

Abstract
Sclerostin is an osteocyte-derived glycoprotein that inhibits Wnt/β-catenin signaling and activation of osteoblast function, thereby inhibiting bone formation. It plays a vital role in the regulation of skeletal growth. In adults, sclerostin secretion is modulated by skeletal loading (increased secretion with immobilization; less with weight bearing) and by hormonal/cytokine actions on the osteocyte. Sclerostin deficiency syndromes in humans and animals are characterized by high bone mass of normal quality. In animal models of osteoporosis, inhibition of sclerostin by monoclonal antibodies induces osteoblast activity and new bone formation, normalizing bone mass and improving bone architecture and strength. In recently completed clinical trials, anti-sclerostin antibody therapy results in marked increases in bone mineral density and rapid and substantial reduction in fracture risk. This review will focus on these recent studies and anticipate the role of anti-sclerostin therapy in the management of patients with osteoporosis.
Keywords: antibody, blosozumab, fractures, romosozumab, sclerostin
Osteoporosis is a disorder of skeletal fragility which is the consequence of bone loss, usually in postmenopausal women or older men, resulting in low bone mass and deranged skeletal architecture that compromises bone strength.1 Most of our current therapies for osteoporosis inhibit bone resorption by osteoclasts and indirectly inhibit bone formation.2 While these therapies can strengthen the skeleton and reduce fracture risk, they do not correct the structural abnormalities that characterize patients with established osteoporosis. Only therapeutic agents that enhance bone formation will be able to do that.
The elucidation of the pivotal role of sclerostin as a modulator of osteoblastic activity and bone formation led to the concept that inhibiting sclerostin would be an attractive strategy to treat osteoporosis, a topic that was thoroughly reviewed in this journal by Dr Lewiecki in 2014.3 Since then, additional preclinical evidence has emerged and several clinical studies, including two phase III fracture endpoint trials, have been completed, making it timely to provide an update on the potential role of anti-sclerostin therapy in the management of patients with osteoporosis.
Clinical disorders of sclerostin deficiency
Homozygous genetic deficiency of sclerostin results in the syndromes of sclerosteosis and van Buchem disease.4 Sclerosteosis is due to loss of function mutations of the SOST gene on chromosome 17q12-21. There are no mutations in the SOST gene with van Buchem disease, but patients have a noncoding deletion, downstream of the SOST gene, that is required for that gene’s transcription in bone. The two diseases are phenotypically similar. In sclerosteosis, affected individuals appear normal at birth except for syndactyly. Bony overgrowth results in mandibular hypertrophy and, in the skull, frontal bossing, narrowing of the foramina causing cranial nerve entrapment with facial palsy and/or deafness, and decreased intracranial volume and cranial basilar stenosis with elevation of intracranial pressure in some cases. Patients with van Buchem disease do not have syndactyly and generally have milder clinical features.
The group led by Dr Papapoulos has demonstrated that heterozygotes of these conditions, with low but not absent levels of sclerostin, have a normal phenotype except for high bone mass.4 In addition, that group has shown improved quality of the bone matrix in sclerostin-deficient mice and humans, with increased heterogeneity of matrix mineralization density and higher levels of matrix proteoglycans.5 Furthermore, unlike most other conditions of high bone mass or osteosclerosis, average matrix mineralization density is decreased relative to normal controls. These alterations in bone composition are beneficial for bone strength, consistent with the lack of bone brittleness in patients with sclerosteosis. These results also imply that the increases in bone mineral density (BMD) observed by dual energy X-ray absorptiometry measurements in clinical trials may underestimate the true increase in bone mass induced with anti-sclerostin treatment. Taken together, this information adds to the promise that inhibiting sclerostin may be an effective and hopefully safe treatment for osteoporosis and other bone disorders.
Preclinical studies
Dr Lewiecki reviewed the early studies in rats and monkeys in which sclerostin-inhibiting antibodies increased bone formation and decreased bone resorption, leading to improved or normalized bone structure, bone mass and bone strength while maintaining bone quality in these animal models of osteoporosis.3 Trabecular and cortical bone thickness was increased, and cortical porosity was reduced. More recent studies have defined the structural changes induced by anti-sclerostin therapy. In cynomolgus monkeys, the anabolic response to anti-sclerostin antibody increased the thickness of trabeculae in all three spatial orientations (axial, oblique, and transverse) and converted rod-like structures into more mechanically sound plate-like structures.6,7 Additionally, the complex kinetics of bone formation and resorption in response to anti-sclerostin therapy have been clarified. The initial response to therapy is a direct activation of quiescent bone lining cells, acutely increasing the number of functioning osteoblasts.8 This results in a marked but transient increase in modeling-based bone formation on previously quiescent trabecular, endocortical as well as periosteal surfaces, accompanied by a decrease in bone resorption. This is then followed by an attenuation of modeling-based bone formation with a persistent decrease in remodeling space and a positive bone balance, resulting in a slower but progressive increase in trabecular BMD with long-term treatment. In cortical bone, anti-sclerostin therapy substantially increased bone formation and bone mass which correlated with increases in estimated strength. No changes in calculated material properties were observed. At the radial diaphysis, a transient, reversible 2% reduction in cortical BMD was observed with therapy despite relative improvements in bone mineral content. High-resolution peripheral quantitative computed tomography (QCT) confirmed that this decline in radial cortical BMD was a due to increased intracortical remodeling but with no increase in cortical porosity. As mentioned previously, this decrease in BMD may reflect the lower mineralization density of the large volume of newly formed bone. Finite element modeling demonstrated maintenance of radial diaphyseal strength and improved strength at metaphyseal sites.
Preclinical studies with particular relevance to the potential clinical use of anti-sclerostin therapy include the observation that the anabolic response to sclerostin antibody therapy was as robust in aged mice as in younger mice.9 Additionally, the anabolic effect of therapy was unaffected by previous or simultaneous treatment with a bisphosphonate and could be restored after a short treatment-free interval.6,10 Relevant to the phase III study design, the increases in bone mass achieved with sclerostin antibody treatment were maintained or improved when that therapy was followed by a receptor activator of nuclear factor κ B inhibitor.6
As Dr Lewiecki pointed out, sclerostin is expressed in chondrocytes and synovial cells, raising the possibility that sclerostin plays a role in inflammatory or degenerative joint diseases.3 Sclerostin-deficient mice developed severe osteoarthritis in response to joint instability, raising the possibility that sclerostin inhibition might be deleterious to cartilage.11 More recently, inhibition of sclerostin has been demonstrated to accelerate disease in a mouse model of rheumatoid arthritis.12
There was also concern about a potential carcinogenic risk with anti-sclerostin therapy since teriparatide, another bone-forming agent, has been associated with dose-related increases in osteosarcoma in rats and since canonical Wnt signaling is involved in the control of cellular proliferation in many tissues. In a rat lifetime carcinogenic toxicity study with romosozumab, an anti-sclerostin antibody being studied in clinical trials, no treatment-related effects on tumor incidence were observed.13
Phase I studies
Dr Lewiecki described in detail the results of single and multiple dose phase I studies with romosozumab, a humanized monoclonal IgG2 antibody with high specificity for human sclerostin, and with blosozumab, an IgG4 humanized monoclonal anti-sclerostin antibody.3 Studies with both agents demonstrated rapid and marked increases in biochemical markers of bone formation, decreases in bone resorption markers and increases in BMD. The effects of romosozumab on trabecular and cortical structural parameters were also assessed by QCT and high-resolution QCT scans of the lumbar and thoracic spine in 48 subjects (32 women, 16 men) with low bone mass in a placebo-controlled phase Ib study.14 Women received active treatment of 1 or 2 mg/kg every 2 weeks or 2 or 3 mg/kg every 4 weeks, while men were given 1 mg/kg every 2 weeks or 3 mg/kg every 2 weeks for 3 months. All active treatment groups were combined for analyses. Subjects were followed off therapy for an additional 3 months. At 3 months, significant increases were observed in trabecular BMD (9.5%) and apparent density-weighted cortical thickness and cortical stiffness (26.9%). These improvements in structural parameters were maintained during the 3-month off-treatment follow-up period.
Phase II studies
The phase II romosozumab dose-ranging study [ClinicalTrials.gov identifier: NCT00896532] evaluated treatment effects in 419 postmenopausal women aged 55–85 with low bone mass who were randomly assigned to receive subcutaneous doses of romosozumab ranging from 70 mg every 3 months to 210 mg every month or placebo injections.15 Other patients were randomly assigned to receive open-label subcutaneous teriparatide 20 µg daily or alendronate 70 mg weekly. At 12 months, the average increases in BMD at the lumbar spine (LS) and total hip (TH) were 11.3% and 4.1% respectively with the 210 mg every month dose of romosozumab. These increases were significantly greater than those observed with teriparatide or alendronate. BMD at the 1/3 radial site decreased by 0.9% with placebo, 1.3% with romosozumab and 1.7% with teriparatide.
Bone formation markers rose promptly, peaked at 1–3 months, returned to baseline by 6 months and were then below baseline for the remainder of the 12-month treatment interval. This pattern of response is very similar to the timing of the anabolic effect of romosozumab in cynomolgus monkeys.8 During the second year of the study, markers of bone formation and resorption remained below baseline in patients who continued romosozumab.16 Consistent with these results, LS BMD increased 3.8% during the second year of romosozumab therapy, which is a smaller increase than had occurred in the first year.
After 2 years, romosozumab therapy was discontinued. Patients were randomized to receive denosumab 60 mg subcutaneously every 6 months or placebo for 12 months. In those who received placebo, BMD values decreased to or toward baseline values.16 Formation markers also returned to baseline values, while serum C-terminal telopeptide of type 1 collagen (CTX) levels rose to values significantly greater than baseline before falling toward pretreatment values. In patients who were switched to denosumab, BMD increased in LS (3.7%) and TH (1.1%), increments that were similar to the increases during the second year of romosozumab therapy.
Volumetric BMD was assessed by QCT in a subset of patients from the romosozumab phase II study.17 Increases in integral volumetric BMD in both the LS and TH were significantly greater after 12 months of therapy with romosozumab compared with teriparatide (Table 1).
Table 1.
Percent changes from baseline in areal and volumetric BMD at 12 months in a subset of patients in the Phase II romosozumab study.17
| Dose | Placebo |
Teriparatide |
Romosozumab |
|---|---|---|---|
| Not applicable | 20 μg/day | 210 mg/month | |
| Areal BMD | |||
| LS | −0.4% | 6.9% | 12.3% |
| TH | −0.7% | 0.8% | 3.9% |
| Volumetric BMD | |||
| LS | 0.8% | 12.9% | 17.7% |
| TH | 0.3% | 1.2% | 4.1% |
BMD, bone mineral density; LS, lumbar spine; TH, total hip.
In a phase II dose-ranging study and its extension evaluating the effect of blosozumab, 120 postmenopausal women with low BMD were randomly assigned to receive that drug in subcutaneous doses of 180 mg every 4 weeks, 180 mg every 2 weeks, 270 mg every 2 weeks or placebo.18 Progressive dose-dependent increases in BMD were observed over the 12-month treatment. Compared with placebo, the average increase in BMD was 17.7% in the LS, 6.2% in the TH and 7.3% in the total body with the dose of 270 mg every 2 weeks. As with romosozumab, the top of the dose–response curve for BMD was not identified. BMD at the 1/3 radius site did not change significantly.
Markers of bone turnover changed in patterns similar to those observed with romosozumab. Serum type 1 procollagen N-terminal peptide increased to a peak of 160% above baseline at 4 weeks with the dose of 270 mg blosozumab every 2 weeks. With continued therapy, the values then gradually decreased, remaining above baseline at 6 months but falling to near baseline at 12 months. Serum CTX decreased promptly after starting therapy to as much as 40% below baseline after 2 weeks of treatment. As was seen with the larger doses of romosozumab, CTX returned to baseline at week 12 and thereafter remained below baseline and lower than values in the placebo group.
After 12 months, blosozumab therapy was discontinued and 106 patients entered a 12-month follow-up study to evaluate the effects of stopping therapy.19 BMD decreased significantly in all treatment groups. In the highest dose group, BMD fell to 6.9% and 3.9% above baseline and was 8.2% and 5.2% greater than in the placebo group in the LS and TH respectively at the end of the follow-up period. Bone formation markers remained near baseline during the year off therapy and serum CTX returned to baseline within 4 weeks of stopping therapy. During the follow-up period, fractures of the rib, humerus or elbow occurred in three patients who had taken blosozumab 180 mg every 4 weeks, but no clinical vertebral fractures were reported.
Phase III studies
The Fracture Study in Postmenopausal Women with Osteoporosis (FRAME) trial [ClinicalTrials.gov identifier: NCT01575834] evaluated the effectiveness of romosozumab 210 mg every month compared with placebo for 12 months followed in both treatment groups by an additional 12 months of denosumab 60 mg every 6 months in 7180 women with postmenopausal osteoporosis whose average age was 71 years.20 About 18% had prevalent vertebral fracture (almost all were mild or grade 1 deformities) and 21.7% had a history of prior nonvertebral fracture. The geographic distribution of patients in this study was different than previous fracture endpoint trials. Forty-three percent of patients were from Latin America and 29% were from Central or Eastern Europe. Eighty-nine percent of patients completed 12 months of the trial and 83.9% completed the 2-year follow up. During 12 months of romosozumab therapy, new vertebral fractures occurred in 1.8% of the placebo group and 0.8% with romosozumab, a relative risk reduction of 73% [95% confidence interval (CI) 5–84%; p < 0.001] (Table 2). As noted in Table 2, the treatment effect was particularly evident during the second 6 months of therapy. At the end of the second year during which all patients had received open-label denosumab therapy, new vertebral facture risk was reduced by 75% in the patents who had initially received romosozumab compared with the group that had received placebo. During the second year, 25 of 3327 in the placebo group experienced a new fracture while only 5 of 3325 patients in the romosozumab group did so (Table 2).
Table 2.
Numbers of patients with new morphometric vertebral fractures in Phase III FRAME study.20
| Placebo n = 3321 |
Romosozumab n = 3322 |
Risk ratio | p value | |
|---|---|---|---|---|
| 0–6 months | 26 | 14 | ||
| 6–12 months | 33 | 2 | ||
| 0–12 months | 59 (1.8%) | 16 (0.5%) | 0.27 | <0.001 |
| n = 3327 | n = 3325 | |||
| 12–24 months | 25 | 5 | ||
| 0–24 months | 84 (2.5%) | 21 (0.6%) | 0.25 | <0.001 |
Clinical fracture risk was reduced by 36% (2.5% with placebo and 1.6% with romosozumab; p = 0.008) at 12 months. Nonvertebral fracture risk decreased by 25%, from 2.1% with placebo to 1.6% with romosozumab (hazard ratio 0.75; 95% CI 0.53–1.05; p = 0.10). In a preplanned subgroup analysis, significant interaction of nonvertebral fracture risk reduction with geography was observed. This was explored in more detail in a post hoc analysis which demonstrated very low fracture risk (assessed by the FRAX fracture risk prediction tool) and fracture incidence in the Latin American subgroup, consistent with previous epidemiological studies from that region.21 In that Latin American cohort, no treatment effect on nonvertebral fracture risk was noted, while in the remainder of the study population, a significant 42% risk reduction was observed. Romosozumab was associated with BMD increases of 13.3% and 6.8% in the LS and TH respectively compared with baseline and with placebo at 12 months (Figure 1). After 12 additional months of denosumab therapy, the total increases in LS and TH BMD from the original baseline were 17.6% and 8.8% respectively (Figure 1). These results of the FRAME study have been filed with American and European regulatory agencies, and decisions about registration are expected soon.
Figure 1.
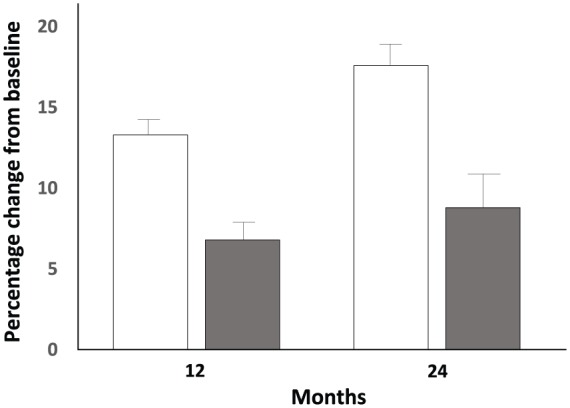
Percent changes from baseline in BMD at 12 and 24 months in Phase III FRAME study.20 Patients received romosozumab 210 mg Q6M during year 1 and denosumab 60 mg Q6M during year 2. Open bars: lumbar spine BMD; Shaded bars: total hip BMD.
A second phase III fracture endpoint trial (ARCH Study) [ClinicalTrials.gov identifier: NCT01631214] compared the effects of subcutaneous romosozumab 210 mg every month with oral alendronate 70 mg weekly for 12 months, followed by another year in which both treatment groups received alendronate. In a report from the company, romosozumab followed by alendronate treatment significantly reduced the risks of new vertebral, clinical and nonvertebral fractures after 24 months compared with the effects of alendronate alone.22 Neither of the phase III studies was powered to evaluate the effect of romosozumab therapy on hip fracture risk, but a nominally significant reduction in hip fracture risk was reported in the ARCH study.
Safety
Except for mild injection site reactions, romosozumab has been well tolerated. One patient in a phase I romosozumab study developed symptomatic hepatitis one day after receiving a dose of 10 mg/kg. Transaminase values rose to 300% above baseline, peaking at day 3 before gradually returning to normal.23 Generalized increases in transaminase values have not been observed.
Mild, transient and asymptomatic decreases in serum calcium and reciprocal increases in parathyroid hormone (PTH) have been noted, especially with the highest doses of both romosozumab and blosozumab.13,15 This is an expected response to the combination of the rapid formation of new bone matrix and inhibition of bone resorption induced by treatment.
In the large phase III FRAME study, injection-site reactions, usually mild in severity, were observed in 5.2% of the romosozumab group and in 2.9% of the placebo group.20 All oral adverse events and femur fractures in the FRAME study were adjudicated by panels of experts to identify those events that met established criteria for osteonecrosis of the jaw or femoral fractures with atypical features. Two patients in the romosozumab group had adverse events consistent with the definition of osteonecrosis of the jaw, both of whom had recognized risk factors. An event consistent with an atypical femoral fracture occurred 3.5 months after the first dose of romosozumab in a patient with a history of prodromal pain at the site of fracture that was present at the time of enrollment.
In FRAME, the frequencies of mortality and of adverse and serious adverse events, including cardiovascular disease, were balanced between the treatment and control groups.20 A cardiovascular safety concern was noted in the preliminary report of the ARCH study.22
A clinical study demonstrated that sclerostin expression was closely associated with the degree of joint damage in patients with osteoarthritis.24 In tibial plateau specimens obtained from patients undergoing total knee arthroplasty, sclerostin expression was elevated from patients with late stage osteoarthritis compared with patients with early stage disease. In FRAME, there were no differences in the incidence of adverse events related to back pain, osteoarthritis or hyperostosis.20
Anti-romosozumab antibodies were detected in 20% of patients during the first year of therapy in the phase II study, and 3% of patients had antibodies with neutralizing activity in vitro.13 Binding and neutralizing antidrug antibodies were found in 18% and 7% respectively of patients receiving romosozumab in the FRAME study.20 The presence of antibodies had no detectable effect on efficacy and did not correlate with injection site reactions or adverse events.
Injections site reactions and the occurrence of antidrug antibodies were also observed with blosozumab.15 One patient in the phase II study developed neutralizing antibodies at week 24 of treatment and titers continued to rise during the 12-month treatment phase. The BMD response in this patient appeared to be blunted, perhaps as a consequence of inactivation of drug by the antibodies. The planned phase III study with blosozumab was not initiated and further development of the drug has been halted, perhaps because of the occurrence of more frequent or severe injection site reactions with a more concentrated preparation of the drug.
Osteoporosis in men
A phase III registration study evaluated changes in BMD, bone turnover markers and adverse events during 12 months of treatment with romosozumab in men with osteoporosis [ClinicalTrials.gov identifier: NCT02186171]. As predicted by the similar pharmacokinetic and BMD responses observed in men and women in phase I studies, the phase III study demonstrated BMD responses in men with osteoporosis similar to those observed in postmenopausal women, providing the data to support registration of the drug to treat osteoporosis in men.25
Role of anti-sclerostin therapy in osteoporosis
The information in hand, including the well characterized genetic defects, the robust preclinical program and the early clinical experience provides optimism that anti-sclerostin therapy, with its unique ability to substantially increase new bone formation while inhibiting resorption, will be a welcome and important addition to our menu of treatment options. The transient anabolic response limits the duration of therapy to 6–12 months at a time. Understanding that mechanisms for the attenuation of bone formation, which may involve depletion of the osteoblast precursor pool or upregulation of other cytokines such as Dkk1, might provide insight into ways to lengthen or to repeat the anabolic response.
The effects of romosozumab therapy in the FRAME study on vertebral and clinical fracture risk, especially after the first 6 months, are consistent with the large and rapid increases in BMD observed. The lack of a significant effect on nonvertebral fracture in comparison with placebo during the first year of therapy was disappointing, but the significant reduction in nonvertebral fracture risk, compared with alendronate, after 12 months of therapy in the ARCH study is impressive. Until that study, nonvertebral fracture risk had never been significantly reduced within the first year of treatment with any therapy.
Whether warnings about limitations to therapy will accompany registration is not yet known. The warning about osteosarcoma in rats with parathyroid hormone analogues does not seem to pertain to romosozumab.13 From a clinical perspective, therapy should be avoided in patients with untreated hypocalcemia and in patients with osteomalacia or severe vitamin D deficiency because of the known effects of anti-sclerostin therapy on serum calcium concentrations at the beginning of treatment. Treatment should also be avoided in patients with known skeletal metastases, other than myeloma, and with neurological compression syndromes until more is known about the safety of therapy in the phase III studies.
We still have much to learn about how this new therapy will be incorporated into daily clinical practice. It is my opinion that romosozumab will be an appropriate initial therapy for patients with severe osteoporosis and very low BMD who are in need of skeletal restoration and reconstruction. On the basis of the FRAME study, romosozumab will likely be approved for the treatment of patients at high risk of fracture for an interval of 12 months to be followed by an antiremodeling agent. Because some patients may then be candidates for another course of romosozumab, it will be important to assess the retreatment responses in patients who took each of the classes of antiremodeling agents.
Conclusion
The novel skeletal effects of anti-sclerostin antibody therapy, especially its unprecedented acute anabolic action, offer great promise for the treatment of osteoporosis. The use of romosozumab followed by potent antiremodeling agents will become an important part of our treatment of patients with severe osteoporosis. Exploring the effects of this therapy in disorders of impaired bone formation will be very interesting.
Acknowledgments
I thank Ms Amy Roth at the Providence Portland Medical Center Medical Library for help with the literature review and accessing articles.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The author has received consulting fees and honoraria from Amgen and Radius Health.
References
- 1. Black DM, Rosen CJ. Postmenopausal osteoporosis. N Engl J Med 2016; 374: 2096–2097. [DOI] [PubMed] [Google Scholar]
- 2. McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med 2013; 126: 13–20. [DOI] [PubMed] [Google Scholar]
- 3. Lewiecki EM. Role of sclerostin in bone and cartilage and its potential as a therapeutic target in bone diseases. Ther Adv Musculoskelet Dis 2014; 6: 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Lierop AH, Appelman-Dijkstra NM, Papapoulos SE. Sclerostin deficiency in humans. Bone 2017; 96: 51–62. [DOI] [PubMed] [Google Scholar]
- 5. Hassler N, Roschger A, Gamsjaeger S, et al. Sclerostin deficiency is linked to altered bone composition. J Bone Miner Res 2014; 29: 2144–2151. [DOI] [PubMed] [Google Scholar]
- 6. Ominsky MS, Boyd SK, Varela A, et al. Romosozumab improves bone mass and strength while maintaining bone quality in ovariectomized cynomolgus monkeys. J Bone Miner Res 2017; 32: 788–801. [DOI] [PubMed] [Google Scholar]
- 7. Matheny JB, Torres AM, Ominsky MS, et al. Romosozumab treatment converts trabecular rods into trabecular plates in male cynomolgus monkeys. Calcif Tissue Int 2017; 101: 82–91. [DOI] [PubMed] [Google Scholar]
- 8. Boyce RW, Niu QT, Ominsky MS. Kinetic reconstruction reveals time-dependent effects of romosozumab on bone formation and osteoblast function in vertebral cancellous and cortical bone in cynomolgus monkeys. Bone 2017; 101: 77–87. pii: S8756-3282(17)30144-8. [DOI] [PubMed] [Google Scholar]
- 9. Thompson ML, Chartier SR, Mitchell SA, et al. Preventing painful age-related bone fractures: anti-sclerostin therapy builds cortical bone and increases the proliferation of osteogenic cells in the periosteum of the geriatric mouse femur. Mol Pain 2016; 12 pii: 1744806916677147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li X, Ominsky MS, Warmington KS, et al. Increased bone formation and bone mass induced by sclerostin antibody is not affected by pretreatment or cotreatment with alendronate in osteopenic, ovariectomized rats. Endocrinology 2011; 152: 3312–3322. [DOI] [PubMed] [Google Scholar]
- 11. Bouaziz W, Funck-Brentano T, Lin H, et al. Loss of sclerostin promotes osteoarthritis in mice via β-catenin-dependent and -independent Wnt pathways. Arthritis Res Ther 2015; 17: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wehmeyer C, Frank S, Beckmann D, et al. Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction. Sci Transl Med 2016; 8: 330ra35. [DOI] [PubMed] [Google Scholar]
- 13. Chouinard L, Felx M, Mellal N, et al. Carcinogenicity risk assessment of romosozumab: a review of scientific weight-of-evidence and findings in a rat lifetime pharmacology study. Regul Toxicol Pharmacol 2016; 81: 212–222. [DOI] [PubMed] [Google Scholar]
- 14. Graeff C, Campbell GM, Peña J, et al. Administration of romosozumab improves vertebral trabecular and cortical bone as assessed with quantitative computed tomography and finite element analysis. Bone 2015; 81: 364–369. [DOI] [PubMed] [Google Scholar]
- 15. McClung MR, Grauer A, Boonen S, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med 2014; 370: 412–420. [DOI] [PubMed] [Google Scholar]
- 16. McClung MR, Chines A, Brown JP, et al. Effects of 2 years of treatment with romosozumab followed by 1 year of denosumab or placebo in postmenopausal women with low bone mineral density. ASBMR 2014 abstract 1152. [Google Scholar]
- 17. Genant HK, Engelke K, Bolognese MA, et al. Effects of romosozumab compared with teriparatide on bone density and mass at the spine and hip in postmenopausal women with low bone mass. J Bone Miner Res 2017; 32: 181–187. [DOI] [PubMed] [Google Scholar]
- 18. Recker RR, Benson CT, Matsumoto T, et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res 2015; 30: 216–224. [DOI] [PubMed] [Google Scholar]
- 19. Recknor CP, Recker RR, Benson CT, et al. The effect of discontinuing treatment with blosozumab: follow-up results of a phase 2 randomized clinical trial in postmenopausal women with low bone mineral density. J Bone Miner Res 2015; 30: 1717–1725. [DOI] [PubMed] [Google Scholar]
- 20. Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med 2016; 375: 1532–1543. [DOI] [PubMed] [Google Scholar]
- 21. Cauley JA, Chalhoub D, Kassem AM, et al. Geographic and ethnic disparities in osteoporotic fractures. Nat Rev Endocrinol 2014; 10: 338–351. [DOI] [PubMed] [Google Scholar]
- 22. Amgen Inc. Amgen and UCB announce top-line phase 3 data from active-comparator study of EVENITY™ (Romosozumab) in postmenopausal women with osteoporosis. https://www.amgen.com/media/news-releases/2017/05/amgen-and-ucb-announce-topline-phase-3-data-from-activecomparator-study-of-evenity-romosozumab-in-postmenopausal-women-with-osteoporosis/ (accessed 15 July 2017).
- 23. Padhi D, Jang G, Stouch B, et al. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res 2011; 26: 19–26. [DOI] [PubMed] [Google Scholar]
- 24. Wu L, Guo H, Sun K, et al. Sclerostin expression in the subchondral bone of patients with knee osteoarthritis. Int J Mol Med 2016; 38: 1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Amgen, Inc. Results from Phase 3BRIDGE Study show romosozumab significantly increases bone mineral density in men with osteoporosis. http://wwwext.amgen.com/media/news-releases/2016/11/results-from-phase-3-bridge-study-show-romosozumab-significantly-increases-bone-mineral-density-in-men-with-osteoporosis/ (accessed 15 July 2017).