

Abstract
Sarcomas are a heterogeneous group of neoplasms of mesenchymal origin. Approximately 80% arise from soft tissue and 20% originate from bone. To date more than 100 sarcoma subtypes have been identified and they vary in molecular characteristics, pathology, clinical presentation and response to treatment. While sarcomas represent <1% of adult cancers, they account for approximately 21% of paediatric malignancies and thus pose some of the greatest risks of mortality and morbidity in children and young adults. Metastases occur in one-third of all patients and approximately 10–20% of sarcomas recur locally. Surgery in combination with preoperative and postoperative therapies is the primary treatment for localized sarcoma tumours and is the most promising curative possibility. Metastasized sarcomas, on the other hand, are treated primarily with single-agent or combination chemotherapy, but this rarely leads to a complete and robust response and often becomes a palliative form of treatment. The heterogeneity of sarcomas results in variable responses to current generalized treatment strategies. In light of this and the lack of curative strategies for metastatic and unresectable sarcomas, there is a need for novel subtype-specific treatment strategies. With the more recent understanding of the molecular mechanisms underlying the pathogenesis of some of these tumours, the treatment of sarcoma subtypes with targeted therapies is a rapidly evolving field. This review discusses the current management of sarcomas as well as promising new therapies that are currently underway in clinical trials.
Keywords: chemotherapy, current management, new therapies, radiation therapy, sarcoma, surgery, targeted therapies
Introduction
Sarcomas are highly diverse mesenchymal malignancies of the bone, cartilage, muscle, peripheral nerves and adipose or fibrous connective tissues.1 Although the ultimate cells of origin of sarcoma subtypes remain unclear, there is increasing evidence that they arise de novo from mesenchymal pluripotent stem cells.2,3 An extension of this theory would be that alteration(s) in mesenchymal stem cell genetics can give rise to several sarcomas, including osteosarcoma, Ewing’s sarcoma, synovial sarcoma, chondrosarcoma, rhabdomyosarcoma, fibrosarcoma and liposarcoma.4 At present, sarcoma classification is based on the tissue type it resembles. This form of classification is, however, challenging for sarcoma subtypes that do not show any clear similarities to normal tissue, such as clear cell and epithelioid sarcomas. More recently, classifications have been revised to include molecular features and genetic profiles of sarcomas.5–7 From a molecular point of view, sarcomas may be broadly classified into two types: (1) sarcomas with simple karyotypes characterized by chromosomal translocations or specific mutations; and (2) sarcomas with complex aneuploidy karyotypes, consisting of numerous losses, gains and amplifications.8 Approximately 15–20% of sarcomas fall into the simple karyotype subgroup, while the vast majority fall into the complex karyotype subgroup.9,10 There is, however, room for improvement in the identification of biomarkers for sarcoma subtypes and determination of optimal subtype-specific treatment strategies. The need for alternative treatments such as targeted therapies and immunotherapies is underscored by observations that several sarcoma subtypes are poorly responsive to chemotherapy and radiation.
This review will focus on the current treatment strategies for sarcoma and the emerging field of targeted therapies for sarcoma.
Local control in sarcoma
Surgery
Complete excision, when possible, is the standard therapy in the management of most subtypes of localized soft tissue sarcoma, and when possible, this confers the greatest possibility of cure. In order to adequately ensure removal of all tumour microsatellites, the tumour must be resected with a perimeter of healthy tissue.11 This lowers the risk of local recurrence and allows for better prognosis. Surgical removal of tumours takes into account limb and function-sparing while accomplishing suitable biological margins.12 The width of margin clearance necessary is subject to controversy and is affected predominantly by the anatomical location of the tumour and its size at presentation. Tumours located in the retroperitoneum, for example, cannot be removed with the same excisional margin as similar neoplasms arising from the thigh or buttock.13 What is unequivocal is that a resection with a microscopically involved margin carries a higher risk of recurrence than another where the margins are clear, even if by a few millimetres. In pursuit of clear margins, surgical resections require aggressive removal of involved tissue and sometimes reconstruction with grafts is necessary. Surgery for sarcoma carries major morbidity and is best performed in centres familiar with sarcoma management and with access to multimodality treatment. In addition, the outcome of patients treated at specialist sarcoma centres is better than patients treated at generalist units.14
The current gold standard for the treatment of bone sarcomas is limb salvage surgery with the aim of preserving a limb with sufficient functionality and without compromising the patient’s overall survival. It implicates clear-margin resection of the tumour followed by reconstruction of the bone defect with endoprosthetics, allografts or autografts.15 Surgery used in combination with chemotherapy increases the overall survival and progression-free survival significantly.16,17 Even though localized sarcomas have a high cure rate with surgery, when they recur and/or metastasize they have a poor prognosis with a median survival of approximately 12 months.18–20
Radiation therapy
Radiation is frequently used to treat soft tissue sarcoma of the extremity. However, although neoadjuvant (before surgery) and adjuvant (after surgery) radiation significantly improve local control of non-metastatic low-grade and high-grade sarcomas of the extremity, in many studies there is no statistically significant benefit for overall survival.21 In this group of patients, radiotherapy (RT) has been used both neoadjuvantly and adjuvantly, with no difference in progression-free survival between these approaches. However, adjuvant RT is associated with a higher incidence of late normal tissue toxicity. For this reason, neoadjuvant RT is often used as an alternative, despite the increased incidence of wound-healing problems.22 The main advantages of neoadjuvant RT are that target volume definition is easier with the visible tumour in situ, normal tissues are displaced out of the field and doses required are lower than with adjuvant treatment. In addition, the tumour may decrease in size, facilitating resection.20
Soft tissue sarcomas of the retroperitoneum present a different problem. These tumours are frequently very large at presentation and are more difficult to remove surgically than extremity tumours. Local control of these tumours remains a challenge and the role of RT in treating these tumours remains controversial. No large randomized trials exist because of the rarity of these tumours, but results from smaller series are at odds, with some showing no benefit from the addition of RT23 and others showing some benefit in terms of delaying local recurrence24 but little effect on overall survival. Both preoperative and postoperative RT are challenging in this site because of the proximity of the tumour to sensitive intra-abdominal organs. Preoperative RT, especially for tumours with borderline resectability, may be associated with fewer complications.
For bone sarcomas, the role of RT is also not well defined. The most common subtypes of bone sarcoma are osteosarcoma and Ewing’s sarcoma. Both of these are considered systemic diseases and the mainstay of treatment involves chemotherapy and surgery. In osteosarcoma, the role of RT is confined to unresectable tumours, postsurgery in the case of positive margins and palliation. In Ewing’s sarcoma, RT has a greater role to play and may be curative in certain patients where tumours are unresectable, clear margins are not achievable or the tumour is deemed high risk.
Advanced RT techniques such as intensity-modulated radiation therapy (IMRT) and volumetric modulated arc therapy may play a role in the current clinical challenges of radiogenic wounds and associated toxicities, and are becoming mainstream in many departments when treating difficult cases. These techniques use a combination of advanced hardware and software which allow radiation dose to conform to complex tumour shapes. The advantage of this is that the optimal amount of radiation can be directed to the tumour or tumour bed, while sparing normal tissue as much as possible.25,26 Sladowska and colleagues and Paumier and colleagues demonstrated that, compared to conventional RT, IMRT improves dose distribution, target coverage and normal tissue sparing in soft tissue sarcomas of the thigh and retroperitoneal sarcoma.27,28
Particle therapy, which uses charged particles such as protons and carbon ions, may also have a role to play in RT for sarcomas. Indeed, there is some evidence to suggest that particle therapy may be a more effective modality in the management of bone and soft tissue sarcomas not eligible for surgical resection, providing good local control and offering a survival advantage without unacceptable morbidity.29–31 Particle therapy utilizes the Bragg peak effect to deliver high-dose radiation to the tumour while minimizing the dose delivered to adjacent normal tissue.32 Particle therapy is, however, extremely expensive, and not routinely available for all cases.
Systemic control in sarcoma
Chemotherapy
The role of chemotherapy in the management of sarcoma is variable and, in some cases, controversial. Significant benefit is seen in a limited group of chemosensitive sarcoma subtypes, including embryonal and alveolar rhabdomyosarcoma, Ewing’s sarcoma and osteosarcoma, and it is thus an integral part of the management of these sarcomas. Indeed, chemotherapy has drastically improved the long-term survival of these patients and offers possibility of cure, even in some cases with metastatic disease.33–36 There are also examples of more recently described chemotherapies that appear effective in specific sarcoma subtypes; these will be discussed later. Other sarcoma subtypes vary from fairly sensitive to completely chemotherapy-unresponsive; in the majority of these cases, patients with metastatic disease face a dismal prognosis.
Single-agent chemotherapy
Doxorubicin (also known as Adriamycin), epirubicin and ifosfamide are the only single-agent chemotherapeutic drugs that consistently achieve response rates of >20% in metastatic sarcomas.37 However, the range of activity of these agents varies greatly for different histological subtypes and there is individual variability for drug efficacy.38
Doxorubicin and epirubicin are anti-cancer antibiotics that belong to the anthracycline class of drugs. They are a four-membered ring system containing an anthraquinone chromophore and an aminoglycoside.39 Doxorubicin (Figure 1) was first used as a single-agent chemotherapeutic treatment in the 1970s.40 Upon intravenous injection, doxorubicin is rapidly taken up into the nucleus of cells, where it binds to DNA with high affinity. It acts by intercalating between DNA base pairs and binding to DNA-associated enzymes, such as topoisomerase enzymes I and II and DNA and RNA polymerases. This induces DNA damage and the cessation of DNA replication and mRNA transcription.41 Furthermore, cells arrest in G1 and G2 in an attempt to repair the damage, but when the damage is irreparable the apoptotic cell death pathway is triggered. Other actions of doxorubicin include the generation of free radicals, causing additional DNA damage, inhibition of macromolecule production, DNA unwinding and an increase in alkylation.42 The reported response rates of sarcomas to doxorubicin vary significantly, ranging from 10% to 25%, with the majority of cases showing a partial response.43–48 In osteosarcoma, studies have demonstrated that cells are sensitized to doxorubicin treatment when autophagy, a process important for cell survival, is inhibited by blocking the high-mobility group box 1 protein (HMGB1).49 It is therefore possible that treatment of sarcomas with doxorubicin may be more successful when combined with an inhibitor of autophagy. Adverse effects of doxorubicin include both acute and chronic cardiotoxicity, reversible myelosuppression, alopecia, mucositis, nausea and vomiting.50,51
Figure 1.
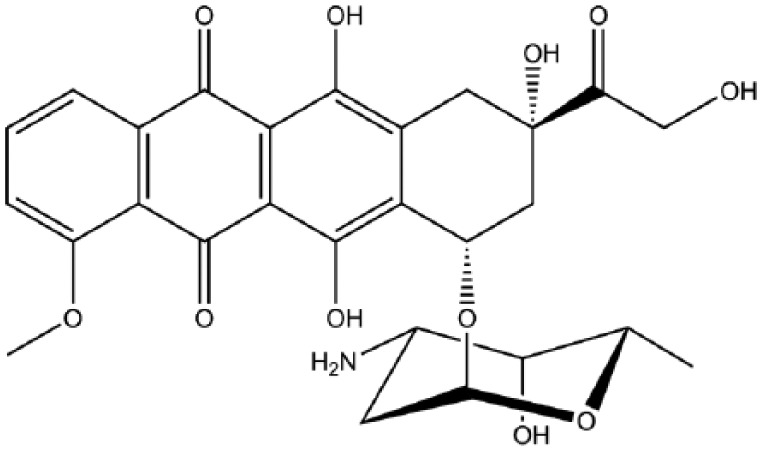
Doxorubicin.
Multiple trials have compared the effects and responses of doxorubicin and epirubicin in sarcoma treatment. In most cases no clear benefit for one over the other drug was seen. However, patients on the doxorubicin schedule demonstrated worse cardiovascular and haematologic toxicity and hence epirubicin is often favoured over doxorubicin.52 While epirubicin (Figure 2) acts in a similar way to doxorubicin, the spatial orientation of the hydroxyl group at the 4′ carbon of the sugar is different and this opposite chirality has been proposed to account for its reduced toxicity.52,53
Figure 2.
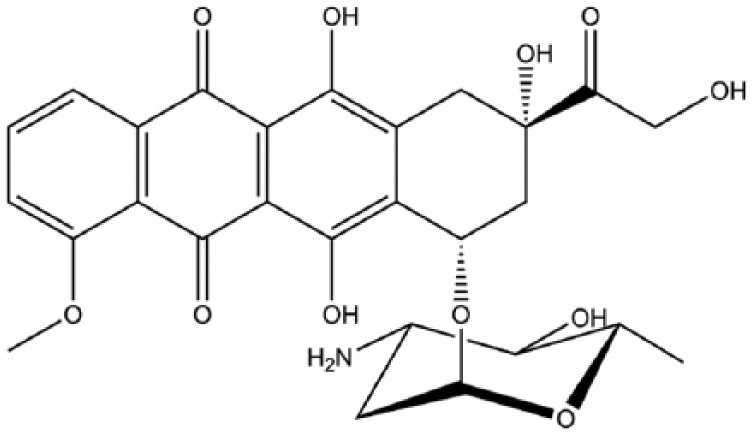
Epirubicin.
Ifosfamide (Figure 3) is a nitrogen mustard alkylating agent that terminates proliferating cancer cells by adding alkyl groups to guanine bases in DNA molecules. This inhibits tumour growth because the guanine nucleobases become cross-linked and prevent DNA double-helix strands from uncoiling and replicating.54 Ifosfamide consistently shows response rates comparable to doxorubicin and it has a 25% average response rate among patients who show limited responses on a doxorubicin-based schedule.55–63 Unlike doxorubicin, which is administered as a single-day infusion,64 ifosfamide is administered intravenously over several days at a time65 and usually requires hospital admission. The toxicities associated with ifosfamide differ to those caused by doxorubicin, and include haemorrhagic cystitis, renal tubular acidosis, salt-wasting nephropathy, central nervous system toxicity and usually encephalopathy.54,66–68 Ifosfamide shows less evidence of cardiotoxicity, thus rendering it an attractive treatment option.58–61,63,69 Ifosfamide-induced urotoxicity can be reduced when it is administered simultaneously with mesna, a thiol compound that binds to the urotoxic ifosfamide metabolite, acrolein, converting it into a non-toxic compound.58,70,71
Figure 3.
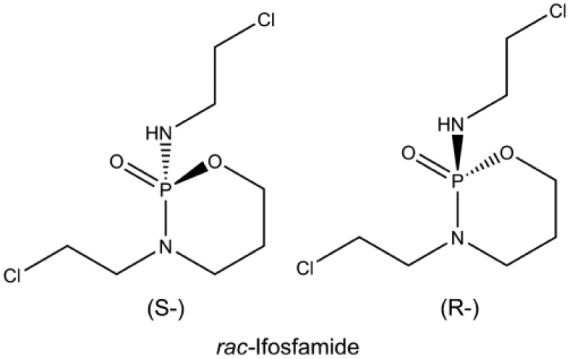
Ifosfamide.
Other single-agent chemotherapeutic drugs that have shown efficacy in some sarcoma subtypes include gemcitabine and topotecan. Gemcitabine (Figure 4) is a cytotoxic agent that has been tested in clinical trials but efficacy data are conflicting.72–75 It is a nucleoside analogue where the hydrogen atoms on the 2′ carbon of deoxycytidine are replaced by fluorine atoms. Gemcitabine arrests tumour growth by replacing cytidine, one of the building blocks of nucleic acids, during DNA replication. As a result, the newly forming DNA strand can no longer be elongated and apoptosis is induced. Gemcitabine also irreversibly inhibits the enzyme ribonucleotide reductase by binding to its active site and preventing the production of deoxyribonucleotides for DNA replication and repair and thus leads to apoptosis.76 It is more successful in leiomyosarcoma of uterine and gastrointestinal origin when used in combination with the anti-mitotic chemotherapeutics docetaxel or vinorelbine or the alkylating agent dacarbazine.77 Topotecan (Figure 5), a quinoline-based alkaloid extracted from the Asian tree Camptotheca acuminate, inhibits topoisomerase-I activity during DNA replication. This causes double-strand breaks as the DNA is not relieved from torsional strain by topoisomerase-I while replicating. These DNA strand breaks cannot be repaired and apoptosis is triggered. This drug generally demonstrates low activity, but response rates appear to be modest in non-uterine leiomyosarcoma.78 Treatment response has also been shown for use of this drug in Ewing’s sarcoma and rhabdomyosarcoma.79–81
Figure 4.
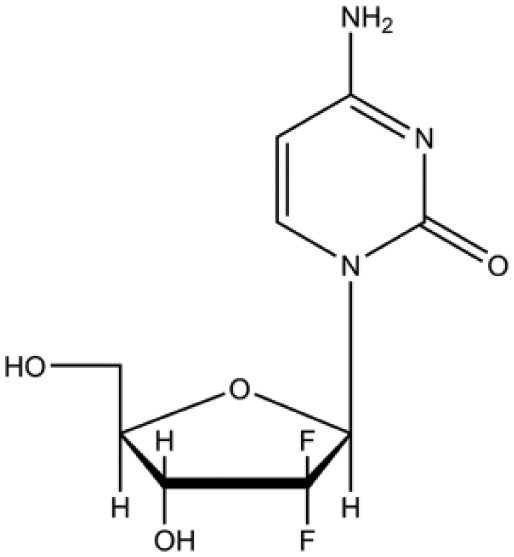
Gemcitabine.
Figure 5.
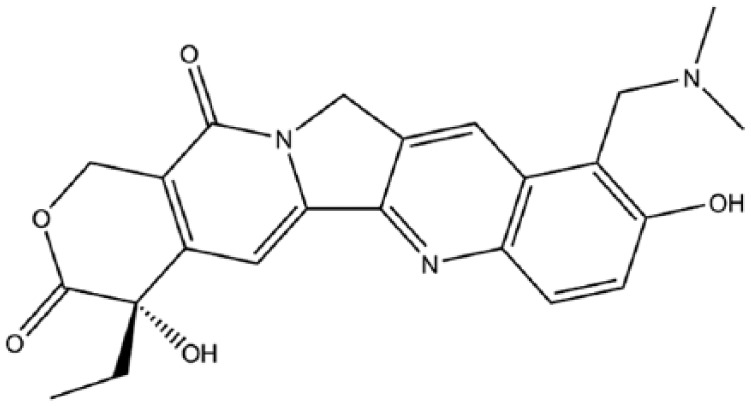
Topotecan.
The taxane agents, paclitaxel and docetaxel, are diterpenes produced by plants of the genus Taxus.82 Although comparatively inactive as single-agent treatment, taxanes, particularly paclitaxel, appear to show significant response rates in angiosarcoma.83–87 This class of drugs functions by disrupting microtubules and thus inhibiting cell division. Other conventional single-agent chemotherapeutic drugs used for the treatment of sarcomas include vinorelbine, methotrexate, dacarbazine, temozolomide, cisplatin and carboplatin, but the response rates for most of them are <20%.88–93
Novel single-agent chemotherapeutics
Trabectedin and eribulin are two novel marine-derived chemotherapeutics which have shown promise for the treatment of leiomyosarcoma and liposarcoma, which together account for approximately 30% of all soft tissue sarcomas.
Trabectedin (Figure 6) is a marine-derived alkaloid that is characterized by three fused tetrahydroisoquinoline rings. Two of these rings covalently interact with the minor groove of the DNA double helix and the third ring interacts with nearby nuclear proteins. These chemical interactions stimulate a cascade of events that compromises DNA binding proteins, including several transcription factors and the DNA nucleotide excision repair machinery. This results in double-strand DNA damage followed by a G2/M cell cycle arrest and ultimately apoptosis.94,95 Trabectedin also targets the tumour microenvironment by triggering apoptosis in monocytes, including tumour-associated macrophages (TAMs), which are known to promote tumour progression and metastasis. Furthermore, trabectedin inhibits the transcription of pro-inflammatory mediators (cytokines and chemokines), which also play a role in tumour growth and progression.96–98
Figure 6.
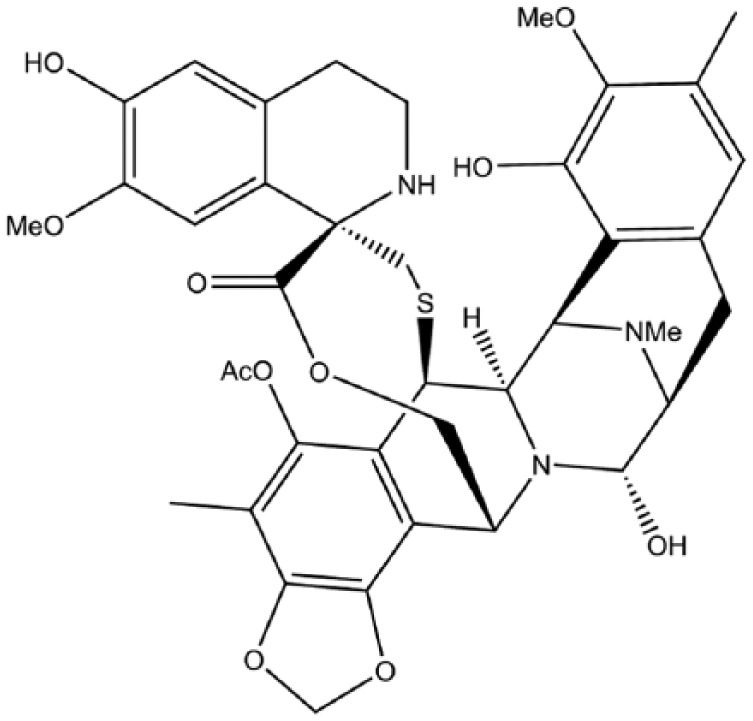
Trabectedin.
Two independent phase II clinical trials in 2004 provided initial analysis of trabectedin in advanced sarcoma subtypes refractory to conventional anthracycline/ifosfamide first-line chemotherapy and a median 6-month progression-free survival of 29% and 24% were achieved. The most profound responses were observed in leiomyosarcoma and synovial sarcoma histologies, with 56% and 61% progression arrest of tumour growth respectively.99,100 Adverse effects of trabectedin include neutropenia, transaminase elevation, fatigue and emesis.101 The success of trabectedin in early clinical trials led to its approval by the European Union for advanced soft tissue sarcoma in 2007 and subsequently the drug has been approved in over 70 countries, especially for patients who have failed to respond to standard therapies.102–104 A recent phase III clinical trial confirmed that advanced liposarcoma and leiomyosarcoma refractory to doxorubicin and ifosfamide showed a 45% reduction in the risk of disease progression or death when treated with trabectedin in comparison with darcarbazine, which prompted the approval of trabectedin by the US Food and Drug Administration (FDA).105 The reason(s) why some sarcomas are particularly sensitive to trabectedin is not fully understood. However, it is likely that it interferes with transcription factors that they are addicted to. The myxoid/round cell subtype of liposarcoma is the most sensitive to trabectedin and 95% of these carry a t(12;16) (q13;p11) chromosome translocation which results in the oncogenic FUS–CHOP fusion-protein. FUS–CHOP functions as a transcription factor that promotes multiple aspects of tumourigenesis, and trabectedin was shown to compete with and displace it from its target promoters.106,107
Recently a phase IIb trial showed that the administration of trabectedin as a first-line chemotherapeutic for advanced soft tissue sarcomas shows no significant improvement in progression-free survival in comparison to doxorubicin, leading to the conclusion that doxorubicin remains the gold standard for first-line treatment of advanced soft tissue sarcomas.108 Although trabectedin clearly demonstrates clinical benefit in anthracycline-resistant advanced leiomyosarcoma and synovial sarcoma histologies as a second- or third-line treatment, its benefit in treating other sarcoma histologies warrants further investigation.
Eribulin (Figure 7) is a novel microtubule-targeting chemotherapeutic drug. It was recently approved by the FDA for the treatment of patients with unresectable or metastatic liposarcoma who have received a prior anthracycline-containing chemotherapy regimen.109 It is a synthetic analogue of halichondrin B that was originally extracted from the marine sponge Halicondria Okaida.110 The anti-cancer properties of eribulin are distinct from other tubulin-targeting agents in that it does not affect microtubule shortening but binds to a unique part of tubulin which results in the suppression of microtubule growth and sequestration into non-functional aggregates.111–113 In this regard, eribulin has been found to be especially efficacious in patients with tumours harbouring beta-tubulin mutations that are refractory to taxanes.114,115 In preclinical models, eribulin was shown to induce an irreversible mitotic arrest, apoptosis and tumour regression in multiple cancer cell lines with a mean IC50 that is 2–4-fold more potent than vinblastine and paclitaxel.114,116,117 A broad range of sarcoma cell lines, including liposarcoma, leiomyosarcoma, Ewing’s sarcoma, synovial sarcoma and fibrosarcoma, have been demonstrated to be sensitive to eribulin.118 The authors showed that it induced cellular differentiation in vitro and reduced tumour formation and vasculature in in vivo xenograft models. In a non-randomized phase II study, the EORTC (European Organisation for Research and Treatment of Cancer) Soft Tissue and Bone Sarcoma Group assessed the efficacy and safety of eribulin. This study showed that eribulin exhibited significant anti-tumour activity in metastatic liposarcoma and leiomyosarcoma patients, but not in synovial sarcoma or any other sarcoma subtype.119 A phase III study also demonstrated that, compared to dacarbazine, eribulin significantly improves overall survival of patients with refractory leiomyosarcoma and liposarcoma.120 The predominant side effects reported for eribulin are neutropenia, anaemia, fatigue, febrile neutropenia, mucositis and sensory neuropathy.121
Figure 7.
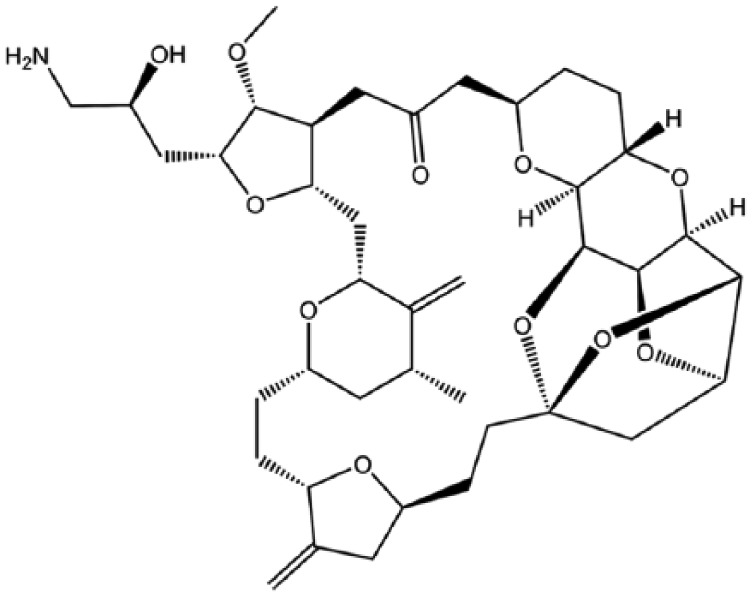
Eribulin.
Combination chemotherapy
Combination chemotherapy has been explored extensively and while not always used as a first-line approach to treating patients with metastatic sarcomas, it is an accepted treatment. Most combination chemotherapy regimens include doxorubicin and an alkylating agent.122,123 Regimens that appear often in the literature include AIM (doxorubicin, ifosfamide and mesna), MAID (mesna, doxorubicin, ifosfamide and dacarbazine) and gemcitabine together with docetaxel, vinorelbine or dacarbazine.45,46,124–129 A variety of bone and soft tissue sarcomas show response to these regimens, with Ewing’s sarcoma and rhabdomyosarcoma showing greater sensitivity to MAID schedules;58,126 myxoid liposarcoma, myxofibrosarcoma and synovial sarcoma showing sensitivity to AIM regimens;130–132 and leiomyosarcoma showing better response rates to the gemcitabine-based regimens.133 Another combination treatment termed VAC/IE for Ewing’s sarcoma and rhabdomyosarcoma includes vincristine, doxorubicin and cyclophosphamide alternating with ifosfamide and etoposide.134,135 This relatively intense chemotherapeutic regimen shows a 16–46% overall response with complete responses occurring in approximately 5–10% of these sarcomas. About one-third of these complete responders are long-term disease-free survivors.126,136–139
Studies comparing single-agent therapy to combination regimens have failed to provide evidence as to which option provides better overall survival benefit.
Targeted therapies
The development of molecular-targeted therapies for sarcomas is a rapidly evolving field. This therapeutic strategy requires identification of key molecular drivers of sarcomas and recent advances in our understanding of sarcoma biology have led to the identification of several molecular determinants of different sarcoma subtypes (Figure 8). This section will review the most relevant targetable pathways in soft tissue and bone sarcomas, as well as discuss findings from preclinical and clinical trials, which are summarized in Figure 9.
Figure 8.

Molecular determinants of different sarcoma subtypes.
Figure 9.

Targeted therapies in sarcomas.
Tyrosine kinase inhibitors
Tyrosine kinase inhibitors have become the most influential targeted therapeutic breakthrough for the treatment of sarcomas. Factors currently targeted in approved treatments include the receptors for the tyrosine kinases c-KIT, platelet-derived growth factor receptor (PDGFR) and vascular endothelial growth factor receptor (VEGFR). Other sarcoma subtype-specific targeted therapies underway include the inhibition of insulin-like growth factor 1 receptor (IGF1R) in Ewing’s sarcoma and MET receptor tyrosine kinase and Src tyrosine-protein kinase in bone sarcomas.
c-KIT, PDGFR and VEGF inhibitors
c-KIT is a class III receptor tyrosine kinase and has been shown to impact on a variety of oncogenic cellular processes such as cell survival, proliferation, differentiation, adhesion and apoptosis by initiating multiple downstream signalling pathways such as the mitogen-activated protein kinases (MAPK), phosphatidylinositol 3-kinase (PI3K) and Janus kinase/signal transducer and activator of transcription (JAK/STAT). PDGF-α is also a class III receptor tyrosine kinase and a key regulator of mesenchymal cell proliferation and migration.140–142 c-KIT and PDGFR kinases play an important role in the pathogenesis of a number of tumours including gastrointestinal stromal tumours (GISTs). GISTs are the most common mesenchymal tumours of the gastrointestinal tract and are usually resistant to chemo- and radiation therapy. Approximately 95% of GISTs arising in adults constitutively express active c-KIT; of these patients, 80% have c-KIT gene mutations which result in ligand-independent constitutive activation of the receptor.143,144 This results in uncontrolled cell proliferation and the stimulation of downstream signalling pathways involving PI3K and MAPK.145 Sarcomas, like other proliferating malignancies, are also dependent on the formation of new blood vessels (angiogenesis) to support their proliferation, invasion and metastasis.146 VEGFR is considered to be one of the most important drivers of angiogenesis and are frequently upregulated in soft tissue sarcomas and are associated with high tumour grade.147,148 A study by Zhang and colleagues also showed that overexpression of VEGF-2 in soft tissue sarcoma cell lines resulted in increased tumour vasculature as well as pulmonary metastases in mice.149 This section will focus on inhibitors of c-KIT, PDGFR and VEGFR, namely imatinib, sunitinib and pazopanib, which have completed all phases of clinical trials and which have been approved as standard treatment for commercial use by the FDA. These drugs have been shown to inhibit tumour growth with improved response rates and reduced toxicity.150–153
Imatinib was originally synthesized to target the fusion-protein Bcr-Abl for treatment of myelogenous leukaemia, but was subsequently found to also inhibit c-KIT and PDGFR. A study in the United States has demonstrated objective response rates in the range of 50–70% for the treatment of GISTs with imatinib.150 Imatinib functions by binding in the cleft between the N- and C-terminal domains of c-KIT (Figure 10) which inhibits its association with downstream cell signalling substrates resulting in cell growth arrest and cell death by apoptosis.154 While most mutations that result in ligand-independent constitutive activation of c-KIT occur in exon 11, there are some less frequent mutations present in exon 9, 13 or 17. These more rare mutations appear to have a different underlying mechanism that results in uncontrolled c-KIT signalling and are less responsive to imatinib treatment.155 Hirota and colleagues also identified mutations causing constitutively active PDGFRs in a minority of GIST cases which induce cytogenetic changes associated with tumour progression.156 Similar to the rare c-KIT mutations, these mutations are characterized by insensitivity to imatinib but they are more sensitive to sunitinib, which inhibits multiple receptor tyrosine kinases including c-KIT, PDGFR, VEGFR, RET (rearranged during transfection) and FLT3 (fms-related tyrosine kin-ase-3).155,157–159 The simultaneous inhibition of these targets results in reduced tumour vascularization and cancer cell death. The ability of sunitinib to target multiple receptors renders it a successful second-line treatment for GIST patients resistant to imatinib. However, this is associated with adverse effects including nausea, diarrhoea, fatigue, hypertension, anorexia, stomatitis, a yellow skin discolouration and hand-foot skin reaction.152
Figure 10.
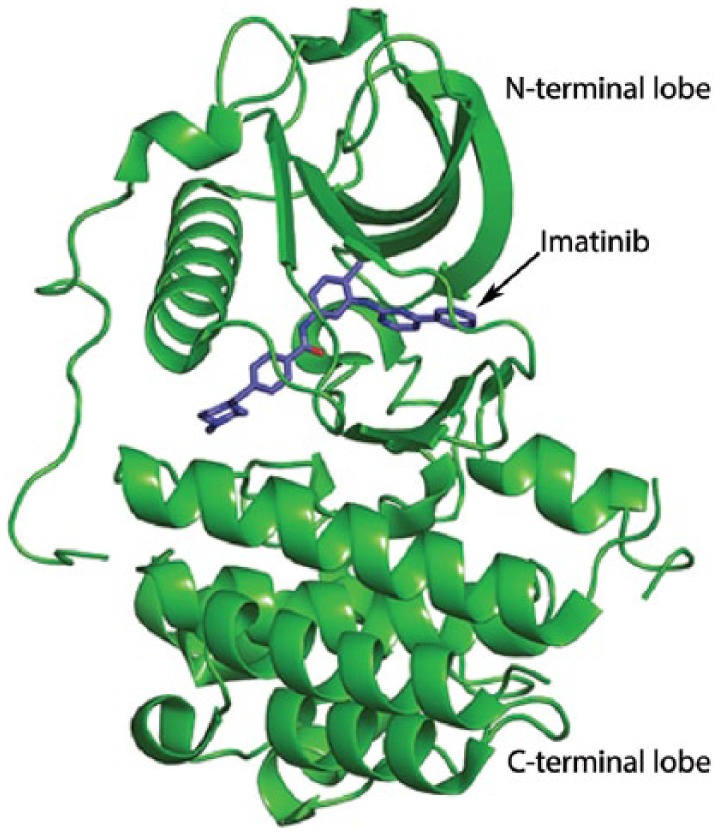
Imatinib, N- and C-terminal domains.
Based on the success rate of imatinib in GISTS, its therapeutic applications have been extended to other sarcoma subtypes that also exhibit aberrant expression of PDGFR or c-KIT. However, the response rates of these cancers to imatinib have been mostly poor.19 A recent phase II clinical trial was conducted by the Children’s Oncology Group (COG) to test the efficacy of imatinib for the treatment of a variety of paediatric sarcomas with high c-KIT or PDGFR levels. Results showed that out of 48 patients comprising 24 Ewing’s sarcoma, 10 osteosarcoma, 10 desmoplastic small round cell sarcoma and 4 synovial sarcoma cases, only 1 patient showed a partial response to imatinib and the COG concluded that the tyrosine kinases targeted by imatinib are not the molecular drivers of these cancers.160,161 It is also possible that these sarcomas escape cell death induced by imatinib by activating alternative signalling pathways such as the PI3K/AKT pathway or through feedback loops. If this is the case then multi-targeted inhibitors may be required.162 Based on the above reports it is clear, with the exception of most GISTS, that no definitive correlations can be drawn based on the expression levels of PDGFR/KIT and the response to imatinib. Acquired resistance to imatinib has also been reported in some GIST patients during chronic therapy and alternative strategies are therefore required to treat or avert imatinib resistance in these cancers.163 IMC-3G3 (olaratumab), a humanized anti-PDGFRα monoclonal antibody, showed promising preclinical efficacy in leiomyosarcoma and osteosarcoma cell lines. Results from a randomized phase Ib/II clinical trial164 led to the accelerated approval of olaratumab by the FDA for the treatment of patients with soft tissue sarcoma not amenable to curative treatment with RT or surgery and with a histologic subtype for which anthracycline-containing regime is appropriate.165
Pazopanib is another multi-targeted tyrosine kinase inhibitor that targets several proteins, including VEGFR and PDGFR, which play important roles in angiogenesis, tumour growth and cell survival.166 It binds to the ATP-binding site which is located in the cleft between the N- and C-terminal lobes of these receptor tyrosine kinases, rendering them inactive (Figure 11).167 Pazopanib has shown promising anti-cancer activity in advanced soft tissue sarcomas with significant responses in leiomyosarcoma and synovial sarcoma.168 The outcome of a randomized, double-blind, placebo-controlled phase III trial led to FDA approval of pazopanib for the treatment of advanced soft tissue sarcomas in patients who had received prior chemotherapy.153 This, however, excluded patients who received treatment for adipocytic tumours or GISTs as these were unresponsive in the trial. The trial showed benefit across many treatment-resistant metastatic sarcoma subtypes with a significant 3-month median increase in progression-free survival in the treated group versus the placebo group. The most common adverse effects for pazopanib reported were fatigue, diarrhoea, nausea, weight loss and hypertension.153,169 The success and approval of this drug lends some evidence to the important role of the VEGFR and related pathways to the growth of diverse sarcoma subtypes.
Figure 11.
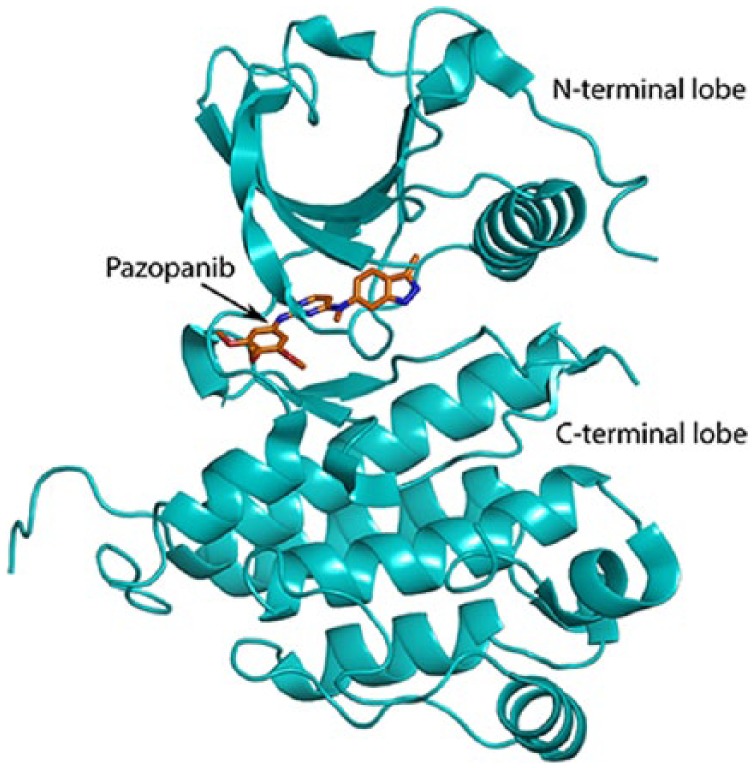
Pazopanib, N- and C-terminal domains.
MET, Src, IGFR inhibitors
The constitutive activation of the MET signalling pathway has been implicated in a wide range of human malignancies, including sarcomas where it promotes cell-matrix dissociation, protease production and consequently invasion and metastasis.170,171 MET overexpression correlates with metastasis in osteosarcoma, and the upregulation of MET in primary human bone-derived cells was sufficient to drive their transformation into osteosarcoma cells in vitro.172,173 The MET inhibitor, crizotinib, inhibits proliferation, survival, invasion and clonogenicity of multiple sarcoma cell lines as well as in vivo growth in osteosarcoma bearing mice.174–176 At present, a phase II clinical trial [ClinicalTrials.gov identifier: NCT01524926] testing crizotinib in patients ⩾15 years with MET-driven sarcomas and lymphomas is underway.
Src has been identified as an oncoprotein in several human cancers and its role in promoting migration is well established in bone sarcomas.177 Indeed, three independent studies have shown that the inhibition of the Src signalling pathway inhibits metastasis of chondrosarcoma cells and the treatment of Ewing’s sarcoma cells with the Src inhibitor dasatinib reduced their migratory and invasive ability.178–181 Clinically, dasatinib has been tested for the treatment of several solid tumours, including sarcomas, but it has been associated with acute side effects.182 Ongoing phase II clinical trials [ClinicalTrials.gov identifier: NCT00788125] are now attempting to further evaluate the use of dasatinib in combination with chemotherapies such as ifosfamide, carboplatin and etoposide.
The IGF signalling pathway promotes cell survival and proliferation by activating the PI3K/AKT/mTOR and Ras/Raf/MAPK pathways.183,184 Elevated levels of IGF Receptor-1 (IGFR-1) and its ligands have been observed in Ewing’s sarcoma, synovial sarcoma, osteosarcoma and chondrosarcoma, as well as some soft tissue sarcoma subtypes such as leiomyosarcoma and rhabdomyosarcoma, where it correlates with tumour aggression and poor prognosis.184–186 IGFR-1 has an established oncogenic role in Ewing’s sarcoma, where it is a direct target gene of the EWS–FLI1 fusion oncoprotein, and it is required for Ewing’s sarcomagenesis.187,188 Preclinical data suggest that targeting the IGF pathway could be a promising molecular therapy for sarcomas. Indeed, humanized monoclonal antibodies targeting IGFR-1 showed promise in phase I and II clinical trials for the treatment of paediatric sarcomas, including osteosarcoma, Ewing’s sarcoma and rhabdomyosarcoma, with a modest clinical benefit in liposarcoma.189–191 Unfortunately, most patients who initially respond to IGFR-1 inhibitors develop resistance to the therapy and suffer from relapse or recurrence within several months. Investigations of additional combination/additive treatments are clearly warranted and the factors/pathways responsible for resistance to IGFR-1 inhibitors remain subjects of investigation.192
mTOR inhibitors
Alterations in the mammalian target of rapamycin (mTOR) pathway are commonly associated with sarcoma formation. Therapies inhibiting this pathway are at preclinical and clinical trial phases. mTOR is a protein kinase and downstream effector of the PI3K/AKT pathway in proliferation, cell survival and migration.193 Several mTOR inhibitors have been evaluated in single-agent clinical trials, with ridaforolimus being the most extensively studied.19 However, while ridaforolimus demonstrated efficacy for the treatment of patients with advanced metastatic sarcoma in phase I and II clinical trials, it did not receive FDA approval because of the results of a larger phase III clinical trial involving 711 patients with advanced sarcoma.194–196 In the latter trial, compared to the placebo, tumour progression was only marginally delayed in patients on ridaforolimus and the median benefit of progression-free survival was low (17.7 weeks for ridaforolimus versus 14.6 weeks for placebo).197 In addition, the mTOR inhibitors, everolimus, sirolimus and temsirolimus have been evaluated in single-agent clinical trials with most of them yielding disappointing results and interpatient variability.19 Due to the possibility of compensatory activation of the PI3K/AKT pathway, combination therapies targeting multiple components of the PI3K/AKT/mTOR pathway have been considered, with one possible drawback being increased cytotoxicity to patients.193
Inhibitors of the cell cycle
The cell cycle is regulated at the most basic level by the ordered expression and activation of the family of Ser/Thr cyclin-dependent kinases (CDKs).198 As their name implies, the activity of CDKs is dependent on their association with cyclins; when activated, cyclin–CDK complexes drive the cell cycle.199 CDK inhibitors (CDKIs) trigger checkpoints which halt the cell cycle and therapeutic agents that inhibit aberrant cell cycle activation are being tested in sarcomas.200 Flavopiridol, a non-selective inhibitor of CDK1, 2, 4, 6 and 7 was tested in a phase II clinical trial for the treatment of advanced, metastatic soft tissue sarcomas including fibrosarcoma, liposarcoma, leiomyosarcoma and synovial sarcoma, but no significant responses were observed.201 Encouraging results have, however, been observed for palbociclib, a selective CDK4/6 inhibitor, in liposarcoma patients with amplified CDK4. In a recent phase II clinical trial, palbociclib was associated with favourable progression-free survival in patients with well-differentiated or dedifferentiated liposarcoma.202 Histone deacetylase (HDAC) inhibitors can induce transcription of key cell cycle regulators including the CDKI p21 which leads to growth arrest and apoptosis in sarcoma cell lines.203,204 For example, treatment of chondrosarcoma cell lines with HDAC inhibitors was shown to result in S-phase arrest and transcriptional activation of p21.205,206 Based on these and other promising results, HDAC inhibitors are also being investigated in early-phase clinical trials in patients with advanced soft tissue sarcomas.207 One emerging phase II study evaluated a single-agent HDAC inhibitor, panabinostat, in patients with translocation-related (myxoid liposarcoma, Ewing’s sarcoma, alveolar soft part sarcoma and synovial sarcoma) and translocation-unrelated (leiomyosarcoma and pleomorphic liposarcoma) soft tissue sarcomas. While this study did not reach its primary endpoint, a subset analysis revealed that six patients with liposarcoma, leiomyosarcoma or Ewing’s sarcoma had prolonged stable disease.208
Mouse double minute 2 homolog (MDM2) is a ubiquitin E3 ligase that mediates the degradation of p53 by the proteasome 26S, and it is frequently amplified and activated in sarcomas.209 The inhibition of MDM2 results in increased levels of p53 and consequently the transcriptional activation of, among other p53 targets, CDKIs, leading to cell cycle arrests and/or senescence and cell death by apoptosis.210 Numerous therapeutic strategies targeting MDM2 have been developed, including nutlin-3 and RG7112. Nutlin-3 activates the p53 signalling pathway and was shown to lead to major tumour regression in osteosarcoma xenografts through activation of apoptosis.184 In a phase I study, sarcoma patients with MDM2-amplified liposarcoma treated with RG7112 showed a significant reduction in tumour growth.19
Aurora kinases (AURKs) constitute a family of serine/threonine kinases that are required for progression through mitosis and cell division. Indeed, they are important in centrosome duplication, spindle formation, alignment of chromosomes on the mitotic spindle, as well as transition through the mitotic checkpoints and cytokinesis.211 Aberrant expression of AURKA has been implicated in many cancers and contributes to chromosome instability and phosphorylation-mediated ubiquitylation and degradation of the tumour suppressor p53.212 In vitro, inhibition of AURKA by shRNA or chemical inhibitors reduces cell proliferation in multiple sarcoma cell lines; results from a recent phase II clinical trial revealed that alisertib, a small-molecule inhibitor of AURKA, significantly improved prolonged stable disease in angiosarcoma and chondrosarcoma patients.184,207,213
Immunotherapy
Immunotherapy is a treatment designed to harness the ability of the body’s immune system to combat infection or disease to either produce an immune response or to enhance immune resistance to disease. With an advanced understanding of the immune system and cancer immunology, immunotherapy has made a significant impact on the oncology world in the last few years, with promising treatments for diverse malignancies including melanoma, leukaemia, prostate cancer, lung cancer and renal cell carcinoma.214–219 Unfortunately, advancements in immunotherapy for the treatment of sarcomas have made limited progress, which can partly be attributed to the rarity and heterogeneity of this cancer type. Recently, however, sarcoma research has turned a corner with numerous immunotherapy research initiatives and multiple early-phase clinical trials underway which include cytokine therapies, adoptive cell therapy, therapeutic cancer vaccines and checkpoint inhibitors/immune modulators.220,221 The efficacy of an immune checkpoint inhibitor in sarcomas has only been evaluated by a phase II study that administered ipilimumab, a CTLA-4 inhibitor, to synovial sarcoma patients. However, the study was closed when none of the patients had an objective tumour response222,223 While the checkpoint inhibitor data have been disappointing, it is anticipated that immunotherapies will improve the prognosis of sarcoma patients.224,225
Mifamurtide, a novel immunomodulator for the treatment of osteosarcoma
Mifamurtide (muramyl tripeptide phosphatidylethanolamine or MTP-PE) (Figure 12) is an immunomodulator that exhibits anti-cancer activity through activation of monocytes and macrophages. It is a synthetic analogue of the immune stimulatory peptidoglycan motif known as muramyl dipeptide (MDP) found in the cell wall of Gram-positive and Gram-negative bacteria.226,227 Mifamurtide is encapsulated in liposomes, which favours targeted delivery and enhances the compound’s ability to activate macrophages and monocytes and also reduces the compound’s toxicity.228–230 Intracellularly, MTP-PE binds to the nucleotide-binding oligomerization domain (Nod) 2 receptor, which is highly expressed in antigen presenting cells.231,232 This binding stimulates the production of pro-inflammatory molecules including interleukin (IL)-1, IL-6, IL-8, tumour necrosis factor alpha, nitric oxide and prostaglandins D23 and E2.233–235 Upregulation of these molecules leads to activation of contact-mediated tumouricidal activity of macrophages and monocytes.236,237 Immunomodulation is of particular relevance in the case of osteosarcoma as there are numerous signalling pathways such as receptor activator of nuclear factor kappa-B ligand (RANKL) signalling, cytokines including IL-1, IL-6, IL-17 and transforming growth factor-β that have overlapping roles in bone and the immune system.238,239 However, there is limited understanding of the cross-talk between osteosarcoma cells, osteoclasts and cells of the immune system and how they may promote tumourigenesis.240,241
Figure 12.

Mifamurtide.
In 2009 mifamurtide was approved by the European Medical Agency for the treatment of high-grade non-metastatic resectable osteosarcoma following surgical removal in children, adolescents and young adults.239,242–244 This approval was prompted by the promising data generated from a large phase III randomized prospective intergroup trial.245 Results showed that intravenous treatment with mifamurtide after complete surgical resection and postoperative multi-agent chemotherapy significantly improved the six-year overall survival from 70% to 78% in patients with newly diagnosed osteosarcoma; patients with metastatic disease showed improvement in five-year overall survival from 40% to 53%.246,247 Mifamurtide is generally well tolerated, with reported adverse effects including fever, chills, nausea, headache, fatigue and myalgias.242,243,248 Although mifamurtide is not yet approved in the US, several trials [ClinicalTrials.gov identifier: NCT014559484] are currently underway to further investigate its efficacy in osteosarcoma.239
Conclusion
Sarcomas continue to present a serious therapeutic challenge, mostly due to the large number of sarcoma subtypes, the heterogeneity within them and their different responses to current treatments. It is anticipated that the identification of the key molecular drivers underlying the various sarcoma subtypes will reveal biomarkers for more reliable diagnosis of sarcomas and lead to the development of more effective targeted therapies.
Footnotes
Funding: This research was supported by grants from the SA Medical Research Council, National Research Foundation (NRF), Cancer Association of South Africa (CANSA) and the University of Cape Town
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Jenna S. Bleloch, Department of Human Biology, University of Cape Town, South Africa
Reyna D. Ballim, Department of Human Biology, University of Cape Town, South Africa
Serah Kimani, Department of Human Biology, University of Cape Town, South Africa.
Jeannette Parkes, Department of Radiation Oncology, University of Cape Town, South Africa.
Eugenio Panieri, Department of Surgery, University of Cape Town, South Africa.
Tarryn Willmer, Department of Human Biology, University of Cape Town, South Africa.
Sharon Prince, Department of Human Biology, Faculty of Health Sciences, University of Cape Town, Anzio Road, Observatory, 7925, South Africa.
References
- 1. Skubitz KM, D’Adamo DR. Sarcoma. Mayo Clin Proc 2007; 82: 1409–1432. [DOI] [PubMed] [Google Scholar]
- 2. Lye KL, Nordin N, Vidyadaran S, et al. Mesenchymal stem cells: from stem cells to sarcomas. Cell Biol Int 2016; 40: 610–618. [DOI] [PubMed] [Google Scholar]
- 3. Xiao W, Mohseny AB, Hogendoorn PCW. Mesenchymal stem cell transformation and sarcoma genesis. Clin Sarcoma Res 2013; 3: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Teicher BA. Searching for molecular targets in sarcoma. Biochem Pharmacol 2012; 84: 1–10. [DOI] [PubMed] [Google Scholar]
- 5. Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer 2003; 3: 685–694. [DOI] [PubMed] [Google Scholar]
- 6. Francis P, Namløs HM, Müller C, et al. Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics 2007; 8: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jain S, Xu R, Prieto VG, et al. Molecular classification of soft tissue sarcomas and its clinical applications. Int J Clin Exp Pathol 2010; 3: 416–428. [PMC free article] [PubMed] [Google Scholar]
- 8. Osuna D, de Alava E. Molecular pathology of sarcomas. Rev Recent Clin Trials 2009; 4: 12–26. [DOI] [PubMed] [Google Scholar]
- 9. Matushansky I, Maki RG, Cormier JN, et al. Mechanisms of sarcomagenesis. Hematol Oncol Clin North Am 2005; 19: 427–449. [DOI] [PubMed] [Google Scholar]
- 10. Oda Y, Tsuneyoshi M. Recent advances in the molecular pathology of soft tissue sarcoma: implications for diagnosis, patient prognosis, and molecular target therapy in the future. Cancer Sci 2009; 100: 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kawaguchi N, Ahmed AR, Manabe J, et al. The concept of curative margin in surgery for bone and soft tissue sarcoma. Clin Orthop Relat Res 2004; 419: 165–172. [DOI] [PubMed] [Google Scholar]
- 12. Trovik CS, Bauer HC, Alvegård TA, et al. Surgical margins, local recurrence and metastasis in soft tissue sarcomas: 559 surgically-treated patients from the Scandinavian Sarcoma Group Register. Eur J Cancer 2000; 36: 710–716. [DOI] [PubMed] [Google Scholar]
- 13. Strauss DC, Hayes AJ, Thway K, et al. Surgical management of primary retroperitoneal sarcoma. Br J Surg 2010; 97: 698–706. [DOI] [PubMed] [Google Scholar]
- 14. Sandrucci S, Trama A, Quagliuolo V, et al. Accreditation for centers of sarcoma surgery. Updates Surg 2017; 69: 1–7. [DOI] [PubMed] [Google Scholar]
- 15. Abed R, Grimer R. Surgical modalities in the treatment of bone sarcoma in children. Cancer Treat Rev 2010; 36: 342–347. [DOI] [PubMed] [Google Scholar]
- 16. Gronchi A, Raut CP. Treatment of localized sarcomas. Hematol Oncol Clin North Am 2013; 27: 921–938. [DOI] [PubMed] [Google Scholar]
- 17. Gronchi A, Verderio P, De Paoli A, et al. Quality of surgery and neoadjuvant combined therapy in the ISG-GEIS trial on soft tissue sarcomas of limbs and trunk wall. Ann Oncol 2013; 24: 817–823. [DOI] [PubMed] [Google Scholar]
- 18. Italiano A, Le Cesne A, Bellera C, et al. GDC-0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann Oncol 2013; 24: 2922–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sborov D, Chen JL. Targeted therapy in sarcomas other than GIST tumors. J Surg Oncol 2015; 111: 632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greto D, Livi L, Saieva C, et al. Neoadjuvant treatment of soft tissue sarcoma. Radiol Med 2014; 119: 195–200. [DOI] [PubMed] [Google Scholar]
- 21. Beane JD, Yang JC, White D, et al. Efficacy of adjuvant radiation therapy in the treatment of soft tissue sarcoma of the extremity: 20-year follow-up of a randomized prospective trial. Ann Surg Oncol 2014; 21: 2484–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 2002; 359: 2235–2241. [DOI] [PubMed] [Google Scholar]
- 23. Ballo MT, Zagars GK, Pollock RE, et al. Retroperitoneal soft tissue sarcoma: an analysis of radiation and surgical treatment. Int J Radiat Oncol 2007; 67: 158–163. [DOI] [PubMed] [Google Scholar]
- 24. Catton CN, O’Sullivan B, Kotwall C, et al. Outcome and prognosis in retroperitoneal soft tissue sarcoma. Int J Radiat Oncol Biol Phys 1994; 29: 1005–1010. [DOI] [PubMed] [Google Scholar]
- 25. Bortfeld T. IMRT: a review and preview. Phys Med Biol 2006; 51: R363–R379. [DOI] [PubMed] [Google Scholar]
- 26. Teh BS, Woo SY, Butler EB. Intensity modulated radiation therapy (IMRT): a new promising technology in radiation oncology. Oncologist 1999; 4: 433–442. [PubMed] [Google Scholar]
- 27. Sladowska A, Hetnał M, Dymek P, et al. Application of IMRT in adjuvant treatment of soft tissue sarcomas of the thigh: preliminary results. Reports Pract Oncol Radiother J Gt Cancer Cent Pozn Polish Soc Radiat Oncol 2011; 16: 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paumier A, Le Péchoux C, Beaudré A, et al. IMRT or conformal radiotherapy for adjuvant treatment of retroperitoneal sarcoma? Radiother Oncol 2011; 99: 73–78. [DOI] [PubMed] [Google Scholar]
- 29. Matsunobu A, Imai R, Kamada T, et al. Impact of carbon ion radiotherapy for unresectable osteosarcoma of the trunk. Cancer 2012; 118: 4555–4563. [DOI] [PubMed] [Google Scholar]
- 30. Serizawa I, Kagei K, Kamada T, et al. Carbon ion radiotherapy for unresectable retroperitoneal sarcomas. Int J Radiat Oncol 2009; 75: 1105–1110. [DOI] [PubMed] [Google Scholar]
- 31. Demizu Y, Jin D, Sulaiman NS, et al. Particle therapy using protons or carbon ions for unresectable or incompletely resected bone and soft tissue sarcomas of the pelvis. Int J Radiat Oncol 2017; 98: 367–374. [DOI] [PubMed] [Google Scholar]
- 32. Kanai T, Furusawa Y, Fukutsu K, et al. Irradiation of mixed beam and design of spread-out Bragg peak for heavy-ion radiotherapy. Radiat Res 1997; 147: 78–85. [PubMed] [Google Scholar]
- 33. Krikelis D, Judson I. Role of chemotherapy in the management of soft tissue sarcomas. Expert Rev Anticancer Ther 2010; 10: 249–260. [DOI] [PubMed] [Google Scholar]
- 34. Luetke A, Meyers PA, Lewis I, et al. Osteosarcoma treatment – where do we stand? A state of the art review. Cancer Treat Rev 2014; 40: 523–532. [DOI] [PubMed] [Google Scholar]
- 35. Carrle D, Bielack SS. Current strategies of chemotherapy in osteosarcoma. Int Orthop 2006; 30: 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grimer R, Judson I, Peake D, et al. Guidelines for the management of soft tissue sarcomas. Sarcoma 2010; 2010: 506182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jain A, Sajeevan KV, Babu KG, et al. Chemotherapy in adult soft tissue sarcoma. Indian J Cancer 2009; 46: 274–287. [DOI] [PubMed] [Google Scholar]
- 38. Lehnhardt M, Muehlberger T, Kuhnen C, et al. Feasibility of chemosensitivity testing in soft tissue sarcomas. World J Surg Oncol 2005; 3: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cutts SM, Nudelman A, Rephaeli A, et al. The power and potential of doxorubicin–DNA adducts. IUBMB Life 2005; 57: 73–81. [DOI] [PubMed] [Google Scholar]
- 40. Benjamin RS, Wiernik PH, Bachur NR. Adriamycin: a new effective agent in the therapy of disseminated sarcomas. Med Pediatr Oncol 1975; 1: 63–76. [DOI] [PubMed] [Google Scholar]
- 41. Hilmer SN, Cogger VC, Muller M, et al. The hepatic pharmacokinetics of doxorubicin and liposomal doxorubicin. Drug Metab Dispos 2004; 32: 794–799. [DOI] [PubMed] [Google Scholar]
- 42. Gewirtz D. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 1999; 57: 727–741. [DOI] [PubMed] [Google Scholar]
- 43. Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987; 5: 840–850. [DOI] [PubMed] [Google Scholar]
- 44. Mouridsen HT, Bastholt L, Somers R, et al. Adriamycin versus epirubicin in advanced soft tissue sarcomas: a randomized phase II/phase III study of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer Clin Oncol 1987; 23: 1477–1483. [DOI] [PubMed] [Google Scholar]
- 45. Edmonson JH, Ryan LM, Blum RH, et al. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993; 11: 1269–1275. [DOI] [PubMed] [Google Scholar]
- 46. Santoro A, Tursz T, Mouridsen H, et al. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995; 13: 1537–1545. [DOI] [PubMed] [Google Scholar]
- 47. Ratan R, Patel SR. Chemotherapy for soft tissue sarcoma. Cancer 2016; 122: 2952–2960. [DOI] [PubMed] [Google Scholar]
- 48. Lorigan P, Verweij J, Papai Z, et al. Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: a European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 2007; 25: 3144–3150. [DOI] [PubMed] [Google Scholar]
- 49. Huang J, Liu K, Yu Y, et al. Targeting HMGB1-mediated autophagy as a novel therapeutic strategy for osteosarcoma. Autophagy 2012; 8: 275–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Demetri GD, Elias AD. Results of single-agent and combination chemotherapy for advanced soft tissue sarcomas: implications for decision making in the clinic. Hematol Oncol Clin North Am 1995; 9: 765–785. [PubMed] [Google Scholar]
- 51. Thorn CF, Oshiro C, Marsh S, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics 2011; 21: 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bonfante V, Bonadonna G, Villani F, et al. Preliminary clinical experience with 4-epidoxorubicin in advanced human neoplasia. Recent Results Cancer Res 1980; 74: 192–199. [DOI] [PubMed] [Google Scholar]
- 53. Nielsen OS, Dombernowsky P, Mouridsen H, et al. High-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas: a study of the EORTC Soft Tissue and Bone Sarcoma Group. Br J Cancer 1998; 78: 1634–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kerbusch T, de Kraker J, Keizer HJ, et al. Clinical pharmacokinetics and pharmacodynamics of ifosfamide and its metabolites. Clin Pharmacokinet 2001; 40: 41–62. [DOI] [PubMed] [Google Scholar]
- 55. Patel SR, Vadhan-Raj S, Papadopolous N, et al. High-dose ifosfamide in bone and soft tissue sarcomas: results of phase II and pilot studies – dose–response and schedule dependence. J Clin Oncol 1997; 15: 2378–2384. [DOI] [PubMed] [Google Scholar]
- 56. Keohan ML, Taub RN. Chemotherapy for advanced sarcoma: therapeutic decisions and modalities. Semin Oncol 1997; 24: 572–579. [PubMed] [Google Scholar]
- 57. Bramwell VH, Mouridsen HT, Santoro A, et al. Cyclophosphamide versus ifosfamide: a randomized phase II trial in adult soft-tissue sarcomas. The European Organization for Research and Treatment of Cancer [EORTC], Soft Tissue and Bone Sarcoma Group. Cancer Chemother Pharmacol 1993; 31(Suppl. 2): S180–S184. [PubMed] [Google Scholar]
- 58. Antman KH, Ryan L, Elias A, et al. Response to ifosfamide and mesna: 124 previously treated patients with metastatic or unresectable sarcoma. J Clin Oncol 1989; 7: 126–131. [DOI] [PubMed] [Google Scholar]
- 59. Nielsen OS, Judson I, van Hoesel Q, et al. Effect of high-dose ifosfamide in advanced soft tissue sarcomas: a multicentre phase II study of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer 2000; 36: 61–67. [DOI] [PubMed] [Google Scholar]
- 60. Cerny T, Leyvraz S, von Briel T, et al. Saturable metabolism of continuous high-dose ifosfamide with mesna and GM-CSF: a pharmacokinetic study in advanced sarcoma patients. Swiss Group for Clinical Cancer Research (SAKK). Ann Oncol 1999; 10: 1087–1094. [DOI] [PubMed] [Google Scholar]
- 61. Buesa JM, López-Pousa A, Martín J, et al. Phase II trial of first-line high-dose ifosfamide in advanced soft tissue sarcomas of the adult: a study of the Spanish Group for Research on Sarcomas (GEIS). Ann Oncol 1998; 9: 871–876. [DOI] [PubMed] [Google Scholar]
- 62. Sutton G, Blessing JA, Park R, et al. Ifosfamide treatment of recurrent or metastatic endometrial stromal sarcomas previously unexposed to chemotherapy: a study of the Gynecologic Oncology Group. Obstet Gynecol 1996; 87: 747–750. [DOI] [PubMed] [Google Scholar]
- 63. van Oosterom AT, Mouridsen HT, Nielsen OS, et al. Results of randomised studies of the EORTC Soft Tissue and Bone Sarcoma Group (STBSG) with two different ifosfamide regimens in first- and second-line chemotherapy in advanced soft tissue sarcoma patients. Eur J Cancer 2002; 38: 2397–2406. [DOI] [PubMed] [Google Scholar]
- 64. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol 2013; 65: 157–170. [DOI] [PubMed] [Google Scholar]
- 65. Tascilar M, Loos WJ, Seynaeve C, et al. The pharmacologic basis of ifosfamide use in adult patients with advanced soft tissue sarcomas. Oncologist 2007; 12: 1351–1360. [DOI] [PubMed] [Google Scholar]
- 66. Küpfer A, Aeschlimann C, Cerny T. Methylene blue and the neurotoxic mechanisms of ifosfamide encephalopathy. Eur J Clin Pharmacol 1996; 50: 249–252. [DOI] [PubMed] [Google Scholar]
- 67. Furlanut M, Franceschi L. Pharmacology of ifosfamide. Oncology 2003; 65: 2–6. [DOI] [PubMed] [Google Scholar]
- 68. Nicolao P, Giometto B. Neurological toxicity of ifosfamide. Oncology 2003; 65: 11–16. [DOI] [PubMed] [Google Scholar]
- 69. Palumbo R, Palmeri S, Antimi M, et al. Phase II study of continuous-infusion high-dose ifosfamide in advanced and/or metastatic pretreated soft tissue sarcomas. Ann Oncol 1997; 8: 1159–1162. [DOI] [PubMed] [Google Scholar]
- 70. Schoenike SE, Dana WJ. Ifosfamide and mesna. Clin Pharm 1990; 9: 179–191. [PubMed] [Google Scholar]
- 71. Siu LL, Moore MJ. Use of mesna to prevent ifosfamide-induced urotoxicity. Support Care Cancer 1998; 6: 144–154. [DOI] [PubMed] [Google Scholar]
- 72. Merimsky O, Meller I, Flusser G, et al. Gemcitabine in soft tissue or bone sarcoma resistant to standard chemotherapy: a phase II study. Cancer Chemother Pharmacol 2000; 45: 177–181. [DOI] [PubMed] [Google Scholar]
- 73. Pautier P, Floquet A, Penel N, et al. Randomized multicenter and stratified phase II study of gemcitabine alone versus gemcitabine and docetaxel in patients with metastatic or relapsed leiomyosarcomas: a Federation Nationale des Centres de Lutte Contre le Cancer (FNCLCC) French Sarcoma Group. Oncologist 2012; 17: 1213–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hartmann JT, Oechsle K, Huober J, et al. An open label, non-comparative phase II study of gemcitabine as salvage treatment for patients with pretreated adult type soft tissue sarcoma. Invest New Drugs 2006; 24: 249–253. [DOI] [PubMed] [Google Scholar]
- 75. Von Burton G, Rankin C, Zalupski MM, et al. Phase II trial of gemcitabine as first line chemotherapy in patients with metastatic or unresectable soft tissue sarcoma. Am J Clin Oncol 2006; 29: 59–61. [DOI] [PubMed] [Google Scholar]
- 76. Cerqueira NMFSA, Fernandes PA, Ramos MJ. Understanding ribonucleotide reductase inactivation by gemcitabine. Chemistry 2007; 13: 8507–8515. [DOI] [PubMed] [Google Scholar]
- 77. Hyman DM, Grisham RN, Hensley ML. Management of advanced uterine leiomyosarcoma. Curr Opin Oncol 2014; 26: 422–427. [DOI] [PubMed] [Google Scholar]
- 78. Bramwell VH, Eisenhauer EA, Blackstein M, et al. Phase II study of topotecan (NSC 609 699) in patients with recurrent or metastatic soft tissue sarcoma. Ann Oncol 1995; 6: 847–849. [DOI] [PubMed] [Google Scholar]
- 79. Blanchette P, Hogg D, Ferguson P, et al. Topotecan and cyclophosphamide in adults with relapsed sarcoma. Sarcoma 2012; 2012: 749067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Farhat R, Raad R, Khoury NJ, et al. Cyclophosphamide and topotecan as first-line salvage therapy in patients with relapsed Ewing sarcoma at a single institution. J Pediatr Hematol Oncol 2013; 35: 356–360. [DOI] [PubMed] [Google Scholar]
- 81. Hunold A, Weddeling N, Paulussen M, et al. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer 2006; 47: 795–800. [DOI] [PubMed] [Google Scholar]
- 82. Hagiwara H, Sunada Y. Mechanism of taxane neurotoxicity. Breast Cancer 2004; 11: 82–85. [DOI] [PubMed] [Google Scholar]
- 83. Skubitz KM, Haddad PA. Paclitaxel and pegylated-liposomal doxorubicin are both active in angiosarcoma. Cancer 2005; 104: 361–366. [DOI] [PubMed] [Google Scholar]
- 84. Fata F, O’Reilly E, Ilson D, et al. Paclitaxel in the treatment of patients with angiosarcoma of the scalp or face. Cancer 1999; 86: 2034–2037. [PubMed] [Google Scholar]
- 85. Penel N, Bui BN, Bay J-O, et al. Phase II trial of weekly paclitaxel for unresectable angiosarcoma: the ANGIOTAX Study. J Clin Oncol 2008; 26: 5269–5274. [DOI] [PubMed] [Google Scholar]
- 86. Schlemmer M, Reichardt P, Verweij J, et al. Paclitaxel in patients with advanced angiosarcomas of soft tissue: a retrospective study of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer 2008; 44: 2433–2436. [DOI] [PubMed] [Google Scholar]
- 87. Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010; 11: 983–991. [DOI] [PubMed] [Google Scholar]
- 88. Casanova M, Ferrari A, Spreafico F, et al. Vinorelbine in previously treated advanced childhood sarcomas: evidence of activity in rhabdomyosarcoma. Cancer 2002; 94: 3263–3268. [DOI] [PubMed] [Google Scholar]
- 89. Anderson SE, Keohan ML, D’Adamo DR, et al. A retrospective analysis of vinorelbine chemotherapy for patients with previously treated soft-tissue sarcomas. Sarcoma 2006; 2006: 15947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zucali PA, Bertuzzi A, Parra HJS, et al. The ‘old drug’ dacarbazine as a second/third line chemotherapy in advanced soft tissue sarcomas. Invest New Drugs 2008; 26: 175–181. [DOI] [PubMed] [Google Scholar]
- 91. Garcia del Muro X, Lopez-Pousa A, Martin J, et al. A phase II trial of temozolomide as a 6-week, continuous, oral schedule in patients with advanced soft tissue sarcoma: a study by the Spanish Group for Research on Sarcomas. Cancer 2005; 104: 1706–1712. [DOI] [PubMed] [Google Scholar]
- 92. Thigpen JT, Blessing JA, Beecham J, et al. Phase II trial of cisplatin as first-line chemotherapy in patients with advanced or recurrent uterine sarcomas: a Gynecologic Oncology Group study. J Clin Oncol 1991; 9: 1962–1966. [DOI] [PubMed] [Google Scholar]
- 93. Goldstein D, Cheuvart B, Trump DL, et al. Phase II trial of carboplatin in soft-tissue sarcoma. Am J Clin Oncol 1990; 13: 420–423. [DOI] [PubMed] [Google Scholar]
- 94. Germano G, Frapolli R, Belgiovine C, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013; 23: 249–262. [DOI] [PubMed] [Google Scholar]
- 95. Herrero AB, Martín-Castellanos C, Marco E, et al. Cross-talk between nucleotide excision and homologous recombination DNA repair pathways in the mechanism of action of antitumor trabectedin. Cancer Res 2006; 66: 8155–8162. [DOI] [PubMed] [Google Scholar]
- 96. Larsen AK, Galmarini CM, D’Incalci M. Unique features of trabectedin mechanism of action. Cancer Chemother Pharmacol 2016; 77: 663–671. [DOI] [PubMed] [Google Scholar]
- 97. D’Incalci M, Galmarini CM. A review of trabectedin (ET-743): a unique mechanism of action. Mol Cancer Ther 2010; 9: 2157–2163. [DOI] [PubMed] [Google Scholar]
- 98. D’Incalci M, Badri N, Galmarini CM, et al. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br J Cancer 2014; 111: 646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yovine A, Riofrio M, Blay JY, et al. Phase II study of ecteinascidin-743 in advanced pretreated soft tissue sarcoma patients. J Clin Oncol 2004; 22: 890–899. [DOI] [PubMed] [Google Scholar]
- 100. Le Cesne A, Blay JY, Judson I, et al. Phase II study of ET-743 in advanced soft tissue sarcomas: a European Organisation for the Research and Treatment of Cancer (EORTC) soft tissue and bone sarcoma group trial. J Clin Oncol 2005; 23: 576–584. [DOI] [PubMed] [Google Scholar]
- 101. Gajdos C, Elias A. Trabectedin: safety and efficacy in the treatment of advanced sarcoma. Clin Med Insights Oncol 2011; 5: 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013; 24: 1703–1709. [DOI] [PubMed] [Google Scholar]
- 103. Brodowicz T. Trabectedin in soft tissue sarcomas. Future Oncol 2014; 10: s1–s5. [DOI] [PubMed] [Google Scholar]
- 104. Peugniez C, Cousin S, Penel N. Trabectedin is an effective second-line treatment in soft tissue sarcoma patients. Ann Oncol 2016; 27: 551–552. [DOI] [PubMed] [Google Scholar]
- 105. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol 2016; 34: 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Quesada J, Amato R. The molecular biology of soft-tissue sarcomas and current trends in therapy. Sarcoma 2012; 2012: 849456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Forni C, Minuzzo M, Virdis E, et al. Trabectedin (ET-743) promotes differentiation in myxoid liposarcoma tumors. Mol Cancer Ther 2009; 8: 449–457. [DOI] [PubMed] [Google Scholar]
- 108. Bui-Nguyen B, Butrynski JE, Penel N, et al. A phase IIb multicentre study comparing the efficacy of trabectedin to doxorubicin in patients with advanced or metastatic untreated soft tissue sarcoma: the TRUSTS trial. Eur J Cancer 2015; 51: 1312–1320. [DOI] [PubMed] [Google Scholar]
- 109. Romero D. Sarcoma: eribulin – a welcomed advance. Nat Rev Clin Oncol 2016; 13: 204. [DOI] [PubMed] [Google Scholar]
- 110. Hirata Y, Uemura D. Halichondrins: antitumor polyether macrolides from a marine sponge. Pure Appl Chem 2009: 17: 2199–2203. [Google Scholar]
- 111. Bai R, Nguyen TL, Burnett JC, et al. Interactions of halichondrin B and eribulin with tubulin. J Chem Inf Model 2011; 51: 1393–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Smith JA, Wilson L, Azarenko O, et al. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry 2010; 49: 1331–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wilson L, Lopus M, Miller HP, et al. Effects of eribulin on microtubule binding and dynamic instability are strengthened in the absence of the βIII tubulin isotype. Biochemistry 2015; 54: 6482–6489. [DOI] [PubMed] [Google Scholar]
- 114. Swami U, Shah U, Goel S. Eribulin in cancer treatment. Mar Drugs 2015; 13: 5016–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Jain S, Cigler T. Eribulin mesylate in the treatment of metastatic breast cancer. Biologics 2012; 6: 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Swami U, Chaudhary I, Ghalib MH, et al. Eribulin: a review of preclinical and clinical studies. Crit Rev Oncol Hematol 2012; 81: 163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Preston JN, Trivedi MV. Eribulin: a novel cytotoxic chemotherapy agent. Ann Pharmacother 2012; 46: 802–811. [DOI] [PubMed] [Google Scholar]
- 118. Kawano S, Asano M, Adachi Y, et al. Antimitotic and non-mitotic effects of eribulin mesilate in soft tissue sarcoma. Anticancer Res 2016; 36: 1553–1561. [PubMed] [Google Scholar]
- 119. Schoffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate (E7389) in patients with soft tissue sarcoma (STS): phase II studies of the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group (EORTC 62052). ASCO Meet Abstr 2010; 28: 10031. [Google Scholar]
- 120. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet 2016; 387: 1629–1637. [DOI] [PubMed] [Google Scholar]
- 121. Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histological subtypes. Lancet Oncol 2011; 12: 1045–1052. [DOI] [PubMed] [Google Scholar]
- 122. Bramwell VHC, Anderson D, Charette ML. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane database Syst Rev 2003; 3: CD003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hannay JAF. Soft tissue sarcoma: biology and therapeutic correlates. PhD thesis, University of Glasgow, UK, 2015. [Google Scholar]
- 124. Worden FP, Taylor JMG, Biermann JS, et al. Randomized phase II evaluation of 6 g/m2 of ifosfamide plus doxorubicin and granulocyte colony-stimulating factor (G-CSF) compared with 12 g/m2 of ifosfamide plus doxorubicin and G-CSF in the treatment of poor-prognosis soft tissue sarcoma. J Clin Oncol 2005; 23: 105–112. [DOI] [PubMed] [Google Scholar]
- 125. Le Cesne A, Judson I, Crowther D, et al. Randomized phase III study comparing conventional-dose doxorubicin plus ifosfamide versus high-dose doxorubicin plus ifosfamide plus recombinant human granulocyte-macrophage colony-stimulating factor in advanced soft tissue sarcomas: a trial of the Europe. J Clin Oncol 2000; 18: 2676–2684. [DOI] [PubMed] [Google Scholar]
- 126. Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993; 11: 1276–1285. [DOI] [PubMed] [Google Scholar]
- 127. Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002; 20: 2824–2831. [DOI] [PubMed] [Google Scholar]
- 128. Leu KM, Ostruszka LJ, Shewach D, et al. Laboratory and clinical evidence of synergistic cytotoxicity of sequential treatment with gemcitabine followed by docetaxel in the treatment of sarcoma. J Clin Oncol 2004; 22: 1706–1712. [DOI] [PubMed] [Google Scholar]
- 129. Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007; 109: 1863–1869. [DOI] [PubMed] [Google Scholar]
- 130. Sleijfer S, Ouali M, van Glabbeke M, et al. Prognostic and predictive factors for outcome to first-line ifosfamide-containing chemotherapy for adult patients with advanced soft tissue sarcomas: an exploratory, retrospective analysis on large series from the European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group (EORTC-STBSG). Eur J Cancer 2010; 46: 72–83. [DOI] [PubMed] [Google Scholar]
- 131. Katz D, Boonsirikamchai P, Choi H, et al. Efficacy of first-line doxorubicin and ifosfamide in myxoid liposarcoma. Clin Sarcoma Res 2012; 2: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Judson I, Verweij J, Gelderblom H, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol 2014; 15: 415–423. [DOI] [PubMed] [Google Scholar]
- 133. Hensley ML, Ishill N, Soslow R, et al. Adjuvant gemcitabine plus docetaxel for completely resected stages I–IV high grade uterine leiomyosarcoma: results of a prospective study. Gynecol Oncol 2009; 112: 563–567. [DOI] [PubMed] [Google Scholar]
- 134. Palumbo R, Palmeri S, Gatti C, et al. Combination chemotherapy using vincristine, adriamycin, cyclophosphamide (VAC) alternating with ifosfamide and etoposide (IE) for advanced soft tissue sarcomas: a phase II study. Oncol Rep 1998; 5: 69–72. [DOI] [PubMed] [Google Scholar]
- 135. Saeter G, Talle K, Solheim OP. Treatment of advanced, high-grade soft-tissue sarcoma with ifosfamide and continuous-infusion etoposide. Cancer Chemother Pharmacol 1995; 36: 172–175. [DOI] [PubMed] [Google Scholar]
- 136. Bramwell V, Rouesse J, Steward W, et al. Adjuvant CYVADIC chemotherapy for adult soft tissue sarcoma: reduced local recurrence but no improvement in survival – a study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1994; 12: 1137–1149. [DOI] [PubMed] [Google Scholar]
- 137. Subramanian S, Wiltshaw E. Chemotherapy of sarcoma: a comparison of three regimens. Lancet 1978; 1: 683–686. [DOI] [PubMed] [Google Scholar]
- 138. Edmonson JH, Long HJ, Kvols LK, et al. Can molgramostim enhance the antitumor effects of cytotoxic drugs in patients with advanced sarcomas? Ann Oncol 1997; 8: 637–641. [DOI] [PubMed] [Google Scholar]
- 139. Jelić S, Kovcin V, Milanović N, et al. Randomised study of high-dose epirubicin versus high-dose epirubicin–cisplatin chemotherapy for advanced soft tissue sarcoma. Eur J Cancer 1997; 33: 220–225. [DOI] [PubMed] [Google Scholar]
- 140. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998; 279: 577–580. [DOI] [PubMed] [Google Scholar]
- 141. Lasota J, Miettinen M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin Diagn Pathol 2006; 23: 91–102. [DOI] [PubMed] [Google Scholar]
- 142. Yu J, Ustach C, Kim HR. Platelet-derived growth factor signaling and human cancer. J Biochem Mol Biol 2003; 36: 49–59. [DOI] [PubMed] [Google Scholar]
- 143. Rubin BP, Singer S, Tsao C, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001; 61: 8118–8121. [PubMed] [Google Scholar]
- 144. Lux ML, Rubin BP, Biase TL, et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 2000; 156: 791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001; 344: 1052–1056. [DOI] [PubMed] [Google Scholar]
- 146. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 147. Chao C, Al-Saleem T, Brooks JJ, et al. Vascular endothelial growth factor and soft tissue sarcomas: tumor expression correlates with grade. Ann Surg Oncol 2001; 8: 260–267. [DOI] [PubMed] [Google Scholar]
- 148. Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol 2002; 20: 4368–4380. [DOI] [PubMed] [Google Scholar]
- 149. Zhang L, Hannay JAF, Liu J, et al. Vascular endothelial growth factor overexpression by soft tissue sarcoma cells: implications for tumor growth, metastasis, and chemoresistance. Cancer Res 2006; 66: 8770–8778. [DOI] [PubMed] [Google Scholar]
- 150. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002; 347: 472–480. [DOI] [PubMed] [Google Scholar]
- 151. Judson IR. Prognosis, imatinib dose, and benefit of sunitinib in GIST: knowing the genotype. J Clin Oncol 2008; 26: 5322–5325. [DOI] [PubMed] [Google Scholar]
- 152. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006; 368: 1329–1338. [DOI] [PubMed] [Google Scholar]
- 153. van der Graaf WTA, Blay J-Y, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012; 379: 1879–1886. [DOI] [PubMed] [Google Scholar]
- 154. Mol CD, Dougan DR, Schneider TR, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem 2004; 279: 31655–31663. [DOI] [PubMed] [Google Scholar]
- 155. Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006; 42: 1093–1103. [DOI] [PubMed] [Google Scholar]
- 156. Hirota S, Ohashi A, Nishida T, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003; 125: 660–667. [DOI] [PubMed] [Google Scholar]
- 157. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003; 299: 708–710. [DOI] [PubMed] [Google Scholar]
- 158. Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005; 23: 5357–5364. [DOI] [PubMed] [Google Scholar]
- 159. Chow LQM, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol 2007; 25: 884–896. [DOI] [PubMed] [Google Scholar]
- 160. Chugh R, Wathen JK, Maki RG, et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a Bayesian hierarchical statistical model. J Clin Oncol 2009; 27: 3148–3153. [DOI] [PubMed] [Google Scholar]
- 161. Sugiura H, Fujiwara Y, Ando M, et al. Multicenter phase II trial assessing effectiveness of imatinib mesylate on relapsed or refractory KIT-positive or PDGFR-positive sarcoma. J Orthop Sci 2010; 15: 654–660. [DOI] [PubMed] [Google Scholar]
- 162. Maki RG, D’Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol 2009; 27: 3133–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Antonescu CR, DeMatteo RP. CCR 20th anniversary commentary: a genetic mechanism of imatinib resistance in gastrointestinal stromal tumor – where are we a decade later? Clin Cancer Res 2015; 21: 3363–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Noujaim J, van der Graaf WTA, Jones RL, et al. Redefining the standard of care in metastatic leiomyosarcoma. Lancet Oncol 2015; 16: 360–362. [DOI] [PubMed] [Google Scholar]
- 165. US Food and Drug Administration Approved Drugs website. Approved Drugs – olaratumab (LARTRUVO), www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm526087.htm (2016, accessed 25 October 2016).
- 166. Schutz FAB, Choueiri TK, Sternberg CN. Pazopanib: clinical development of a potent anti-angiogenic drug. Crit Rev Oncol Hematol 2011; 77: 163–171. [DOI] [PubMed] [Google Scholar]
- 167. Harris PA, Boloor A, Cheung M, et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem 2008; 51: 4632–4640. [DOI] [PubMed] [Google Scholar]
- 168. Heudel P, Cassier P, Derbel O, et al. Pazopanib for the treatment of soft-tissue sarcoma. Clin Pharmacol 2012; 4: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Bible KC, Suman VJ, Molina JR, et al. Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol 2010; 11: 962–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Oda Y, Naka T, Takeshita M, et al. Comparison of histological changes and changes in nm23 and c-MET expression between primary and metastatic sites in osteosarcoma: a clinicopathologic and immunohistochemical study. Hum Pathol 2000; 31: 709–716. [DOI] [PubMed] [Google Scholar]
- 171. Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4: 915–925. [DOI] [PubMed] [Google Scholar]
- 172. Rong S, Jeffers M, Resau JH, et al. Met expression and sarcoma tumorigenicity. Cancer Res 1993; 53: 5355–5360. [PubMed] [Google Scholar]
- 173. Dani N, Olivero M, Mareschi K, et al. The MET oncogene transforms human primary bone-derived cells into osteosarcomas by targeting committed osteo-progenitors. J Bone Miner Res 2012; 27: 1322–1334. [DOI] [PubMed] [Google Scholar]
- 174. Coltella N, Manara MC, Cerisano V, et al. Role of the MET/HGF receptor in proliferation and invasive behavior of osteosarcoma. FASEB J 2003; 17: 1162–1164. [DOI] [PubMed] [Google Scholar]
- 175. Liao AT, McCleese J, Kamerling S, et al. A novel small molecule Met inhibitor, PF2362376, exhibits biological activity against osteosarcoma. Vet Comp Oncol 2007; 5: 177–196. [DOI] [PubMed] [Google Scholar]
- 176. Sampson ER, Martin BA, Morris AE, et al. The orally bioavailable met inhibitor PF-2341066 inhibits osteosarcoma growth and osteolysis/matrix production in a xenograft model. J Bone Miner Res 2011; 26: 1283–1294. [DOI] [PubMed] [Google Scholar]
- 177. Chen Q, Chen Q, Zhou Z, et al. The importance of Src signaling in sarcoma (review). Oncol Lett 2015; 10: 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Horng C-T, Shieh P-C, Tan T-W, et al. Paeonol suppresses chondrosarcoma metastasis through up-regulation of miR-141 by modulating PKCδ and c-Src signaling pathway. Int J Mol Sci 2014; 15: 11760–11772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Villacis RAR, Silveira SM, Barros-Filho MC, et al. Gene expression profiling in leiomyosarcomas and undifferentiated pleomorphic sarcomas: SRC as a new diagnostic marker. PLoS One 2014; 9: e102281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Wu C-M, Li T-M, Tan T-W, et al. Berberine reduces the metastasis of chondrosarcoma by modulating the α v β 3 integrin and the PKC δ, c-Src, and AP-1 signaling pathways. Evidence-Based Complement Altern Med 2013; 2013: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Shor AC, Keschman EA, Lee FY, et al. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res 2007; 67: 2800–2808. [DOI] [PubMed] [Google Scholar]
- 182. Gnoni A, Marech I, Silvestris N, et al. Dasatinib: an anti-tumour agent via Src inhibition. Curr Drug Targets 2011; 12: 563–578. [DOI] [PubMed] [Google Scholar]
- 183. Frith AE, Hirbe AC, Van Tine BA. Novel pathways and molecular targets for the treatment of sarcoma. Curr Oncol Rep 2013; 15: 378–385. [DOI] [PubMed] [Google Scholar]
- 184. Heymann D, Rédini F. Targeted therapies for bone sarcomas. Bonekey Rep. Epub ahead of print 17 July 2013. DOI: 10.1038/bonekey.2013.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Liang J, Li B, Yuan L, et al. Prognostic value of IGF-1R expression in bone and soft tissue sarcomas: a meta-analysis. Onco Targets Ther 2015; 8: 1949–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Rikhof B, de Jong S, Suurmeijer AJH, et al. The insulin-like growth factor system and sarcomas. J Pathol 2009; 217: 469–482. [DOI] [PubMed] [Google Scholar]
- 187. Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol 2010; 11: 184–192. [DOI] [PubMed] [Google Scholar]
- 188. Olmos D, Martins AS, Jones RL, et al. Targeting the insulin-like growth factor 1 receptor in Ewing’s sarcoma: reality and expectations. Sarcoma 2011; 2011: 402508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Kolb EA, Gorlick R, Houghton PJ, et al. Initial testing (stage 1) of a monoclonal antibody (SCH 717454) against the IGF-1 receptor by the pediatric preclinical testing program. Pediatr Blood Cancer 2008; 50: 1190–1197. [DOI] [PubMed] [Google Scholar]
- 190. Olmos D, Tan DSW, Jones RL, et al. Biological rationale and current clinical experience with anti-insulin-like growth factor 1 receptor monoclonal antibodies in treating sarcoma. Cancer J 2010; 16: 183–194. [DOI] [PubMed] [Google Scholar]
- 191. Olmos D, Postel-Vinay S, Molife LR, et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol 2010; 11: 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Toretsky JA, Gorlick R. IGF-1R targeted treatment of sarcoma. Lancet Oncol 2010; 11: 105–106. [DOI] [PubMed] [Google Scholar]
- 193. Vemulapalli S, Mita A, Alvarado Y, et al. The emerging role of mammalian target of rapamycin inhibitors in the treatment of sarcomas. Target Oncol 2011; 6: 29–39. [DOI] [PubMed] [Google Scholar]
- 194. Chawla SP, Tolcher AW, Staddon AP, et al. Survival results with AP23573, a novel mTOR inhibitor, in patients (pts) with advanced soft tissue or bone sarcomas: update of phase II trial. ASCO Meet Abstr 2007; 25: 10076. [Google Scholar]
- 195. Quek R, Wang Q, Morgan JA, et al. Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res 2011; 17: 871–879. [DOI] [PubMed] [Google Scholar]
- 196. Wan X, Helman LJ. The biology behind mTOR inhibition in sarcoma. Oncologist 2007; 12: 1007–1018. [DOI] [PubMed] [Google Scholar]
- 197. Demetri GD, Chawla SP, Ray-Coquard I, et al. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol 2013; 31: 2485–2492. [DOI] [PubMed] [Google Scholar]
- 198. Morgan DO. Principles of CDK regulation. Nature 1995; 374: 131–134. [DOI] [PubMed] [Google Scholar]
- 199. Murray A, Hunt T. The cell cycle: an introduction. New York: Oxford University Press. [Google Scholar]
- 200. Jacobs TW. Cell cycle control. Annu Rev Plant Physiol Plant Mol Biol 1995; 46: 317–339. [Google Scholar]
- 201. Morris DG, Bramwell VHC, Turcotte R, et al. A phase II study of flavopiridol in patients with previously untreated advanced soft tissue sarcoma. Sarcoma 2006; 2006: 64374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol 2016; 2: 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene 2007; 26: 5420–5432. [DOI] [PubMed] [Google Scholar]
- 204. Mork CN, Faller DV, Spanjaard RA. A mechanistic approach to anticancer therapy: targeting the cell cycle with histone deacetylase inhibitors. Curr Pharm Des 2005; 11: 1091–1104 [DOI] [PubMed] [Google Scholar]
- 205. Zhu J, Gu J, Ma J, et al. Histone deacetylase inhibitors repress chondrosarcoma cell proliferation. J BUON 2015; 20: 269–274. [PubMed] [Google Scholar]
- 206. Sakimura R, Tanaka K, Yamamoto S, et al. The effects of histone deacetylase inhibitors on the induction of differentiation in chondrosarcoma cells. Clin Cancer Res 2007; 13: 275–282. [DOI] [PubMed] [Google Scholar]
- 207. Demicco EG, Maki RG, Lev DC, et al. New therapeutic targets in soft tissue sarcoma. Adv Anat Pathol 2012; 19: 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Cassier PA, Lefranc A, Amela EY, et al. A phase II trial of panobinostat in patients with advanced pretreated soft tissue sarcoma: a study from the French Sarcoma Group. Br J Cancer 2013; 109: 909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Sdek P, Ying H, Chang DLF, et al. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell 2005; 20: 699–708. [DOI] [PubMed] [Google Scholar]
- 210. Yu J, Zhang L. The transcriptional targets of p53 in apoptosis control. Biochem Biophys Res Commun 2005; 331: 851–858. [DOI] [PubMed] [Google Scholar]
- 211. Carvajal RD, Tse A, Schwartz GK. Aurora kinases: new targets for cancer therapy. Clin Cancer Res 2006; 12: 6869–6875. [DOI] [PubMed] [Google Scholar]
- 212. Fu J, Bian M, Jiang Q, et al. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res 2007; 5: 1–10. [DOI] [PubMed] [Google Scholar]
- 213. Dickson MA, Mahoney MR, Tap WD, et al. Phase II study of MLN8237 (Alisertib) in advanced/metastatic sarcoma. Ann Oncol 2016; 27: 1855–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat Rev Immunol 2010; 10: 580–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Britten CM, Schuler G, Grabbe S. Immunotherapy of malignant melanoma. In: Britten CM, Kreiter S, Diken M, et al. (eds) Cancer immunotherapy meets oncology. Cham: Springer International Publishing, pp.139–154. [Google Scholar]
- 216. Rambaldi A, Biagi E, Bonini C, et al. Cell-based strategies to manage leukemia relapse: efficacy and feasibility of immunotherapy approaches. Leukemia 2015; 29: 1–10. [DOI] [PubMed] [Google Scholar]
- 217. Anagnostou VK, Brahmer JR. Cancer immunotherapy: a future paradigm shift in the treatment of non-small cell lung cancer. Clin Cancer Res 2015; 21: 976–984. [DOI] [PubMed] [Google Scholar]
- 218. Massari F, Santoni M, Ciccarese C, et al. PD-1 blockade therapy in renal cell carcinoma: current studies and future promises. Cancer Treat Rev 2015; 41: 114–121. [DOI] [PubMed] [Google Scholar]
- 219. Rosenberg SA. Decade in review: cancer immunotherapy – entering the mainstream of cancer treatment. Nat Rev Clin Oncol 2014; 11: 630–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Mitsis D, Francescutti V, Skitzki J. Current immunotherapies for sarcoma: clinical trials and rationale. Sarcoma 2016; 2016: 9757219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. D’Angelo SP, Shoushtari AN, Keohan ML, et al. Combined KIT and CTLA-4 blockade in patients with refractory GIST and other advanced sarcomas: a phase Ib study of dasatinib plus ipilimumab. Clin Cancer Res, http://clincancerres.aacrjournals.org/content/early/2017/03/19/1078-0432.CCR-16-2349 (2017, accessed 6 June 2017). [DOI] [PMC free article] [PubMed]
- 222. Ghosn M, El Rassy E, Kourie HR. Immunotherapies in sarcoma: updates and future perspectives. World J Clin Oncol 2017; 8: 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Maki RG, Jungbluth AA, Gnjatic S, et al. A pilot study of anti-CTLA4 antibody ipilimumab in patients with synovial sarcoma. Sarcoma 2013; 2013: 168145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Uehara T, Fujiwara T, Takeda K, et al. Immunotherapy for bone and soft tissue sarcomas. Biomed Res Int 2015; 2015: 820813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Mitsis D, Francescutti V, Skitzki J. Current immunotherapies for sarcoma: clinical trials and rationale. Sarcoma 2016; 2016: 9757219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Schroit AJ, Fidler IJ. Effects of liposome structure and lipid composition on the activation of the tumoricidal properties of macrophages by liposomes containing muramyl dipeptide. Cancer Res 1982; 42: 161–167. [PubMed] [Google Scholar]
- 227. Asano T, Matsushima K, Kleinerman ES. Liposome-encapsulated muramyl tripeptide up-regulates monocyte chemotactic and activating factor gene expression in human monocytes at the transcriptional and post-transcriptional levels. Cancer Immunol Immunother 1994; 38: 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Fogler WE, Fidler IJ. Comparative interaction of free and liposome-encapsulated nor-muramyl dipeptide or muramyl tripeptide phosphatidylethanolamine (3H-labelled) with human blood monocytes. Int J Immunopharmacol 1987; 9: 141–150. [DOI] [PubMed] [Google Scholar]
- 229. Sone S, Mutsuura S, Ogawara M, et al. Potentiating effect of muramyl dipeptide and its lipophilic analog encapsulated in liposomes on tumor cell killing by human monocytes. J Immunol 1984; 132: 2105–2110. [PubMed] [Google Scholar]
- 230. Fidler IJ, Brown NO, Hart IR. Species variability for toxicity of free and liposome-encapsulated muramyl peptides administered intravenously. J Biol Response Mod 1985; 4: 298–309. [PubMed] [Google Scholar]
- 231. Strober W, Murray PJ, Kitani A, et al. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol 2006; 6: 9–20. [DOI] [PubMed] [Google Scholar]
- 232. Girardin SE. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 2003; 278: 8869–8872. [DOI] [PubMed] [Google Scholar]
- 233. Dieter P, Ambs P, Fitzke E, et al. Comparative studies of cytotoxicity and the release of TNF-alpha, nitric oxide, and eicosanoids of liver macrophages treated with lipopolysaccharide and liposome-encapsulated MTP-PE. J Immunol 1995; 155: 2595–2604. [PubMed] [Google Scholar]
- 234. Asano T, McWatters A, An T, et al. Liposomal muramyl tripeptide up-regulates interleukin-1 alpha, interleukin-1 beta, tumor necrosis factor-alpha, interleukin-6 and interleukin-8 gene expression in human monocytes. J Pharmacol Exp Ther 1994; 268: 1032–1039. [PubMed] [Google Scholar]
- 235. Maeda M, Knowles RD, Kleinerman ES. Muramyl tripeptide phosphatidylethanolamine encapsulated in liposomes stimulates monocyte production of tumor necrosis factor and interleukin-1 in vitro. Cancer Commun 1991; 3: 313–321. [DOI] [PubMed] [Google Scholar]
- 236. Meyers PA. Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma. Expert Rev Anticancer Ther 2009; 9: 1035–1049. [DOI] [PubMed] [Google Scholar]
- 237. Kager L, Pötschger U, Bielack S. Review of mifamurtide in the treatment of patients with osteosarcoma. Ther Clin Risk Manag 2010; 6: 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Kansara M, Teng MW, Smyth MJ, et al. Translational biology of osteosarcoma. Nat Rev Cancer 2014; 14: 722–735. [DOI] [PubMed] [Google Scholar]
- 239. Shaikh A, Li F, Li M, et al. Present advances and future perspectives of molecular targeted therapy for osteosarcoma. Int J Mol Sci 2016; 17: 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol 2014; 5: 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol 2007; 7: 292–304. [DOI] [PubMed] [Google Scholar]
- 242. Huh WW, Egas-Bejar D, Anderson PM. New treatment options for nonmetastatic osteosarcoma: focus on mifamurtide in adolescents. Clin Oncol Adolesc Young Adults 2012; 2: 1–6. [Google Scholar]
- 243. Frampton JE. Mifamurtide. Pediatr Drugs 2010; 12: 141–153. [DOI] [PubMed] [Google Scholar]
- 244. Kager L, Tamamyan G, Bielack S. Novel insights and therapeutic interventions for pediatric osteosarcoma. Futur Oncol 2017; 13: 357–368. [DOI] [PubMed] [Google Scholar]
- 245. Anderson PM, Meyers P, Kleinerman E, et al. Mifamurtide in metastatic and recurrent osteosarcoma: a patient access study with pharmacokinetic, pharmacodynamic, and safety assessments. Pediatr Blood Cancer 2014; 61: 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Chou AJ, Kleinerman ES, Krailo MD, et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma. Cancer 2009; 115: 5339–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Meyers PA, Schwartz CL, Krailo MD, et al. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival: a report from the Children’s Oncology Group. J Clin Oncol 2008; 26: 633–638. [DOI] [PubMed] [Google Scholar]
- 248. Creaven PJ, Cowens JW, Brenner DE, et al. Initial clinical trial of the macrophage activator muramyl tripeptide-phosphatidylethanolamine encapsulated in liposomes in patients with advanced cancer. J Biol Response Mod 1990; 9: 492–498. [PubMed] [Google Scholar]