

Abstract
Native polyacrylamide gel electrophoresis is a fundamental tool of molecular biology that has been used extensively for the biochemical analysis of RNA-protein interactions. These interactions have been traditionally analyzed with polyacrylamide gels generated between two glass plates and samples electrophoresed vertically. However, polyacrylamide gels cast in trays and electrophoresed horizontally offers several advantages. For example, horizontal gels used to analyze complexes between fluorescent RNA substrates and specific proteins can be imaged multiple times as electrophoresis progresses. This provides the unique opportunity to monitor RNA-protein complexes at several points during the experiment. In addition, horizontal gel electrophoresis makes it possible to analyze many samples in parallel. This can greatly facilitate time course experiments as well as analyzing multiple reactions simultaneously to compare different components and conditions. Here we provide a detailed protocol for generating and using horizontal native gel electrophoresis for analyzing RNA-Protein interactions.
Keywords: Biochemistry, Issue 125, Native gel electrophoresis, RNA-protein interactions, mobility shift assays
Introduction
Electrophoretic mobility shift assays (EMSAs) have proven to be an invaluable biochemical tool to analyze specific protein-nucleic acid interactions1,2,3. These assays can provide important information regarding the binding affinity of proteins to RNA or DNA3, the component stoichiometry of nucleic acid-protein complexes1 and provide important new insights about the binding specificity of RNA binding proteins via substrate competition experiments1.
The traditional experimental setup for these assays consists of mixing purified protein with a radiolabeled RNA substrate. The resulting complexes are then analyzed with non-denaturing (native) polyacrylamide gels poured between two glass plates followed by sample electrophoresis in a vertical apparatus3. While this approach has been used exhaustively to provide important insights in the biochemical mechanisms that underlie the binding of proteins to nucleic acids, it also has several limitations. For example, this basic strategy has relatively low throughput and it is not readily adaptable for applications that require analyzing many binding reactions in parallel. In addition, with the traditional vertical apparatus it is challenging to potentially monitor complexes at multiple times during electrophoresis3,4.
Here we present an adaptation of the EMSA assay that uses native polyacrylamide gels cast in a flatbed apparatus, horizontal electrophoresis and fluorescently labeled RNA substrates4,5,6,7. The incorporation of these relatively simple modifications to the basic strategy provides some powerful advantages. In particular, the horizontal flatbed format easily lends itself to analyzing dozens of samples simultaneously4. Also, for some RNA-protein complexes, such as those formed between the Bicaudal-C protein and its RNA substrate electrophoresis in a horizontal gel provides an increased ability to resolve distinct RNA-protein complexes and discriminate these from unbound RNA substrate.
Protocol
1. Preparation of the Horizontal Native Polyacrylamide Gel4.
- Preparation of the materials needed.
- For the horizontal gel apparatus, use a gel box (37 cm x 24 cm) with a 27 cm x 21 cm tray and a capacity for two 24-well combs. This setup provides a total of 48 samples that can be analyzed simultaneously.
- Prepare the following reagents: 40% Acrylamide-Bis 19:1, 5x TBE (Tris-Borate-EDTA pH 8.0), TEMED, and 10% APS (ammonium persulfate).
- Generation of a 10% native acrylamide gel.
- In a 500 mL flask, add 80 mL of 5x TBE, 100 mL of 40% acrylamide, and water to a final volume of 400 mL.
- Add 4 mL of 10% APS and 400 µL of TEMED. Mix well.
- Pour the mix into the horizontal casting tray and allow the gel to polymerize for 30 min. The gel will be approximately 2 cm thick. NOTE: Gels can cast in a fume hood to speed up polymerization. After polymerization, a thin layer of liquid will remain on top of the gel.
- Pour the liquid off and rinse with running buffer. Remove the comb(s) carefully and rinse the wells with running buffer using a syringe.
- Preparation of running buffer.
- Prepare 2 L of 1x TBE running buffer for the gel box. Dilute 400 mL of 5XTBE with 1600 mL and mix. Place 1x TBE buffer in cold room to cool.
- Pre-Running the gel
- Add 2 L of cold running buffer to the gel box and insert the gel.
- Pre-run the gel at 120 V for 1 h at 4 °C in a cold room. During this time, prepare the binding reaction.
2. Binding Reaction8
2.1.Prepare the following reaction components. 20 mM DTT 10 mM BSA 10 mM yeast tRNAs 5X binding buffer (50 mM HEPES pH 8.0, 5 mM EDTA, 250 mM KCl, 0.2% Tween-20) 100 nM fluorescein labeled RNA purchased commercially Concentrated Bicaudal-C protein 50% glycerol in DEPC-treated H2O DEPC-treated H2O
- Assemble the binding reaction components in an RNase free microcentrifuge tube.
- To the tube, add 5 µL of 10 mM BSA, 5 µL of 20 mM DTT, 5 µL of 10 mM yeast tRNAs, 10 µL of 5x Binding Buffer.
Add protein to a final concentration of 250 nM in 50 µL. Add water to a final volume of 45 µL.
Add 5 µL of fluorescently labeled RNA substrate. Use a commercially purchased 3' fluorescein labeled 32-nucleotide substrate.
Mix and incubate in the dark for 30 min at room temperature.
3. Analysis of Samples on the Native Horizontal Polyacrylamide Gel
To each reaction, add 15 µL of 50% glycerol and mix gently.
Carefully add 30 µL of each reaction to the gel. The amount of sample that can be loaded into a single well varies depending on the thickness of the gel and the size of the wells. We have loaded volumes as little as 15 µL and as much as 45 µL into a single well of gels created with a 24-well comb and poured to a thickness of 2 cm.
- After connecting the power supply to the gel apparatus switch on the power and adjust the voltage. Run the gel in 4 °C at 120 V. NOTE: For the horizontal gel apparatus, this ends up being approximately 5 V/cm. For the vertical gel apparatus, this is 16 V/cm.
- To prevent damaging or bleaching of the fluorescently labeled RNA, conduct electrophoresis in the dark.
- Vary electrophoresis times depending upon the specifics of the RNA-protein complex being analyzed. NOTE: A major advantage of the horizontal EMSA analysis is that electrophoresis can be stopped, the gel imaged and then the gel can be placed back into the apparatus and electrophoresis can be continued for longer times.
4. Imaging the Gel
Perform analysis with any instrument designed for detection and imaging fluorescent substrates.
Place the gel on the imager.
Adjust imager settings. For fluorescein-labeled RNA ligands, use the following settings: FAM (wavelength 473 nm), 750 Photomultiplier voltage (PMT), 100 µm pixel size.
Scan the gel to capture image.
Representative Results
To demonstrate the power and versatility of the horizontal native gel electrophoresis we analyzed the binding of the Xenopus Bicaudal-C (Bicc1) protein to a fluorescently labeled RNA containing a Bicc1 binding site. Bicc1 proteins function as mRNA-specific translational repressors to control cell fate decisions during the maternal stages of animal development9,10,11,12 as well as the functions of specific organs in adults13,14. Our work in Xenopus identified the first binding site for a Bicc1 protein8: a 32-nucleotide region from the 3'UTR of the Cripto-1 mRNA8,12. This binding site was defined using a combination of techniques: RNase protection and footprinting assays, in vivo binding experiments and in vitro EMSA assays8.
Purified Xenopus Bicc1 protein was mixed with a fluorescently labeled 32 nucleotide RNA; the Bicc1 binding site from the Cripto-1 mRNA8. In a separate reaction Bicc1 protein was mixed with a fluorescently labeled 32 nucleotide RNA derived from the 3'UTR of the cyclin B1 mRNA. Bicc1 does not bind to the cyclin B1 mRNA8,12 and this RNA serves as a negative control. Half of each reaction was analyzed on a vertical native polyacrylamide gel (Figure 1A) while the other portion was analyzed in parallel using horizontal native gel (Figure 1B). It is readily apparent using either method that Bicc1 binds to the RNA Cripto-1 RNA and forms a specific complex and it does not bind the cyclin B1 negative control RNA. However, the Bicc1-Cripto-1 complexes are significantly more distinct when analyzed with the horizontal gel, both facilitating their detection and allowing the discrimination of protein-RNA complexes from unbound RNA. After imaging, the gel was placed back into the gel apparatus and electrophoresed for an additional three and a half hours. After re-imaging the gel the complexes had migrated further and were still readily detectable, indicating their stability (Figure 1C). This ability to continue electrophoresis and analysis after initial imaging would be challenging, if not impossible, with the vertical gel format.
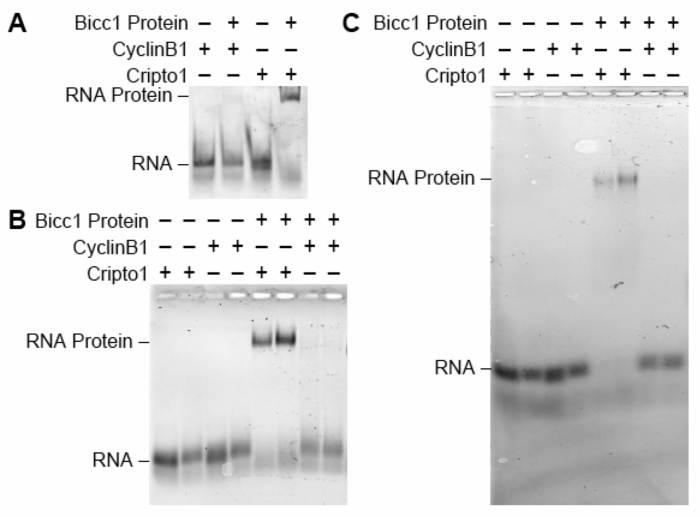
Figure 1: A comparison of the vertical and the horizontal native gels for analyzing the binding of Bicc1 to Cripto1 RNA. Bicc1 binding experiments were conducted with a 32-nucleotide fluorescent CyclinB1 RNA negative control or a fluorescent 32-nucleotide Cripto1 RNA positive control. Each reaction contained either 0 nM or 250 nM of SUMO-Bicc1 N-terminal and analyzed on either a (A) vertical polyacrylamide native gel run for 30 min at 120 V at 4 °C or as duplicate samples in the horizontal polyacrylamide native gel for (B) 2 h at 120 V at 4 °C or (C) 5.5 h at 120 V at 4 °C. Please click here to view a larger version of this figure.
Discussion
Native polyacrylamide gels are an invaluable tool for investigating protein-RNA interactions and traditionally these gels are electrophoresed vertically2,3. We have used a modification of the protocol that substitutes native polyacrylamide gels created and electrophoresed horizontally1,4,6,7,15. These changes used in combination with fluorescently labeled RNA substrates provide numerous advantages for the biochemical analysis of protein-RNA binding interactions. These advantages include the following: mixing of buffers is simplified compared to a vertical apparatus which requires a pump to transfer buffer from the lower chamber to the upper, horizontal gels can be generated with larger wells that provide an increased capacity for analyzing larger samples, electrophoresis on horizontal gels often provide improved resolution of RNA-protein complexes compared to vertical gels, and horizontal gel electrophoresis provides the ability to image experiments at multiple time points during analysis.
Another major advantage is that the horizontal format lends itself to creating gels with a large number of wells that provides the capacity to analyze multiple samples in parallel1. This feature significantly expands their use as a biochemical tool for analyzing RNA-protein interactions. For example, the ability to analyze multiple samples facilitates investigating the quantitative parameters of protein-RNA complex formation through the use of competition experiments15, koff analysis15 as well as experiments designed to define the stoichiometry of the binding complex components15. In addition, the use of a fluorescent RNA substrates makes it possible to perform multiple analyses, such as fluorescence polarization binding assays from a single reaction and obtain both quantitative and qualitative binding data5,15.
To obtain optimal results from the horizontal gel setup, there are a variety of parameters that can be adjusted to improve the quality of the complexes for imaging. These include changing the acrylamide percentage of the gel in order to make it easier to handle and enhance the separation of protein-RNA complexes from free RNA more. However, it can be difficult to handle low percentage acrylamide gels due to the size. We find it easiest to use gels that have an acrylamide concentration of 7.5% or greater. Also, if necessary the electrophoresis conditions can be changed to improve stability of protein-RNA complexes; adjusting the electrophoresis voltage, running the gel at room temperature instead of at 4 °C and adjusting the pre-run conditions.
Despite the many advantages there are also some limitations of horizontal native gel electrophoresis assays. For example, its utility is maximized by using a fluorescent RNA substrates. Depending on the fluorophore chosen and the size of RNA, obtaining such substrates can be expensive. Additionally, horizontal gels are generally much larger than vertical gels and therefore require larger amounts of material such as polyacrylamide and DEPC-treated solutions.
Horizontal native gel electrophoresis has the potential to be used as a validation tool for molecular screens that seek to identify compounds that target clinically relevant RNA-protein interactions16. Such screens typically use solution biochemical assays to identify relevant compounds for further study. The higher throughput potential of the horizontal format compared to the vertical format provides an opportunity to apply native gel electrophoresis as a tool for secondary validation of potential candidates.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Laura Vanderploeg for preparing figures. Work in the Sheets lab is supported by NSF grant 1050395 and NIH grant (R21HD076828). Work in the Ryder lab is supported by NIH grants R01GM117237 and R01GM117008. Megan Dowdle is supported by a SciMed GRS Advanced Opportunity Fellowship through University of Wisconsin-Madison Graduate School and Biotechnology Training Program through the National Institute of General Medical Sciences of the National Institutes of Health (T32GM008349).
References
- Ryder SP, Recht MI, Williamson JR. Quantitative analysis of protein-RNA interactions by gel mobility shift. Methods Mol Biol. 2008;488:99–115. doi: 10.1007/978-1-60327-475-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg AE, Dingman CW, Peacock AC. Electrophoretic characterization of bacterial polyribosomes in agarose-acrylamide composite gels. J Mol Biol. 1969;41(1):139–147. doi: 10.1016/0022-2836(69)90131-4. [DOI] [PubMed] [Google Scholar]
- Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc. 2007;2(8):1849–1861. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano JM, Farley BM, Essien KI, Ryder SP. RNA recognition by the embryonic cell fate determinant and germline totipotency factor MEX-3. Proc Natl Acad Sci USA. 2009;106(48):20252–20257. doi: 10.1073/pnas.0907916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer CA, Scarlata SF. Fluorescence approaches to quantifying biomolecular interactions. Meth Enzymol. 2008;450:79–106. doi: 10.1016/S0076-6879(08)03405-8. [DOI] [PubMed] [Google Scholar]
- Su C, Wang F, Ciolek D, Pan YC. Electrophoresis of proteins and protein-protein complexes in native polyacrylamide gels using a horizontal gel apparatus. Anal Biochem. 1994;223:93–98. doi: 10.1006/abio.1994.1552. [DOI] [PubMed] [Google Scholar]
- Buczek P, Horvath MP. Thermodynamic Characterization of Binding Oxytricha nova Single Strand Telomere DNA with the Alpha Protein N-terminal Domain. J Mol Biol. 2006;359(5):1217–1234. doi: 10.1016/j.jmb.2006.02.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Park S, Blaser S, Sheets MD. Determinants of RNA binding and translational repression by the Bicaudal-C regulatory protein. J Biol Chem. 2014;289(11):7497–7504. doi: 10.1074/jbc.M113.526426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamberi C, Lasko P. The Bic-C family of developmental translational regulators. Comp Funct Genomics. 2012. p. 141386. [DOI] [PMC free article] [PubMed]
- Saffman EE, et al. Premature translation of oskar in oocytes lacking the RNA-binding protein bicaudal-C. Mol Cell Biol. 1998;18(8):4855–4862. doi: 10.1128/mcb.18.8.4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicoine J, et al. Bicaudal-C recruits CCR4-NOT deadenylase to target mRNAs and regulates oogenesis, cytoskeletal organization, and its own expression. Dev Cell. 2007;13(5):691–704. doi: 10.1016/j.devcel.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. Bicaudal-C spatially controls translation of vertebrate maternal mRNAs. RNA. 2013;19(11):1575–1582. doi: 10.1261/rna.041665.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonneuve C, et al. Bicaudal C, a novel regulator of Dvl signaling abutting RNA-processing bodies, controls cilia orientation and leftward flow. Development. 2009;136(17):3019–3030. doi: 10.1242/dev.038174. [DOI] [PubMed] [Google Scholar]
- Tran U, et al. The RNA-binding protein bicaudal C regulates polycystin 2 in the kidney by antagonizing miR-17 activity. Development. 2010;137(7):1107–1116. doi: 10.1242/dev.046045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano JM, Clingman CC, Ryder SP. Quantitative approaches to monitor protein-nucleic acid interactions using fluorescent probes. RNA. 2011;17(1):14–20. doi: 10.1261/rna.2428111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley TL, et al. Platform to Enable the Pharmacological Profiling of Small Molecules in Gel-Based Electrophoretic Mobility Shift Assays. J Biomol Screen. 2016;21(10):1125–1131. doi: 10.1177/1087057116652895. [DOI] [PMC free article] [PubMed] [Google Scholar]