

Abstract
Background and Purpose
Intracerebral hemorrhage (ICH) leads to disability or death with few established treatments. Adverse outcomes following ICH result from irreversible damage to neurons resulting from primary and secondary injury. Secondary injury has been attributed to hemoglobin and its oxidized product hemin from lysed red blood cells. The aim of this study was to identify the underlying cell death mechanisms attributable to secondary injury by hemoglobin and hemin to broaden treatment options.
Methods
We investigated cell death mechanisms in cultured neurons exposed to hemoglobin or hemin. Chemical inhibitors implicated in all known cell death pathways were employed. Identified cell death mechanisms were confirmed using molecular markers and electron microscopy.
Results
Chemical inhibitors of ferroptosis and necroptosis protected against hemoglobin- and hemin-induced toxicity. By contrast, inhibitors of caspase-dependent apoptosis, protein or mRNA synthesis, autophagy, mitophagy or parthanatos had no effect. Accordingly, molecular markers of ferroptosis and necroptosis were increased following ICH in vitro and in vivo. Electron microscopy showed that hemin induced a necrotic phenotype. Necroptosis and ferroptosis inhibitors each abrogated death by greater than 80% and had similar therapeutic windows in vitro.
Conclusion
Experimental ICH shares features of ferroptotic and necroptotic cell death, but not caspase-dependent apoptosis or autophagy. We propose that ferroptosis or necroptotic signaling induced by lysed blood is sufficient to reach a threshold of death that leads to neuronal necrosis and that inhibition of either one of these pathways can bring cells below that threshold to survival.
Keywords: intracerebral hemorrhage, cell death, ferroptosis, necrosis, apoptosis
Introduction
Although intracerebral hemorrhage (ICH) accounts for about 15% of all strokes, it has the highest mortality rates among stroke subtypes with up to 50% within 30 days after the insult.1, 2 Treatment options are lacking and translation of findings from the lab bench to the human bedside has been limited.
In ICH, a hydrostatic jet of blood emerging from the ruptured vessel causes direct tissue destruction immediately. In contrast, secondary injury including perihematoma edema formation, inflammation, and cell death occurs hours to days following the hemorrhagic event.3 Increasing evidence suggests that hemoglobin from the hematoma contributes to cell autonomous and non-cell autonomous neuronal injury.4 Specifically, infusion of lysed red blood cells into the striatum of rats leads to an increase in brain water content accompanied by neurological deficits in the forelimb placing task.5 Similarly, infusion of hemoglobin as well as its oxidized product hemin increases brain water content, inflammatory responses, and neuronal cell death.4, 6 Furthermore, hemoglobin induces cell death both in vivo and in vitro.7, 8
In the blood, haptoglobin and hemopexin act as hemoglobin and heme scavengers during hemolysis.9 Following experimental ICH, expression of haptoglobin increases. Consequently, deletion of haptoglobin worsens neurological outcome, while mice overexpressing human haptoglobin 2 show less severe deficits following ICH.10 In vitro, haptoglobin prevents hemoglobin-induced neuronal toxicity.11 Similarly, knockout of hemopexin leads to an increase in lesion volumes and degenerating neurons and aggravates behavioral deficits following ICH in vivo.12
Despite an increasing understanding of the role of hemoglobin in ICH, it remains unclear how cells die in response to bleeding in the brain, which is crucial to develop effective therapeutic strategies. Classically, the research community distinguished between two modes of cell death, i.e. apoptosis and necrosis. Both apoptotic and necrotic cells have been identified in animal models of intracerebral hemorrhage13, 14 as well as in the perihematoma region after surgical evacuation in humans.15, 16 However, caution must be taken when attributing apoptosis or necrosis to single biochemical markers as these markers are not specific for one type of cell death.17, 18 Moreover, recent studies discovered other forms of regulated non-apoptotic cell death such as necroptosis and ferroptosis.19, 20 To date, it is unclear to what extent the different forms of cell death contribute to ICH-induced toxicity.
In this study, we performed an unbiased screen of known pharmacological cell death inhibitors in hemoglobin- and hemin-induced toxicity in primary cortical neurons. We validated the identified cell death mechanisms using molecular markers in vitro and in vivo and performed electron microscopy to identify the morphological characteristics of ICH-induced cell death in vitro.
Material and Methods
Chemicals and reagents are listed in the supplement.
Animals
Experimental procedures on mice were approved by the Weill Cornell Medicine Institutional Animal Care and Use Committee and conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and ARRIVE guidelines. Mice were purchased from Charles River Laboratories and housed at 68°–72°F, 30–70% humidity, under 12-hour light/dark cycle, with food (PicoLab Rodent diet 5053, LabDiet) and water freely accessible.
Mouse Model of Intracerebral Hemorrhage
ICH was induced in mice as previously described.21 Male C57BL/6 mice (8 to 10 weeks old) were randomly assigned to groups.
Cell Culture
Primary cortical neurons were obtained from CD-1/ICR mice at embryonic day 15 as previously described22 and cultured at 37°C in a humidified 5% CO2 atmosphere (density: 105 cells/well). Immortalized hippocampal neuroblasts (HT22 cells) were cultured in Dulbecco’s modified Eagle's medium containing 10% fetal bovine serum and 1% penicillin/streptomycin and treated when 70% confluent.
Cell Viability
We determined cell viability as previously described,23 at 14–18 hours following hemin or at 24–28 hours following hemoglobin exposure. Plates were measured at SpectraMax Plus Microplate Reader using SoftMax Pro v4.7.1 (both Molecular Devices, Sunnyvale, USA). We confirmed MTT assays by LIVE/DEAD assay and fluorescence microscopy at Nikon Eclipse TS100 microscope using Nikon DS-L3 (Nikon Instruments, Melville, USA).
Immunoblot Analysis
We prepared protein extracts using 1% Triton buffer (25mM Tris pH 7.4, 100mM NaCl, 1mM EGTA, 1% Triton X-100, protease inhibitors, 2.5mM sodium orthovanadate). Samples were electrophoresed under reducing conditions on NuPAGE gels and transferred to nitrocellulose membrane. Antibodies against phospho-ERK1/2, total ERK1/2, γ-tubulin, phospho-S166 RIP1, and β-actin were incubated overnight at 4°C. Secondary antibodies were incubated for 1 hour at room temperature. Proteins were detected using Odyssey infrared imaging system (LI-COR Biosciences).
RNA Extraction and Real-time PCR
Total RNA was prepared using NucleoSpin RNA isolation kit according to established protocols. We performed Real-time PCR using Taqman RNA-to-CT 1-Step Kit for mouse RIP1 (#Mm00436354_m1) and RIP3 (Mm00444947_m1) at a 7500 Real-Time PCR System (Applied Biosystems). Expression levels were normalized to mouse β-actin endogenous control.
Electron Microscopy
Primary neurons were grown on poly-l-lysine-coated aclar plastic coverslips. We fixed the cells overnight at 4°C in 0.05M phosphate buffer (PB; pH 7.4) containing 2 % glutaraldehyde and 0.1M sucrose. The coverslips were processed for electron microscopy as previously described with modifications.24 We incubated the coverslips in 2% osmium tetroxide in PB for 1 hour followed by embedding in Epon-812. Ultrathin sections (70nm) using a Leica UC6 ultratome were collected on 400-mesh thin-bar copper grids (Electron Microscopy Sciences, Fort Washington, PA, USA) and counterstained with 5% uranyl acetate and Reynold’s lead citrate. Micrographs were taken on a Tecnai Biotwin transmission electron microscope (FEI, Hillsboro, Oregon, USA). We quantified the percentage of cells displaying necrotic or apoptotic morphology (n=33–43/condition and replicate). Using ImageJ v.1.49 (http://imagej.nih.gov/ij/), we measured mitochondrial size as percentage area of total area of the cytoplasm25 (comprising >1800 mitochondria in total). Analysis was performed by an investigator blinded to treatment group assignment.
Statistical Analysis
We evaluated normality by Kolmogorov-Smirnov test and variance homogeneity using Levené test. For normally distributed data with homogeneous variance, one-way ANOVA followed by posthoc Bonferroni test was performed. When one of the criteria was not met, Kruskal-Wallis test was performed followed by posthoc Mann-Whitney U test with α-correction according to Bonferroni to adjust for the inflation of type I error due to multiple testing. Data are represented as mean ± standard deviation (S.D.) except for nonparametric data, in which case medians are given. For electron microscopy data, mean ± standard error of the mean (S.E.M.) is given as this is the convention in this field. A value of p<0.05 was considered statistically significant. For Kruskal-Wallis test followed by Mann-Whitney U, p=0.05/k was used, with k as number of single hypotheses. To analyze contingency tables, Fisher’s exact test was used. For detailed statistical analyses see supplemental data. We performed all statistical analyses with IBM SPSS version 21.
Results
Hemorrhagic Stroke In Vitro Shares Features of Ferroptotic and Necroptotic Cell Death
Lysis of red blood cells after ICH leads to release of hemoglobin, which is further broken down into heme or its oxidized form hemin. Hemoglobin or hemin have been used in vitro to study cell death in primary neurons and neuronal cell lines.7, 21 Incubation of primary neurons with 100μM hemin led to a 50% reduction in cell survival (Figure 1A). This concentration is below the concentration estimated in vivo following ICH in humans.26
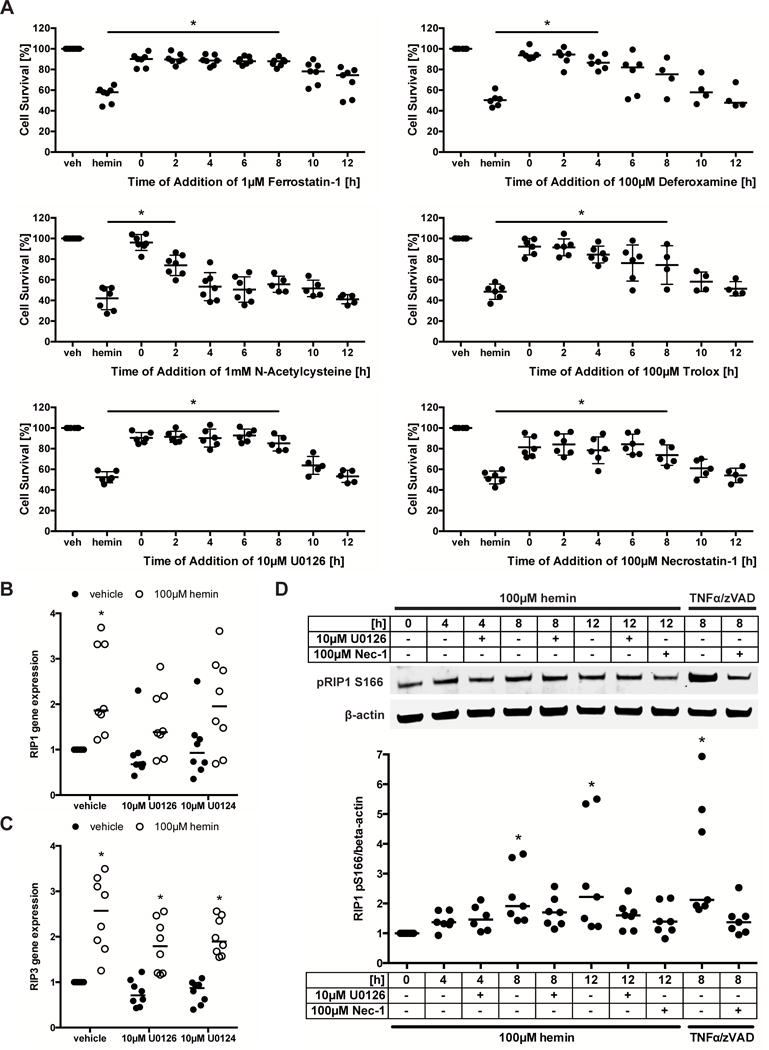
Figure 1. Systematic pharmacologic characterization of hemin-induced cell death in primary cortical neurons. Cell death is inhibited by chemical inhibitors of ferroptosis or necroptosis.
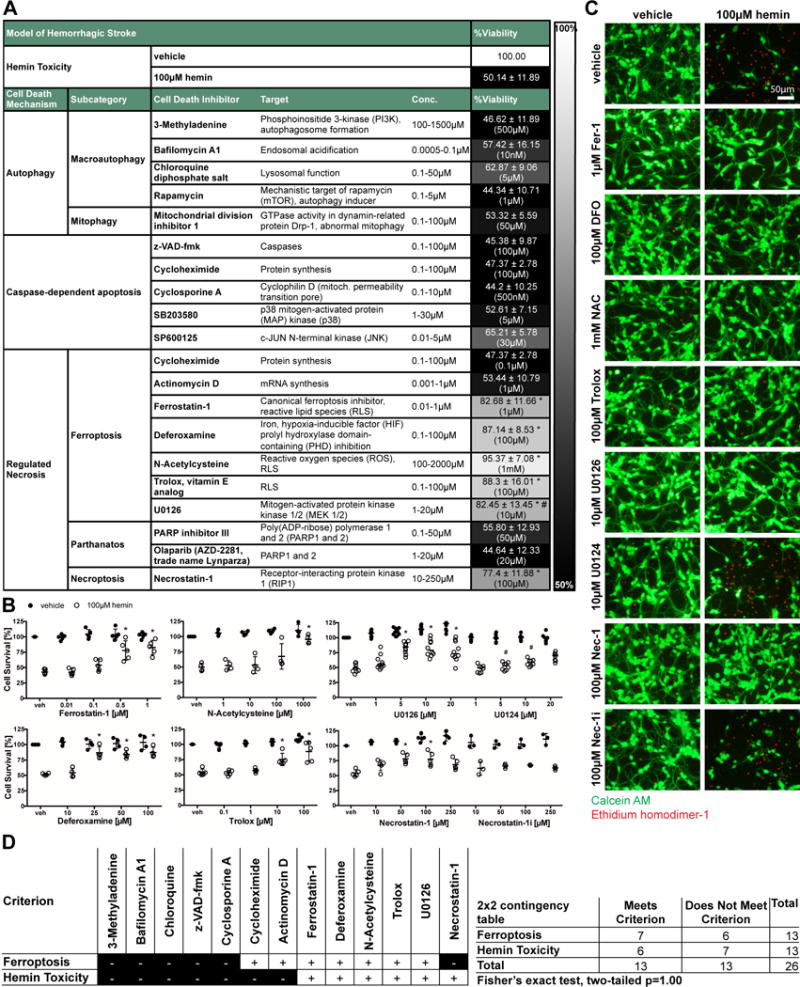
(A) Primary neurons treated with 100μM hemin (LD50) and chemical inhibitors effective in distinct cell death models (e.g. apoptosis, autophagy, ferroptosis) were examined. Values show mean ± SD at representative concentration in brackets. Grayscale coding indicates the continuum from no protection in the presence of hemin (black) to maximal cell viability (white). (B) Concentration-responses of inhibitors that inhibit hemin-induced death. Values represent mean ± SD, except for U0126 where medians are given. * p<0.05 versus hemin, # p<0.05 versus U0124. (C) Representative live/dead staining are shown. Scale bar = 50μm. (D) Statistical analysis of profile of chemical inhibitors between operationally defined ferroptosis20, 27 and hemin-induced toxicity revealed that hemin toxicity in primary neurons can be considered ferroptotic.
In a systematic pharmacological characterization, we found that several chemical inhibitors that collectively define a ferroptotic form of cell death inhibited hemin-induced death (Figure 1A). As with classically defined ferroptosis,20, 27 the canonical ferroptosis inhibitor Ferrostatin-1 protected against hemin toxicity. Moreover other inhibitors of ferroptosis, including the iron chelator Deferoxamine, the glutathione prodrug N-acetylcysteine, the water soluble lipid peroxidation inhibitor Trolox, and the extracellular-signaling kinase 1/2 (ERK1/2) inhibitor U0126 (but not its inactive control) abrogated hemin toxicity (Figure 1B).
Consistent with ferroptosis being a dominant mode of death, caspase inhibitors (that by definition inhibit apoptotic death17), inhibitors of protein or mRNA synthesis, inhibitors or activators of autophagy did not increase cell viability in the presence of hemin (Figure 1A). Live/dead assays, which represent a visible measure of cell death in single cells, revealed results that were similar to quantitative measurements of cell death performed with MTT assays (Figure 1C).
To statistically address whether hemin-induced neuronal death is ferroptotic, we compared the profile of chemical inhibitors of hemin toxicity with operationally defined ferroptosis.20, 27 We found a large overlap; statistical analysis revealed that cell death induced by hemin in primary neurons can be considered ferroptotic (Fisher’s exact test, two-tailed p=1.00).
Unexpectedly, an inhibitor of necroptosis (Necrostatin-1), a regulated form of necrosis19 and previously implicated in ischemic stroke, also blocked hemin-induced death (Figure 1A–C). The specificity of this inhibitor against receptor-interacting kinase 1 (RIP1) was suggested by the observation that a structural analog (Necrostatin-1i), with no activity toward RIP1, did not block hemin-induced death.
Together, these studies suggested that hemin-induced death in cortical neurons shares features of ferroptosis and necroptosis. Convergence of these distinct modes of cell death in a single cell death paradigm has not been described.
To confirm that the present profile of protective inhibitors can protect generally from other potential toxins of lysed blood in addition to hemin, we examined whether inhibitors of ferroptosis and necroptosis could abrogate hemoglobin-induced death. As expected from our hemin studies, inhibitors of ferroptosis and necroptosis abrogated hemoglobin-induced cell death (Figure 2).
Figure 2. Inhibitors capable of preventing hemin-induced death also inhibit hemoglobin-induced-cell death.
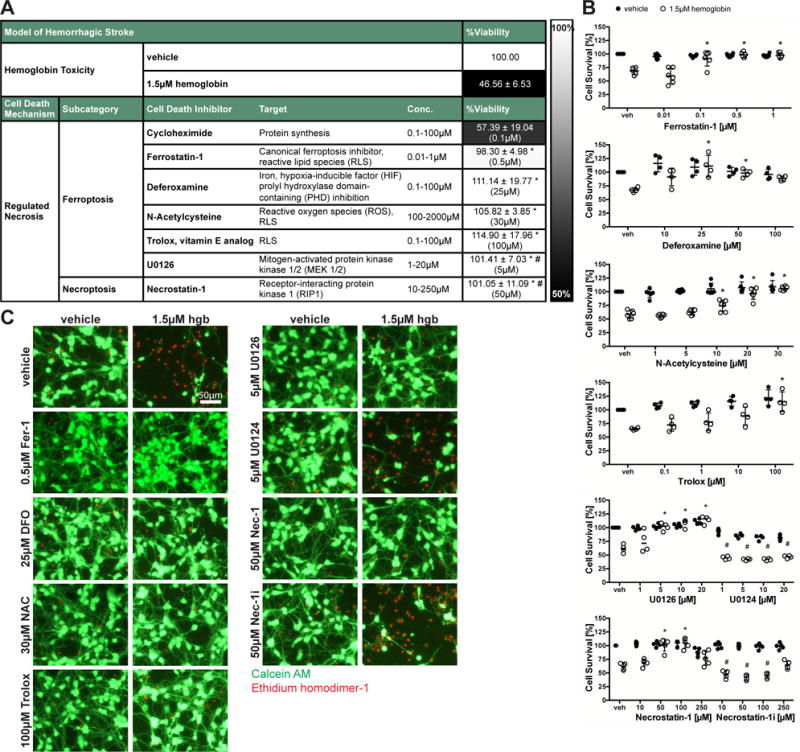
(A) Primary neurons were exposed to 1.5μM hemoglobin (LD50) and ferroptotic and necroptotic inhibitors. Values reflect mean ± SD at representative concentration in brackets. Grayscale coding reflects a continuum from no protection from hemoglobin toxicity by a designated chemical inhibitor (black) to maximal possible cell viability (white). (B) Concentrations-responses of protective inhibitors in hemoglobin toxicity. Values represent mean ± SD, except for U0126 where medians are given. * p<0.05 versus hemoglobin, # p< 0.05 versus inactive analogs U0124 or Necrostatin-1i. (C) Representative live/dead staining are shown. Scale bar = 50μm.
Together, these findings suggest that neurons exposed to hemoglobin or hemin die via a similar ferroptotic and necroptotic pathway.
Hemorrhagic Stroke In Vitro and In Vivo Shows Molecular Features of Ferroptosis and Necroptosis
We thus hypothesized that cell death following ICH in vitro is a mixture of ferroptosis and necroptosis. To establish the specificity of our chemical inhibitors, we assessed whether signaling molecules required for ferroptotic or necroptotic death were activated using biochemical measurements.
Activation of MAP kinase signaling involving phospho-ERK1/2 is important in ferroptosis induced by erastin in tumor cells bearing oncogenic Ras.28 We assessed whether ERK1/2 are activated by hemin in primary neurons evaluating total and phospho-ERK1/2 protein levels. We found an increase in phospho-ERK1/2 starting at 4 hours of hemin treatment (Figure 3A). As expected, this phosphorylation was inhibited by U0126, but not its inactive control U0124 (Figure 3B). Next, we wanted to confirm whether our in vitro findings translate to ICH in vivo. We found that phospho-ERK1/2 was significantly elevated at 6 and 24 hours following ICH in mice (Figure 3C).
Figure 3. Hemorrhagic stroke in vitro and in vivo shows molecular features of ferroptosis or necroptosis.
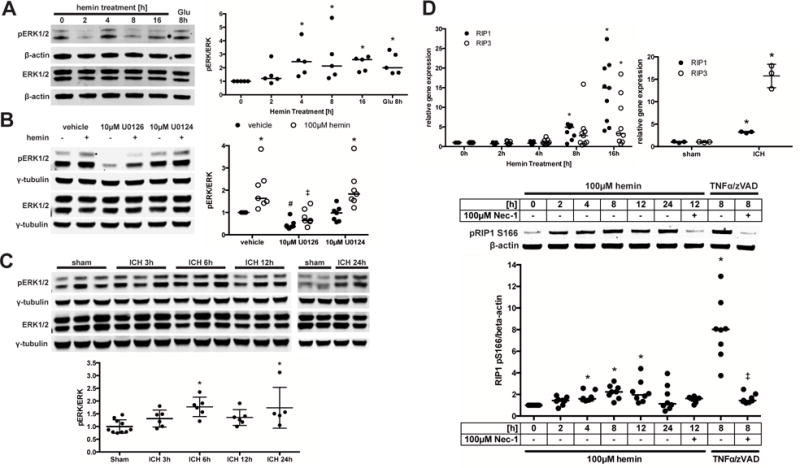
(A) Total and phospho-ERK1/2 protein levels were assessed in primary neurons exposed to hemin or Glutamate (positive control for oxidative stress-induced ERK phosphorylation). Values represent medians of ratios of phospho- by total ERK1/2 normalized to β-actin loading control that does not change in response to death stimuli. * p<0.05 versus 0 hours. (B) Primary neurons were exposed to hemin and U0126 or its inactive analog U0124 for 8 hours. Total and phospho-ERK1/2 protein levels were normalized to γ-tubulin. Values represent medians. * p<0.05 versus without hemin, # p<0.05 versus vehicle without hemin, ‡ p<0.05 versus vehicle or U0124 with hemin. (C) Total and phospho-ERK1/2 expression is shown in striatum of mice following ICH. Values represent mean±SD of ratios of phospho- by total ERK1/2 normalized to γ-tubulin. * p<0.05 versus sham. (D) RIP1 and RIP3 mRNA expression was measured in primary neurons exposed to hemin or in striatum of mice following 24 hours of ICH. Values represent medians for hemin and mean±SD for ICH. * p<0.05 versus 0 hours or sham. Levels of phospho-RIP1 (normalized to β-actin) were measured in HT22 cells exposed to hemin or 100ng/ml TNFα + 5μM z-VAD-fmk for 8 hours (positive control). Necrostatin-1 served as confirmation for specificity of RIP1 kinase activity. Values represent median. * p<0.05 versus 0 hours hemin, ‡ p<0.05 versus 8h TNFα/zVAD.
RIP1 and RIP3 mediate necroptotic cell death and prior studies demonstrated an increase in RIP1 and RIP3 message in association with this type of death.29, 30 Both RIP1 and RIP3 mRNA levels increased following hemin treatment and after ICH (Figure 3D). However, increases in RIP1 and RIP3 mRNA do not necessarily indicate enhanced necroptosis, as both proteins, especially RIP1, have other functions that do not lead to necroptosis.31, 32 It is established that RIP1 kinase activity is required to execute necroptosis.33 Using an antibody against a known RIP1 autophosphorylation site at serine 166,34 we found a Necrostatin-1-sensitive increase in phospho-RIP1 following hemin treatment (Figure 3D).
Hemorrhagic Stroke In Vitro Presents a Necrotic Morphology
Historically, distinct mechanisms of cell death were discriminated by their morphology at an ultrastructural level. Cells undergoing necrosis/necroptosis display swelling of organelles, structural loss of mitochondria, disruption of the plasma membrane and subsequent release of intracellular content. By contrast, cells that die by apoptosis show cell shrinkage, nuclear fragmentation and formation of apoptotic bodies.17 In ferroptosis, shrunken mitochondria have been observed while the plasma membrane remained intact and formation of apoptotic bodies was absent.20
To establish whether hemin toxicity involves ultrastructural features of necr(opt)osis and/or ferroptosis, we performed electron microscopy of primary neurons at 16 hours following hemin treatment. We chose this time as it is eight hours before cell death has reached a steady state (24 hours) thus increasing the likelihood to observe morphologies reflecting a continuum from early to late stages of death. Compared to vehicle-treatment (Figure 4A upper row), some of the hemin-treated neurons displayed a necrotic morphology, as reflected by a loss of plasma membrane integrity and disintegration of organelles (Figure 4A middle row, left and middle). Other hemin-treated neurons presented an intact plasma membrane, but larger mitochondria (Figure 4A lower row, left and middle). Neuronal processes of hemin-treated cells were no longer intact (Figure 4A middle and lower row, right pictures). When we quantified the number of cells with either necrotic or apoptotic morphology, we found a significant increase in necrotic cells after hemin treatment, while we did not detect any apoptotic bodies (Figure 4B).
Figure 4. Hemin-induced death in cortical neurons shows ultrastructural features of necrosis/necroptosis.
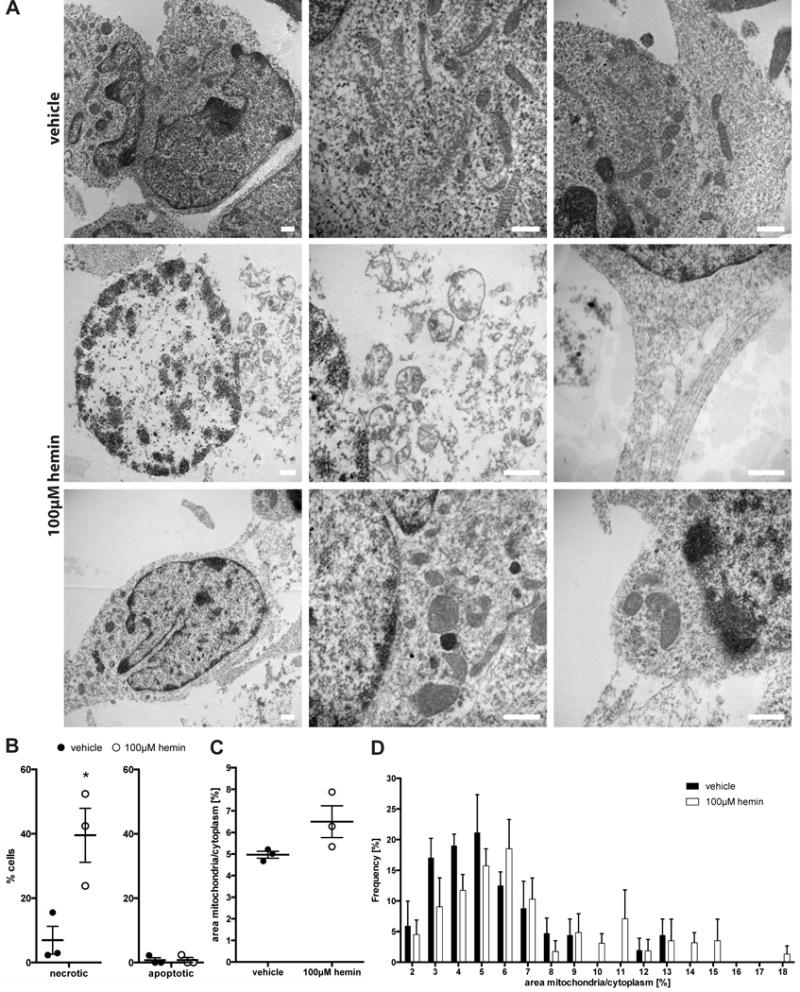
(A) Electron microscopic images of vehicle- (upper row) or hemin-treated cells (for 16 hours, middle and lower row). Some of the hemin-treated cells presented a necrotic phenotype with loss of plasma membrane integrity and disintegration of organelles (middle row, left and middle picture). Other hemin-treated cells displayed intact plasma membranes, but larger mitochondria (lower row, left and middle picture). Neuronal processes were no longer intact (middle and lower row, right pictures). Scale bars = 500 nm. (B) Hemin treatment increased the number of necrotic cells in culture. Note: we did not detect any apoptotic bodies in hemin-treated cells. Values represent mean ± S.E.M. (as this is the convention in the electron microscopy field). * p<0.05 versus vehicle. (C) Hemin treatment increased the mean percentage area of cytoplasm covered by mitochondria (left). Similarly, the frequency of appearance of larger mitochondria increased while the frequency of smaller mitochondria decreased (right). Values represent mean ± S.E.M.
As the sole positive morphologic criterion for ferroptosis is shrunken mitochondria, we quantified mitochondrial size following hemin treatment. We did not find any shrunken mitochondria in any of the cells examined. In contrast, mean mitochondrial size was larger in hemin-treated cells, although not statistically significant (Figure 4C); i.e. the proportion of larger mitochondria increased, while that of smaller mitochondria decreased (Figure 4D).
Hemin Induces Regulated Necrosis in Cortical Neurons by Recruiting Both Ferroptotic and Necroptotic Cell Death Mechanisms
As hemin-induced cell death shares pharmacological and molecular features of both ferroptosis and necroptosis, in the presence of ultrastructural features of necr(opt)osis, but not ferroptosis at 16 hours, we hypothesized that ferroptosis may induce an affector phase of death leading to a necroptotic effector phase. If this were true, we predicted that the necroptosis inhibitor should have a longer therapeutic window than the ferroptosis inhibitors and that inhibiting ferroptosis should abrogate the increase in RIP1 and RIP3 gene expression and phospho-RIP1.
To assess the therapeutic window, primary neurons were treated with chemical inhibitors starting every 2 hours between 0 and 12 hours following hemin treatment. Necrostatin-1 was protective when added up to 8 hours. Similarly, Ferrostatin-1, Trolox, and U0126 abrogated hemin toxicity up to 8 hours. In contrast, the ferroptosis inhibitors N-acetylcysteine and Deferoxamine had shorter protective windows of 2 and 4 hours, respectively (Figure 5A).
Figure 5. Ferroptosis is not upstream of necroptotic death signaling following hemin toxicity.
(A) Therapeutic window of protective ferroptosis and necroptosis inhibitors are shown. Primary neurons were treated with chemical inhibitors starting every 2 hours between 0 and 12 hours following hemin treatment and cell viability assessed at 16 hours. Values represent mean ± SD, except for Deferoxamine where medians are given. * p<0.05 versus hemin. (B) RIP1 and (C) RIP3 mRNA expression was measured in primary neurons exposed to hemin and U0126 or its inactive analog U0124 for 8 hours. Values represent medians. * p<0.05 versus vehicle without hemin. (D) Levels of phospho-RIP1 (normalized to β-actin) were measured in HT22 cells exposed to hemin and U0126 or 100ng/ml TNFα + 5μM z-VAD-fmk for 8 hours (positive control). Necrostatin-1 served as confirmation for specificity of RIP1 kinase activity. Values represent median. * p<0.05 versus 0 hours.
In line with the finding that the therapeutic window of Necrostatin-1 is not wider than that of the ferroptosis inhibitors, U0126 also failed to abrogate the increases in RIP1 (Figure 5B) and RIP3 (Figure 5C) gene expression and phospho-RIP1 (Figure 5D) following hemin treatment. Together, these data suggest that ferroptosis is not followed by downstream necroptosis in hemorrhagic stroke in vitro.
Alternatively, hemoglobin or hemin might elicit necroptotic (RIP1/3) or ferroptotic (phospho-ERK1/2) cell death mechanisms in parallel. If this is true, we expected that inhibition of both simultaneously would have an additive effect. Therefore, we investigated whether a sub-threshold dose of U0126 would have an additive or synergistic effect on the dose-response of Necrostatin-1 protection. We found that addition of 0.5μM U0126 did not change the profile of protection by Necrostatin-1 (Figure 6A,B). This suggests that there might be a common yet to be identified denominator onto which both ferroptosis and necroptosis converge which then leads to neuronal necrosis (Figure 6C).
Figure 6. Blood breakdown products induce regulated necrosis in cortical neurons by recruiting both ferroptotic and necroptotic cell death mechanisms.
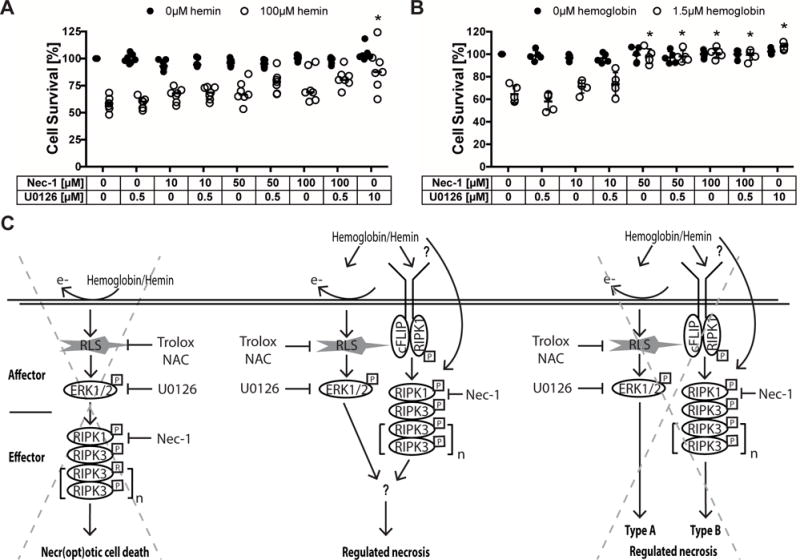
(A–B) Concentration-responses of Necrostatin-1 cotreated with U0126 (sub-threshold dose) in hemin (A) and hemoglobin (B) toxicity. Values represent medians for hemin and mean ± SD for hemoglobin. * p<0.05 versus hemin or hemoglobin alone. (C) Three alternative hypotheses suggesting how hemoglobin or its breakdown product hemin induces cell death with ultrastructural features of necrosis: (1) Hemoglobin or hemin generate reactive lipid species (RLS), which increase phospho-ERK1/2. Phospho-ERK1/2 induces RIP1 that executes necroptotic cell death via RIP3. In this case, hemorrhagic stroke would induce ferroptosis as the affector phase of death leading to a necroptotic effector phase. (2) Hemoglobin or hemin induces necroptotic (RIP1/3) and ferroptotic (phospho-ERK) cell death mechanisms independently and these then converge at a yet to be identified common denominator into a necrotic morphology. (3) Hemoglobin or hemin induces necroptotic (RIP1/3) and ferroptotic (phospho-ERK) cell death mechanisms that independently lead to different types of regulated necrosis. Our data supports the second hypothesis. NAC – N-acetylcysteine, Nec-1 – Necrostatin-1.
Discussion
The objective of this study was to use the leverage of an in vitro model of secondary injury in hemorrhagic stroke in cultured neurons to enhance understanding of cell death mechanisms following ICH that will allow more targeted and effective therapeutic strategies for this often fatal and highly morbid disease. We conclude that established toxic mediators from lysed blood, hemin and hemoglobin, induced death in primary cortical neurons via features of ferroptotic and/or necroptotic cell death based on several criteria.
First, with the exception of cycloheximide, a panel of inhibitors of ferroptosis abrogated hemin- and hemoglobin-induced death (Figures 1 and 2). The overlap between hemorrhagic stroke in vitro and ferroptosis in terms of pharmacological inhibitors was large enough that cell death induced by hemin in primary neurons can be considered ferroptotic. Second, biochemical features consistent with ferroptosis (enhanced phospho-ERK1/2) were observed in both in vitro and in vivo models of ICH (Figure 3). Of note, while shrunken mitochondria, a putative marker of ferroptosis, were not observed in our electron microscopy studies, we did observe ultrastructural features of necrosis/necroptosis (Figure 4). In line with these morphological changes, the canonical necroptosis inhibitor, Necrostatin-1, but not its inactive analog prevented hemin- or hemoglobin-induced death in cortical neurons (Figures 1 and 2) and biochemical features of necroptosis (enhanced RIP1 and RIP3 mRNA expression and phospho-RIP1) were detected (Figure 3). Altogether, these data support the conclusion that experimental ICH induces cell death with features of ferroptosis and necroptosis.
Our studies raised the possibility that hemin or hemoglobin induce a ferroptotic affector pathway of death leading to a necroptotic effector phase of cell death (Figure 6C). Contrary to this model, however, the therapeutic window for the necroptosis inhibitor Necrostatin-1 was not wider than the ferroptotic inhibitors Ferrostatin-1 and U0126. Moreover, U0126 did not abrogate the increase in RIP1 and RIP3 gene expression or phospho-RIP1 following hemin treatment (Figure 5). While we cannot definitively exclude the possibility that the therapeutic window reflects differences in the time required for ferroptotic and necroptotic inhibitors to reach steady-state intracellular concentrations in vitro, the data suggest that necroptotic or ferroptotic signaling induced by hemin or hemoglobin is sufficient to reach a threshold of death that leads to neuronal necrosis. Inhibition of either one of these pathways can bring neurons below that threshold to survival (Figure 6). To our knowledge hemin and hemoglobin are the first death stimuli described to involve a mixture of necroptotic and ferroptotic features.
Ferroptosis has not previously been shown to mediate ICH-induced cell death. However, four genes induced by a ferroptotic stimulus in cancer cells (i.e., iron response element binding protein 2 (Ireb2), ATP synthase F0 complex subunit C3 (ATP5G3), citrate synthase (Cs), and ATP ribosomal protein L8 (Rpl8)20 are also induced following ICH in mice.35 Furthermore, Regan and colleagues demonstrated that hemin activates ERK1/2 in astrocytes.36 This is in line with our findings demonstrating an increase in phospho-ERK1/2 in neurons in vitro following hemin exposure and in vivo following ICH (Figure 3).
Other studies support the idea that necroptosis is at least partly involved in ICH. In vitro, pretreatment with Necrostatin-1 protects astrocytes subjected to hemin treatment for 5 hours.37 In a mouse model of ICH, intracerebroventricular administration of necrostatin-1 reduces cell death, brain edema, hematoma volumes, and neurologic score deficits.38 Moreover, expression of RIP3 increases following ICH, while pretreatment with Necrostatin-1 diminishes the interaction of RIP3 with RIP1 and ameliorates cell death following ICH.39 Mice deficient in RIP3 show decreased cell death following ICH.40 This is consistent with our findings that RIP1 and RIP3 gene expression as well as phospho-RIP1 levels are increased following ICH in vitro and in vivo (Figure 3).
The apoptotic contribution to ICH-induced cell death has so far been unclear. Regan and colleagues showed that z-VAD-fmk reduced cell death of cortical astrocytes at 4 hours but not at 8 hours after hemin treatment.36 Conversely, Laird and colleagues demonstrated that pretreatment with z-VAD-fmk failed to protect after 5 hours of hemin toxicity in cortical astrocytes.37 Co-treatment with z-VAD-fmk increased cellular viability of microvascular cells after 18 hours of hemin treatment.41 In contrast, in human neuron-like cells exposed to hemin, z-VAD-fmk had no effect on cell viability.42 While, hemoglobin treatment of rat cortical neurons induced upregulation of cleaved caspase-3 and increased caspase-3-like enzyme activity, pretreatment with z-VAD-fmk and other caspase inhibitors did not prevent cell death.43 After ICH in vivo, immunohistochemical expression of cleaved caspase-3 increased in rats44 and mice38 as did expression of apoptosis-related genes in rats.45
Similarly, the role of autophagy following ICH is not clear. Yang and colleagues found that treatment of microglia with lysed blood in vitro resulted in TLR4-mediated autophagy. Activated microglia caused neuronal death that was attenuated by 3-methyladenine. When administered intracerebroventricularly immediately after ICH onset, 3-methyladenine reduced microglia activation and brain damage.46 Furthermore, ICH induced immunohistochemical expression of the autophagosome marker LC3-II in mice.38 Similarly, He and colleagues reported an increase in LC3-II, the lysosomal marker cathepsin D, and vacuole formation after ICH in rats.47
Surprisingly, when we investigated the morphological phenotype of cell death following hemin-induced neuronal toxicity, we found a necrotic phenotype with swelling of organelles and cell bodies and disruption of the plasma membrane. We did not observe any shrunken mitochondria, characteristic for ferroptosis, or apoptotic bodies (Figure 4). However, in animal models of ICH and in the perihematoma region after surgical evacuation in humans, both apoptotic and necrotic cells have been described.13, 14 Since our morphological analysis was confined to hemin toxicity in neurons, the contribution of other cell death mechanisms in other cell types in vivo cannot be excluded.
The lack of protection by canonical, broad caspase inhibitors in neurons exposed to hemin, along with the necrotic morphology seems to exclude apoptosis as a mechanism of death in vitro. Future studies will provide a head-to-head comparison of necroptotic, apoptotic, and ferroptotic inhibitors in vivo. It is likely that these studies will require intraventricular injections of drugs as the blood-brain barrier penetrability of many inhibitors is unclear. Our prediction is that ferroptotic and necroptotic inhibitors will be most effective. Consistent with this notion, the ferroptosis inhibitors Deferoxamine48 and N-acetylcysteine (unpublished results) provide significant functional recovery in some models of ICH.
Supplementary Material
Acknowledgments
We thank Linda M. Gerber for her advice on the statistical analysis.
Sources of Funding
This work was supported by grant DFG Zi 1613/1-1 to M.Z. from the German Research Foundation, funding from the Sheldon G and Dr. Miriam Adelson Medical Research Foundation to R.R.R., the Sperling Center for Hemorrhagic Stroke Recovery to R.R.R. and S.S.K., and NIH grants DA08259 and HL098351 to T.A.M.
Footnotes
Conflict of Interest
The authors declare the following competing financial interest: P.J.G., J.B., and J.F. are all employees and shareholders of GlaxoSmithKline.
Contribution of authors
M.Z. and R.R.R. designed all experiments, M.Z. performed most experiments, S.S.K. and Y.C. performed ICH surgery, S.S.K. performed gene expression experiments in vivo. P.J.G., J.B., and J.F. characterized and provided S166 phospho-RIP1 antibody. T.A.M. gave advice for and performed part of the electron microscopy experiment. E.A.J. advised in the electron microscopy analysis. M.Z. analyzed the data, performed statistical analysis and graphical artwork. M.Z. and R.R.R. wrote and edited the paper. All authors discussed the results and commented on the manuscript.
References
- 1.Feigin VL, Lawes CM, Bennett DA, Barker-Collo SL, Parag V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: A systematic review. The Lancet. Neurology. 2009;8:355–369. doi: 10.1016/S1474-4422(09)70025-0. [DOI] [PubMed] [Google Scholar]
- 2.van Asch CJ, Luitse MJ, Rinkel GJ, van der Tweel I, Algra A, Klijn CJ. Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: A systematic review and meta-analysis. The Lancet. Neurology. 2010;9:167–176. doi: 10.1016/S1474-4422(09)70340-0. [DOI] [PubMed] [Google Scholar]
- 3.Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. The Lancet. Neurology. 2012;11:720–731. doi: 10.1016/S1474-4422(12)70104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang YC, Zhou Y, Fang H, Lin S, Wang PF, Xiong RP, et al. Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann Neurol. 2014;75:876–889. doi: 10.1002/ana.24159. [DOI] [PubMed] [Google Scholar]
- 5.Wu J, Hua Y, Keep RF, Schallert T, Hoff JT, Xi G. Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Res. 2002;953:45–52. doi: 10.1016/s0006-8993(02)03268-7. [DOI] [PubMed] [Google Scholar]
- 6.Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: Role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- 7.Regan RF, Panter SS. Neurotoxicity of hemoglobin in cortical cell culture. Neurosci Lett. 1993;153:219–222. doi: 10.1016/0304-3940(93)90326-g. [DOI] [PubMed] [Google Scholar]
- 8.Zhao F, Song S, Liu W, Keep RF, Xi G, Hua Y. Red blood cell lysis and brain tissue-type transglutaminase upregulation in a hippocampal model of intracerebral hemorrhage. Acta Neurochir Suppl. 2011;111:101–105. doi: 10.1007/978-3-7091-0693-8_16. [DOI] [PubMed] [Google Scholar]
- 9.Deuel JW, Vallelian F, Schaer CA, Puglia M, Buehler PW, Schaer DJ. Different target specificities of haptoglobin and hemopexin define a sequential protection system against vascular hemoglobin toxicity. Free Radic Biol Med. 2015;89:931–943. doi: 10.1016/j.freeradbiomed.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 10.Zhao X, Song S, Sun G, Zhang J, Strong R, Zhang L, et al. Cytoprotective role of haptoglobin in brain after experimental intracerebral hemorrhage. Acta Neurochir Suppl. 2011;111:107–112. doi: 10.1007/978-3-7091-0693-8_17. [DOI] [PubMed] [Google Scholar]
- 11.Zhao X, Song S, Sun G, Strong R, Zhang J, Grotta JC, et al. Neuroprotective role of haptoglobin after intracerebral hemorrhage. J Neurosci. 2009;29:15819–15827. doi: 10.1523/JNEUROSCI.3776-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma B, Day JP, Phillips H, Slootsky B, Tolosano E, Dore S. Deletion of the hemopexin or heme oxygenase-2 gene aggravates brain injury following stroma-free hemoglobin-induced intracerebral hemorrhage. J Neuroinflammation. 2016;13:e26. doi: 10.1186/s12974-016-0490-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hickenbottom SL, Grotta JC, Strong R, Denner LA, Aronowski J. Nuclear factor-kappab and cell death after experimental intracerebral hemorrhage in rats. Stroke. 1999;30:2472–2477. doi: 10.1161/01.str.30.11.2472. discussion 2477–2478. [DOI] [PubMed] [Google Scholar]
- 14.Qureshi AI, Ling GS, Khan J, Suri MF, Miskolczi L, Guterman LR, et al. Quantitative analysis of injured, necrotic, and apoptotic cells in a new experimental model of intracerebral hemorrhage. Crit Care Med. 2001;29:152–157. doi: 10.1097/00003246-200101000-00030. [DOI] [PubMed] [Google Scholar]
- 15.Wang KY, Wu CH, Zhou LY, Yan XH, Yang RL, Liao LM, et al. Ultrastructural changes of brain tissues surrounding hematomas after intracerebral hemorrhage. Eur Neurol. 2015;74:28–35. doi: 10.1159/000434631. [DOI] [PubMed] [Google Scholar]
- 16.Qureshi AI, Suri MF, Ostrow PT, Kim SH, Ali Z, Shatla AA, et al. Apoptosis as a form of cell death in intracerebral hemorrhage. Neurosurgery. 2003;52:1041–1047. discussion 1047–1048. [PubMed] [Google Scholar]
- 17.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: Recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zille M, Farr TD, Przesdzing I, Muller J, Sommer C, Dirnagl U, et al. Visualizing cell death in experimental focal cerebral ischemia: Promises, problems, and perspectives. J Cereb Blood Flow Metab. 2012;32:213–231. doi: 10.1038/jcbfm.2011.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 20.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karuppagounder SS, Alim I, Khim SJ, Bourassa MW, Sleiman SF, John R, et al. Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates atf4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci Transl Med. 2016;8:e328ra329. doi: 10.1126/scitranslmed.aac6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 23.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 24.Milner TA, Waters EM, Robinson DC, Pierce JP. Degenerating processes identified by electron microscopic immunocytochemical methods. Methods Mol Biol. 2011;793:23–59. doi: 10.1007/978-1-61779-328-8_3. [DOI] [PubMed] [Google Scholar]
- 25.Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB, 3rd, Goebbels S, et al. Bcl-x l increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009;184:707–719. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen-Roetling J, Lu X, Regan RF. Targeting heme oxygenase after intracerebral hemorrhage. Ther Targets Neurol Dis. 2015;2:e474. doi: 10.14800/ttnd.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. Ras-raf-mek-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, et al. Identification of rip1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Declercq W, Vanden Berghe T, Vandenabeele P. Rip kinases at the crossroads of cell death and survival. Cell. 2009;138:229–232. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Christofferson DE, Li Y, Yuan J. Control of life-or-death decisions by rip1 kinase. Annu Rev Physiol. 2014;76:129–150. doi: 10.1146/annurev-physiol-021113-170259. [DOI] [PubMed] [Google Scholar]
- 32.Silke J, Rickard JA, Gerlic M. The diverse role of rip kinases in necroptosis and inflammation. Nat Immunol. 2015;16:689–697. doi: 10.1038/ni.3206. [DOI] [PubMed] [Google Scholar]
- 33.Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, et al. Cutting edge: Rip1 kinase activity is dispensable for normal development but is a key regulator of inflammation in sharpin-deficient mice. J Immunol. 2014;192:5476–5480. doi: 10.4049/jimmunol.1400499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, et al. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 2015;17:243–251. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang CF, Cho S, Wang J. (−)-epicatechin protects hemorrhagic brain via synergistic nrf2 pathways. Ann Clin Transl Neurol. 2014;1:258–271. doi: 10.1002/acn3.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Regan RF, Wang Y, Ma X, Chong A, Guo Y. Activation of extracellular signal-regulated kinases potentiates hemin toxicity in astrocyte cultures. J Neurochem. 2001;79:545–555. doi: 10.1046/j.1471-4159.2001.00590.x. [DOI] [PubMed] [Google Scholar]
- 37.Laird MD, Wakade C, Alleyne CH, Jr, Dhandapani KM. Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Radic Biol Med. 2008;45:1103–1114. doi: 10.1016/j.freeradbiomed.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 38.Chang P, Dong W, Zhang M, Wang Z, Wang Y, Wang T, et al. Anti-necroptosis chemical necrostatin-1 can also suppress apoptotic and autophagic pathway to exert neuroprotective effect in mice intracerebral hemorrhage model. Journal of molecular neuroscience : MN. 2014;52:242–249. doi: 10.1007/s12031-013-0132-3. [DOI] [PubMed] [Google Scholar]
- 39.Su X, Wang H, Kang D, Zhu J, Sun Q, Li T, et al. Necrostatin-1 ameliorates intracerebral hemorrhage-induced brain injury in mice through inhibiting rip1/rip3 pathway. Neurochem Res. 2015;40:643–650. doi: 10.1007/s11064-014-1510-0. [DOI] [PubMed] [Google Scholar]
- 40.Zhu X, Tao L, Tejima-Mandeville E, Qiu J, Park J, Garber K, et al. Plasmalemma permeability and necrotic cell death phenotypes after intracerebral hemorrhage in mice. Stroke. 2012;43:524–531. doi: 10.1161/STROKEAHA.111.635672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sukumari-Ramesh S, Laird MD, Singh N, Vender JR, Alleyne CH, Jr, Dhandapani KM. Astrocyte-derived glutathione attenuates hemin-induced apoptosis in cerebral microvascular cells. Glia. 2010;58:1858–1870. doi: 10.1002/glia.21055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldstein L, Teng ZP, Zeserson E, Patel M, Regan RF. Hemin induces an iron-dependent, oxidative injury to human neuron-like cells. J Neurosci Res. 2003;73:113–121. doi: 10.1002/jnr.10633. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Mori T, Sumii T, Lo EH. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: Caspase activation and oxidative stress. Stroke. 2002;33:1882–1888. doi: 10.1161/01.str.0000020121.41527.5d. [DOI] [PubMed] [Google Scholar]
- 44.Zhang D, Yuan D, Shen J, Yan Y, Gong C, Gu J, et al. Up-regulation of vcam1 relates to neuronal apoptosis after intracerebral hemorrhage in adult rats. Neurochemical research. 2015;40:1042–1052. doi: 10.1007/s11064-015-1561-x. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Q, Tang Q, Li X, Li J, Zhang L, Yan C, et al. Effects of intracerebral hemorrhage and subsequent minimally invasive hematoma aspiration on expression of apoptosisrelated genes in rats. International journal of clinical and experimental pathology. 2015;8:5371–5378. [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Z, Liu B, Zhong L, Shen H, Lin C, Lin L, et al. Toll-like receptor-4-mediated autophagy contributes to microglial activation and inflammatory injury in mouse models of intracerebral haemorrhage. Neuropathol Appl Neurobiol. 2015;41:e95–106. doi: 10.1111/nan.12177. [DOI] [PubMed] [Google Scholar]
- 47.He Y, Wan S, Hua Y, Keep RF, Xi G. Autophagy after experimental intracerebral hemorrhage. J Cereb Blood Flow Metab. 2008;28:897–905. doi: 10.1038/sj.jcbfm.9600578. [DOI] [PubMed] [Google Scholar]
- 48.Okauchi M, Hua Y, Keep RF, Morgenstern LB, Schallert T, Xi G. Deferoxamine treatment for intracerebral hemorrhage in aged rats: Therapeutic time window and optimal duration. Stroke. 2010;41:375–382. doi: 10.1161/STROKEAHA.109.569830. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.