999;265:501–523. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- 33.Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramanan D, Tang MS, Bowcutt R, et al. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity. 2014;41:311–324. doi: 10.1016/j.immuni.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bloom SM, Bijanki VN, Nava GM, et al. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe. 2011;9:390–403. doi: 10.1016/j.chom.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forbes JD, Van Domselaar G, Bernstein CN. Microbiome Survey of the Inflamed and Noninflamed Gut at Different Compartments Within the Gastrointestinal Tract of Inflammatory Bowel Disease Patients. Inflamm Bowel Dis. 2016;22:817–825. doi: 10.1097/MIB.0000000000000684. [DOI] [PubMed] [Google Scholar]
- 37.Ott SJ, Musfeldt M, Wenderoth DF, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–693. doi: 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zoetendal EG, Raes J, van den Bogert B, et al. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012;6:1415–1426. doi: 10.1038/ismej.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartman AL, Lough DM, Barupal DK, et al. Human gut microbiome adopts an alternative state following small bowel transplantation. Proceedings of the National Academy of Sciences. 2009;106:17187–17192. doi: 10.1073/pnas.0904847106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Booijink CCGM, El-Aidy S, Rajilić-Stojanović M, et al. High temporal and inter-individual variation detected in the human ileal microbiota. Environmental microbiology. 2010;12:3213–3227. doi: 10.1111/j.1462-2920.2010.02294.x. [DOI] [PubMed] [Google Scholar]
- 41.Chassaing B, Srinivasan G, Delgado MA, et al. Fecal Lipocalin 2, a Sensitive and Broadly Dynamic Non-Invasive Biomarker for Intestinal Inflammation. PLOS ONE. 2012;7:e44328. doi: 10.1371/journal.pone.0044328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oikonomou KA, Kapsoritakis AN, Theodoridou C, et al. Neutrophil gelatinase-associated lipocalin (NGAL) in inflammatory bowel disease: association with pathophysiology of inflammation, established markers, and disease activity. J Gastroenterol. 2012;47:519–530. doi: 10.1007/s00535-011-0516-5. [DOI] [PubMed] [Google Scholar]
- 43.Moschen Alexander R, Gerner Romana R, Wang J, et al. Lipocalin 2 Protects from Inflammation and Tumorigenesis Associated with Gut Microbiota Alterations. Cell Host & Microbe. 19:455–469. doi: 10.1016/j.chom.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 44.Shen F, Ruddy MJ, Plamondon P, et al. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-α-induced genes in bone cells. Journal of Leukocyte Biology. 2005;77:388–399. doi: 10.1189/jlb.0904490. [DOI] [PubMed] [Google Scholar]
- 45.Mirza AH, Berthelsen CH, Seemann SE, et al. Transcriptomic landscape of lncRNAs in inflammatory bowel disease. Genome Medicine. 2015;7:39. doi: 10.1186/s13073-015-0162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hedrick TL, Friel CM. Colonic Crohn Disease. Clinics in Colon and Rectal Surgery. 2013;26:84–89. doi: 10.1055/s-0033-1348046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morgan XC, Kabakchiev B, Waldron L, et al. Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome biology. 2015;16:67. doi: 10.1186/s13059-015-0637-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buenrostro JD, Giresi PG, Zaba LC, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qu K, Zaba LC, Giresi PG, et al. Individuality and variation of personal regulomes in primary human T cells. Cell systems. 2015;1:51–61. doi: 10.1016/j.cels.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.