

Abstract
Development of the mammalian secondary palate involves highly dynamic morphogenetic processes, including outgrowth of palatal shelves from the oral side of the embryonic maxillary prominences, elevation of the initially vertically oriented palatal shelves to the horizontal position above the embryonic tongue, and subsequently adhesion and fusion of the paired palatal shelves at the midline to separate the oral cavity from the nasal cavity. Perturbation of any of these processes could cause cleft palate, a common birth defect that significantly affects patients’ quality of life even after surgical treatment. In addition to identifying a large number of genes required for palate development, recent studies have begun to unravel the extensive cross-regulation of multiple signaling pathways, including Sonic hedgehog, bone morphogenetic protein, fibroblast growth factor, transforming growth factor β, and Wnt signaling, and multiple transcription factors during palatal shelf growth and patterning. Multiple studies also provide new insights into the gene regulatory networks and/or dynamic cellular processes underlying palatal shelf elevation, adhesion, and fusion. Here we summarize major recent advances and integrate the genes and molecular pathways with the cellular and morphogenetic processes of palatal shelf growth, patterning, elevation, adhesion, and fusion.
Keywords: cell signaling, cleft palate, morphogenesis, growth factor, growth/development, transcription factor
Introduction
In humans and other mammals, the palate acts as a mechanical barrier separating the mouth from the nasal cavity, enabling simultaneous breathing and food intake. Structurally, the palate consists of bony hard palate anteriorly and muscular soft palate posteriorly, with the soft palate also functioning as a valve that closes off the nasal airway for swallowing and directs airflow for sound variation during speech. According to embryological origins, the palate is divided into primary and secondary palates; the primary palate contains the philtrum and the upper incisor region of the upper jaw anterior to the incisive foramen, and the secondary palate includes the rest of the hard and soft palate (Bush and Jiang 2012). The primary palate arises from the embryonic frontonasal prominence situated at the rostral boundary of the primitive mouth, whereas the secondary palate forms from outgrowths from the oral side of the paired maxillary prominences flanking the primitive mouth. The palatal outgrowths expand vertically to become palatal shelves flanking the tongue that forms from the oral side of the mandible. Subsequently, palatal shelves elevate to the horizontal position above the dorsum of the tongue, and they grow toward and fuse with each other at the midline (Fig. 1). The palatal shelves also fuse anteriorly with the primary palate and nasal septum to form the intact roof of the oral cavity. Genetic or environmental factors that directly interfere with the growth, elevation, or fusion of the palatal shelves and those that disrupt the growth or morphogenesis of the surrounding craniofacial structures, such as the mandible and tongue, often cause cleft palate, one of the most common structural birth defects in humans (Bush and Jiang 2012). Extensive human genetic studies have identified many gene mutations associated with cleft palate phenotypes (reviewed by Dixon et al. 2011). In addition, analyses of laboratory mice carrying spontaneous or induced mutations, as well as targeted null alleles or tissue-specific gene inactivation, have implicated more than 250 genes in cleft palate pathogenesis (Bush and Jiang 2012; Lane and Kaartinen 2014; an updated list of genotypes associated with cleft palate phenotypes in mice can be found by querying the Mammalian Phenotype Browser at www.informatics.jax.org/searches/MP_form.shtml). These studies have revealed involvement of extensive gene regulatory networks consisting of multiple signaling pathways and transcription factors in the control of palatal shelf growth, patterning, and fusion (recently reviewed by Bush and Jiang 2012; Lane and Kaartinen 2014; Lan et al. 2015). Here we discuss recent progress in palate development studies, focusing on integrating the genes and molecular pathways with cellular and morphogenetic mechanisms of palatogenesis.
Figure 1.

Stages of mouse palate development. (A–C) Representative hematoxylin and eosin (HE)–stained coronal sections through the middle of the anterior-posterior axis of the developing palatal shelves at E13.5 (A), E14.0 (B), and E14.5 (C). p, palatal shelf; t, tongue. Dashed box in C marks the region shown in higher magnification in D. (D, E) Higher magnification views of HE-stained coronal sections through the middle of the palatal shelves undergoing fusion at E14.5 (D) and E15.5 (E). Arrows in D point to the epithelial triangles formed from displacement of epithelial cells in the oronasal axis during formation of the midline epithelial seam (MES). Arrowhead in D points to the single cell layer MES at the midline. Arrows in E point to epithelial islands during MES dissolution. Scale bars: 100 µm.
Molecular Regulation of Palatal Shelf Growth and Patterning
Palatal shelves are composed of mesenchyme derived mainly from cranial neural crest cells covered by a layer of oral epithelium (Bush and Jiang 2012). A series of genetic and explant culture studies revealed that Sonic hedgehog (Shh), a secreted protein expressed by the early embryonic oral epithelium, is a key signal for palatal shelf outgrowth (Jeong et al. 2004; Lan and Jiang 2009). Shh signals to palatal mesenchyme through the Smoothened transmembrane protein and regulates expression of fibroblast growth factor 10 (Fgf10) through a positive feedback loop to coordinate palatal epithelial and mesenchymal proliferation (Rice et al. 2004; Lan and Jiang 2009). Shh signaling is required for activation or maintenance of expression of several transcription factors, including Foxf1, Foxf2, and Osr2, in the palatal mesenchyme (Lan and Jiang 2009). Osr2 is an intrinsic regulator of palatal mesenchyme cell proliferation, and its expression in palatal mesenchyme is also dependent on function of the Pax9 transcription factor (Lan et al. 2004; Zhou et al. 2013). Both Osr2–/– and Pax9–/– embryos exhibit cleft palate and significant reduction in Fgf10 expression in the developing palatal mesenchyme (Lan et al. 2004; Zhou et al. 2013), indicating that these transcription factors act in a gene regulatory network with Shh and Fgf10 signaling pathways (Fig. 2A).
Figure 2.
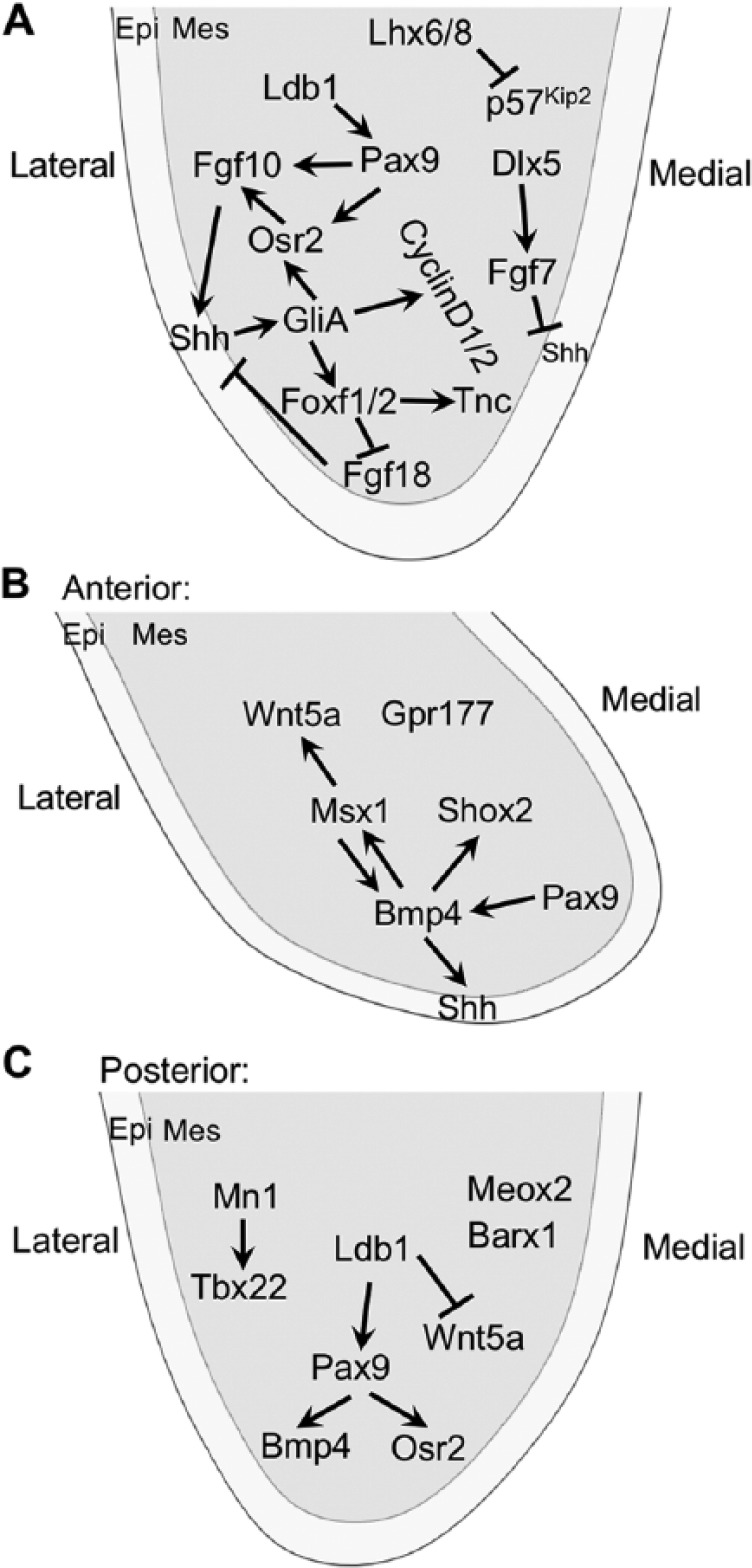
Molecular regulation of palatal shelf growth and patterning. (A) Sonic hedgehog (Shh) is a central node in the signaling networks regulating palatal shelf growth and patterning throughout the anterior-posterior axis. (B, C) Molecular pathways specific for regulating growth in the anterior and posterior regions of the palatal shelf, respectively. Arrows represent inductive relationships, and blunt arrows indicate inhibition. epi, epithelium; mes, mesenchyme.
Whereas Fgf10 function is required for maintenance of Shh expression in the palatal epithelium (Rice et al. 2004), a closely related fibroblast growth factor (FGF) family member, Fgf7, inhibits Shh expression in palatal epithelium (Han et al. 2009). Dlx5-regulated expression of Fgf7 in the medial side of the palatal mesenchyme restricts Shh expression to the lateral palatal epithelium during palatal shelf outgrowth (Han et al. 2009). Remarkably, recent genetic and explant culture studies revealed a novel Shh-Foxf1/2-Fgf18-Shh molecular circuit critical for palatal shelf outgrowth (Xu et al. 2016). Foxf2–/– mouse embryos exhibit impairment in palatal shelf growth accompanied by ectopic activation of Fgf18 expression in specific subdomains of the palatal mesenchyme and loss of Shh expression in corresponding domains of palatal epithelium (Xu et al. 2016). Moreover, mouse embryos with neural crest–specific inactivation of both Foxf1 and Foxf2 exhibit only rudimentary palatal outgrowths accompanied by ectopic Fgf18 expression throughout palatal mesenchyme (Xu et al. 2016). Explant culture assays show that exogenous Fgf18 protein inhibits Shh expression in the palatal epithelium (Xu et al. 2016). Thus, Shh signaling orchestrates several subnetworks involving distinct FGF ligands and multiple transcription factors in the regulation of palatal shelf growth and patterning (Fig. 2A). In addition, Shh signaling in the palatal mesenchyme is indirectly modulated by transforming growth factor β (TGFβ) signaling through regulation of lipid metabolism (Iwata et al. 2014).
The developing palatal shelves exhibit both morphological and molecular heterogeneity along the anterior-posterior (A/P) axis, and several pathways operate specifically in the anterior and posterior compartments, respectively (reviewed by Bush and Jiang 2012; Lan et al. 2015; Fig. 2B, C). The transcription factors Meox2 and Tbx22 are restricted to and play important roles in the development of the posterior palate (Li and Ding 2007; Pauws et al. 2009). The transcription factors Msx1 and Shox2 are restricted to anterior palatal mesenchyme, and each plays a critical role in anterior palatal mesenchyme proliferation (Zhang et al. 2002; Yu et al. 2005). Msx1 function is required for maintenance of Shh expression in the anterior palatal epithelium through maintaining Bmp4 expression in the anterior palatal mesenchyme (Zhang et al. 2002). A recent study showed that canonical Wnt signaling in developing palatal mesenchyme, detected by using the BATGAL transgenic reporter, is restricted to the anterior-most region and dependent on Gpr177-mediated Wnt secretion by the neural crest–derived mesenchyme (Liu et al. 2015). In addition, Wnt5a is preferentially expressed in the anterior palatal mesenchyme and regulates palatal mesenchyme migration and palatal shelf elongation (He et al. 2008). A transposable element-derived enhancer has been shown to confer Msx1-mediated activation of Wnt5a expression in the anterior palatal mesenchyme (Nishihara et al. 2016). On the other hand, neural crest–specific inactivation of Ldb1, encoding a cofactor for LIM domain-containing transcription factors, causes ectopic expression of Wnt5a in the posterior palatal mesenchyme (Almaidhan et al. 2014), suggesting that some LIM domain-containing transcription factors are important for palatal shelf growth and A/P patterning. Cesario et al. (2015) showed that Lhx6 and Lhx8 function partly redundantly to regulate maxillary arch and palatal mesenchyme proliferation through repression of expression of the cell cycle inhibitor gene Cdkn1c (also known as p57Kip2) and of genes encoding several FOX family transcription factors, including Foxc1, Foxd1, Foxd2, Foxp1, and Foxp2. Given the recent discovery of the Shh-Foxf1/2-Fgf18-Shh molecular circuit in early palate development (Xu et al. 2016), it remains to be determined whether Lhx6/8 also modulates the Shh and FGF signaling network during palatal shelf growth.
Palatal Shelf Elevation/Reorientation
In a specific time window during palate development, palatal shelves change rapidly from vertical orientation to the horizontal position above the developing tongue (Fig. 1A, B). Extensive histomorphological analyses in animal models indicate that the mechanism of palatal shelf elevation is heterogeneous along the A/P axis, whereby the anterior region simply flips up, whereas the middle and posterior regions reorient through protruding horizontally from the medial wall and concomitantly regressing from the ventral wall (Jin et al. 2010; Yu and Ornitz 2011). Recent analyses of expression patterns of several molecular markers associated with distinct regions of palatal shelf epithelium and mesenchyme, respectively, support this region-specific remodeling model (Chiquet et al. 2016; Fig. 3). Specifically, comparison of gene expression patterns in the middle and posterior regions of the palatal shelves before and after elevation suggests that the entire lingual portion of the vertical shelves moves close to the midline, whereas the mesenchyme at the distal tip of the vertical shelves ends up ventrolaterally (Jin et al. 2010; Chiquet et al. 2016). Moreover, analyses of cell nucleus–Golgi orientations in serial sections of developing mouse palatal shelves spanning palatal shelf growth and elevation stages showed that mesenchyme cells in the posterior palatal shelf switch their direction by almost 180°, whereas mesenchyme cells in the anterior region switch direction by about 90° at the onset of palatal shelf elevation (Brock et al. 2016). Data from combined analyses of distribution of intracellular actin fibers, extracellular matrix (ECM) meshwork, and cellular nuclear elongation suggest that tensile stress builds up in the palatal mesenchyme and actin fibers align toward the upper medial wall in the middle and posterior regions of the palatal shelves prior to elevation (Chiquet et al. 2016; Fig. 3). These data validate the decades-old hypothesis that palatal shelf elevation is driven by an “internal force” (reviewed by Ferguson 1988).
Figure 3.

Schematic representation of patterns of expression of several extracellular matrix molecules in the mid-posterior region of the palatal shelves before and after palatal shelf elevation. (A, B) The lateral and medial sides of the vertically oriented palatal shelf before elevation (A) correspond to the oral and nasal sides of the horizontally oriented palatal shelf (B) after elevation. Arrows in A indicate alignment of F-actin fibers toward the medial wall prior to shelf elevation. No actin fiber is depicted in B because the palatal mesenchyme cells in already elevated palatal shelves have less organized actin network (Chiquet et al. 2016).
Several hypotheses have been put forward to explain the mechanism generating the forces for palatal shelf elevation. One prevailing hypothesis is that hyaluronic acid (HA), an extracellular glycosaminoglycan that is capable of binding a large amount of water, accumulates at higher levels in specific regions of the palatal mesenchyme and generates osmotic pressure to drive medial-ward remodeling movement of the palatal shelf (Brinkley and Morris-Wiman 1987; reviewed by Ferguson 1988). Direct detection of HA using biotin-labeled HA-binding peptides has confirmed differential distribution of HA in the developing palatal mesenchyme (Lan et al. 2016). Moreover, mouse embryos deficient in the Golgi-associated protein Golgb1 exhibit failure of palatal shelf elevation accompanied by a significant reduction in accumulation of HA in palatal mesenchyme (Lan et al. 2016). However, whether HA or other glycosaminoglycans are involved in generating the force for palatal shelf elevation remains to be proven. On the other hand, a recent study showed that F-actin stress fibers align with nuclear orientation of palatal mesenchyme cells between the distal tip and the newly forming medial wall protrusion in the middle and posterior regions of the elevating shelf, which led to the hypothesis that palatal shelf reorientation in the middle and posterior regions is driven by actin-based cell contraction, whereas differences in ECM composition and stiffness within the palatal shelf contribute to reshaping of the tissues (Chiquet et al. 2016). Further studies are needed to elucidate whether and how actin-based cell contraction is involved in generating the forces for palatal shelf elevation/reorientation.
In addition to HA, several other ECM components are differentially expressed in developing palatal tissues. Tenascin-C is preferentially expressed in the medial half of middle and posterior regions but throughout the anterior part of palatal mesenchyme prior to shelf elevation, whereas tenascin-W expression is restricted to the nasomedial region of the palatal mesenchyme (Chiquet et al. 2016). Foxf2–/– mutant mice exhibit failure of palatal shelf elevation accompanied by significantly reduced expression of several ECM components, including tenascin-C and fibronectin, and reduced expression of an ECM receptor integrin-β1 (Nik et al. 2016).
Several transcription factors, including Osr2 and Pax9, are differentially expressed along the medial-lateral axis of palatal mesenchyme prior to palatal shelf elevation (Lan et al. 2004; Zhou et al. 2013). Both Osr2–/– and Pax9–/– embryos exhibit defects in palatal shelf elevation in addition to impaired palatal mesenchyme proliferation (Lan et al. 2004; Zhou et al. 2013). Ldb1–/– embryos exhibit reduction in expression of both Osr2 and Pax9 in the palatal mesenchyme and failure of palatal shelf elevation (Almaidhan et al. 2014). These data indicate that molecular control of palatal shelf elevation is intimately linked to the regulation of palatal shelf growth and patterning.
Palatal shelf elevation defects have been consistently observed in mouse embryos deficient in one of several components of the Wnt–planar cell polarity (PCP) pathway (He et al. 2008; Yu et al. 2010; Yang et al. 2014). Both Wnt5a and its receptor, Ror2, are required for anterior-ward directional migration of palatal mesenchyme cells during palatal shelf growth (He et al. 2008). Wnt5a–/– and Ror2–/– mice exhibit failure of palatal shelf elevation (He et al. 2008). Ror2 forms coreceptor complexes with specific frizzled proteins such as Fzd2. About 50% of Fzd2–/– mice and all Fzd1–/–Fzd2–/– mutant mice exhibit cleft palate (Yu et al. 2010). More recently, Yang et al. (2014) showed that mice deficient in Pricke1, another component of the PCP pathway, exhibit cleft palate and defect in palatal shelf elevation. Neural crest–specific inactivation of Gpr177, which impairs Wnt secretion in the palatal mesenchyme, also disrupted palatal shelf elevation (Liu et al. 2015).
Intracellular signaling of the PCP pathway is mediated by small GTPases Rac1, RhoA, and Cdc42 (reviewed by Sedgwick and D’Souza-Schorey 2016). A recent study showed that Rac1 protein levels are regionally differentially regulated in palatal mesenchyme prior to palatal shelf elevation and that overexpression of Rac1 in embryonic maxillary explant cultures disrupted palatal shelf elevation (Tang et al. 2015). Altogether, these data indicate that Wnt-PCP pathway-directed morphogenetic movements of the palatal mesenchyme are critical for palatal shelf elevation.
Palatal Shelf Adhesion and Fusion
Proper Periderm Differentiation and Maintenance Are Key to Preventing Pathological Adhesion of Palatal Shelves to Other Oral Structures
Prior to elevation, palatal shelves inhabit the space between the embryonic tongue and oral side of the mandible. Mice lacking each of Jag2, Fgf10, Irf6, and Grhl3 gene function exhibit cleft palate and aberrant adhesion or fusion of palatal shelves to mandible and/or tongue (Alappat et al. 2005; Casey et al. 2006; Ingraham et al. 2006; Richardson et al. 2006; Peyrard-Janvid et al. 2014). Analyses of these as well as Irf6/Jag2 and Irf6/p63 compound mutants have revealed a molecular network involving Jag2/Notch and Fgf10/Fgfr2 signaling, as well as the Irf6, ∆Np63, and Grhl3 transcription factors in driving differentiation of the periderm, a monolayer of flattened epithelial cells covering the external and internal surfaces of the developing embryo (reviewed by Lan et al. 2015; Kousa and Schutte 2016). The central player in this molecular network is Irf6, whose loss of function has been associated with both syndromic and nonsyndromic cleft palate in humans (reviewed by Kousa and Schutte 2016). Following initial oral epithelial stratification, ∆Np63 activates expression of Irf6, which in turn downregulates ∆Np63 protein levels in the suprabasal cell layer by promoting proteasome-mediated degradation to induce p21-mediated cell cycle exit (Richardson et al. 2009, 2014; Moretti et al. 2010; reviewed by Lan et al. 2015). Direct genetic ablation of periderm cells caused aberrant oral epithelial adhesions, indicating that the periderm acts as a nonsticking barrier to prevent pathological epithelial adhesion (Richardson et al. 2014).
Molecular Regulation of Palatal Shelf Adhesion
After elevation, the palatal shelves grow toward and make contact with each other at the midline. Whereas an intact periderm layer is key to preventing aberrant oral epithelial adhesions, the periderm covering the medial edge of palatal shelves must be removed to initiate palatal shelf fusion. Although multiple studies have detected death of the periderm cells during palatal fusion (reviewed by Nawshad 2008), the molecular mechanism controlling periderm removal during palatal fusion is still not completely understood. Extensive genetic and explant culture studies have revealed an essential role for Tgfβ3 signaling in this process (reviewed by Bush and Jiang 2012; Lan et al. 2015). Tgfβ3 expression is specifically activated in medial edge epithelial (MEE) cells and the overlying periderm cells prior to palatal shelf elevation (Lane et al. 2014). Mice lacking Tgfβ3 exhibit failure of palatal fusion (Kaartinen et al. 1995; Proetzel et al. 1995). Even when placed in direct contact in explant culture, Tgfβ3–/– mutant palatal shelves adhere poorly (Kaartinen et al. 1997). Recent studies show that Tgfβ3–/– embryos exhibit persistent periderm cells covering the palatal shelves (Hu et al. 2015). Intracellularly, Tgfβ signaling activates Smad2 and/or Smad3, which partners with Smad4 to activate expression of downstream target genes. Tgfβ signaling also activates Tgfβ-activated kinase 1 (Tak1) independently of the Smad pathway, leading to activation of the p38 MAPK pathway, and both pathways act redundantly to drive palatal fusion (Xu et al. 2008; reviewed by Bush and Jiang 2012). In addition, Lane et al. (2015) recently demonstrated that Trim33, a chromatin reader, partners with Smad2 and acts partly redundantly with Smad4 to mediate Tgfβ3 signaling during palate fusion. Remarkably, expression of the critical periderm differentiation factor Irf6 is activated in the basal layer of the MEE cells, in addition to the periderm, prior to palatal shelf fusion (Iwata et al. 2013). Whereas Tgfbr2fl/fl;K14-Cre mutant embryos exhibit failure of palatal fusion accompanied by significantly diminished MEE expression of Irf6, transgenic expression of Irf6 in basal epithelial cells was able to rescue palatal fusion in Tgfbr2fl/fl;K14-Cre embryos (Iwata et al. 2013). Similar to its function during periderm differentiation, Irf6 expression causes downregulation of ∆Np63 and an increase in p21 expression in MEE cells, which likely contributes to cell cycle exit and subsequent degeneration of the MEE (Iwata et al. 2013; Fig. 4A). In addition, Tgfβ3 is required for downregulation of Jag2 in the MEE and inhibition of Notch signaling partly rescued fusion between paired Tgfβ3–/– palatal shelves in explant culture (Jin et al. 2014). Since Jag2-Notch signaling is required for maintenance of oral periderm integrity (Casey et al. 2006), the downregulation of Jag2 in the MEE is likely an important part of the mechanism mediating Tgfβ3-dependent disruption of the periderm for palatal shelf adhesion.
Figure 4.

Molecular and cellular mechanisms of palatal adhesion and fusion. (A) Tgfβ3 signaling acts through multiple intracellular pathways and transcription factors in the medial edge epithelial (MEE) and overlying periderm to induce cell cycle exit, disrupt epithelial adhesion, and break down extracellular matrix (ECM). (B) Rho-kinase (ROCK)- and myosin light-chain kinase (MLCK)-activated nonmuscle myosin II (NMII)–mediated actomyosin contractility drives MEE cell intercalation, displacement, and extrusion during midline epithelial seam formation and breakdown. epi, epithelium; mes, mesenchyme.
Mutant mouse studies indicate that the Snail family transcription factors play a crucial role in the initiation of palatal shelf adhesion. Whereas Snai1–/– embryos die during early embryogenesis and Snai2–/– mice exhibit incomplete penetrance of cleft palate, Snai1+/–Snai2–/– compound mutant embryos exhibit persistence of palatal periderm cells and lack of palatal fusion (Murray et al. 2007). Although expression of Tgfβ3 in the MEE was unaffected in Snai1+/–Snai2–/– embryos (Murray et al. 2007), exogenous Tgfβ3 induced Snai1 messenger RNA (RNA) expression in cultured primary MEE cells via a Smad-independent pathway (Jalali et al. 2012). Recently, Ke et al. (2015) showed that Irf6 positively regulated Snai2 expression in MEE cells and that Snai2 knockdown delayed palatal fusion in explant cultures. In addition, Serrano et al. (2015) showed that experimental activation of ephrin reverse signaling increased MEE expression of Snai1 in palatal explant cultures and partly rescued palatal shelf fusion in the presence of Tgfβ3 function-blocking antibodies, suggesting that ephrin reverse signaling might act synergistically with Tgfβ3 signaling to control palatal fusion. Snai1 is a transcriptional repressor of E-cadherin expression and a crucial regulator of epithelial-mesenchymal transition (EMT) during development and cancer pathogenesis (reviewed by de Herreros and Baulida 2012). It is possible that Snai1 and Snai2 contribute to periderm desquamation by acting downstream of Tgfβ3 signaling to loosen MEE and periderm cell adhesion through downregulation of E-cadherin (Fig. 4A).
Tgfβ3 and Irf6 are also required for the activation of MMP13 expression in the palatal MEE (Richardson et al. 2009; Lane et al. 2015), which might contribute to periderm desquamation by breaking down the basement membrane (Fig. 4A). In addition, Mima et al. (2013) recently showed that expression of carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) is specifically upregulated in the MEE periderm cells prior to palatal fusion, and Ceacam1–/– mutant mouse embryos exhibit delay in completion of palatal fusion. Expression of Tgfβ3 in palatal MEE is unaffected in Ceacam1–/– embryos (Mima et al. 2013), but whether CEACAM1 acts downstream of Tgfβ3 to regulate periderm desquamation and/or palatal shelf adhesion requires further investigation. In addition, whether the molecular processes leading to desquamation also trigger apoptosis and what other molecules are involved in Tgfβ3-induced periderm cell apoptosis remain to be resolved.
Formation and Dissolution of the Midline Epithelial Seam
Upon adhesion of palatal shelves at the midline, the intervening MEE cells quickly converge to form the midline epithelial seam (MES) (Fig. 1D), which is subsequently removed to complete palatal fusion (Fig. 1E). Using genetic lineage labeling and live imaging of MEE cells in palatal explant cultures, Kim et al. (2015) showed that the initially multilayered intershelf epithelial cells converge toward the midline by cell intercalation and concurrent orthogonal cell displacement in the oronasal axis. The converging epithelial cells form rosettes, and cells in the center of rosettes are extruded by multicellular actin cable-mediated contraction (Kim et al. 2015). Through a combination of tissue-specific genetic inactivation and pharmacological inhibition assays, Kim et al. (2015) further demonstrated that Rho kinase (ROCK)-mediated and myosin light-chain kinase (MLCK)-mediated activation of contractility of nonmuscle myosin II, involving Myh9 and Myh10, is crucial for MES formation and progression through normal palatal fusion.
Whereas many cells extruded during MES formation were undergoing apoptosis, apoptotic cells were frequently detected outside of the multicellular rosettes as well (Kim et al. 2015). Tgfβ3 signaling and Irf6 function are critical for induction of MEE apoptosis and dissolution of the MES (Richardson et al. 2009; Iwata et al. 2013; Lane et al. 2015). Irf6 acts to downregulate ∆Np63 to induce p21-mediated cell cycle exit in the MEE (Iwata et al. 2013). Recently, Noda et al. (2016) showed that ectopic activation of ACVR1-mediated bone morphogenetic protein (BMP) signaling in palatal epithelium caused MEE persistence and subsequently submucous cleft palate through upregulating ∆Np63 in the MEE, indicating that downregulation of ∆Np63 is an important part of the molecular mechanism of palatal fusion. In addition, Tgfβ3 signaling affects expression of other cell cycle regulators, such as p16, in MEE cells (Lane et al. 2015). However, current understanding of the cellular and molecular processes from Tgfβ3-induced cell cycle arrest to MEE apoptosis is incomplete. Earlier studies of palatal fusion suggested that EMT is also an important mechanism for MES disintegration (reviewed by Nawshad 2008). Although several genetic lineage studies have concluded that MEE cells do not contribute to the postfusion mesenchyme cells that are maintained in the secondary palate (reviewed by Bush and Jiang 2012), the requirement for Snai1 and Snai2, known transcriptional regulators of EMT, in palatal fusion and the findings that Tgfβ3 induces expression of Snai1 and downregulation of E-cadherin in cultured MEE cells (Jalali et al. 2012) suggest that Snai1/2-mediated downregulation of E-cadherin contributes to disruption of MEE integrity. In addition, Kim et al. (2015) demonstrate that both apoptotic and crowding-induced live-cell extrusion occur during MES formation and breakdown. Taken together, combinatorial actions of Tgfβ3-Irf6 induced cell cycle arrest, Snai1/2-mediated disruption of epithelial adhesion, MMP13-mediated ECM breakdown, and actomyosin contractility-driven cell displacement and extrusion together lead to killing and/or apoptosis of the MEE cells to remove the MES and achieve palatal fusion (Fig. 4).
Concluding Remarks
A large number of genes and molecular pathways required for palatogenesis have been identified. In addition, recent studies have begun to unravel the extensive crosstalk of various signaling pathways and gene regulatory subnetworks and to integrate molecular mechanisms with cellular processes and cell behavior during palatal shelf growth, patterning, and fusion (Lane and Kaartinen 2014; Kim et al. 2015; Lan et al. 2015; Brock et al. 2016). The abundance of signaling molecules and transcription factors in and the heterogeneity and dynamics of cellular and molecular processes across the developing palatal shelves provide an outstanding paradigm for genetic regulation of mammalian organogenesis. Whereas creative combination of live imaging techniques with detailed genetic studies has provided unprecedented insight into the cellular mechanisms of palatal fusion (Kim et al. 2015), direct measurement of cell behavior during palatal shelf growth and elevation requires new technological advances since palatal explants under current culturing conditions do not fully recapitulate in vivo growth patterns (Brock et al. 2016). In addition, major gaps remain in the molecular mechanisms of cleft palate palatogenesis, particularly regarding the roles of and mechanisms involving microRNAs, various environmental factors, and gene-environment interactions (Seelan et al. 2012). As increasingly high-throughput molecular and biochemical studies, coupled with unprecedented ability to directly dissect the in vivo functions of genes, proteins, RNAs, and other factors, are rapidly improving our knowledge of the molecular networks underlying palate development, it is essential to integrate the molecular processes with cellular and tissue morphogenetic processes to gain comprehensive understanding of the genotype-phenotype functional relationships to develop effective prevention strategies for cleft palate disorders.
Author Contributions
C. Li, R. Jiang, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; Y. Lan, contributed to data acquisition, analysis, and interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Acknowledgments
Due to space limitations, we apologize to the authors whose relevant publications are not cited or discussed in this article.
Footnotes
Palate development research in the Lan and Jiang laboratories was supported by the National Institutes of Health/National Institute of Dental and Craniofacial Research (NIH/NIDCR) grants R03D E023864 (Y.L.) and R01DE013681 (R.J.) and by Shriners Hospitals for Children grant 85900 (R.J.).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Alappat SR, Zhang Z, Suzuki K, Zhang X, Liu H, Jiang R, Yamada G, Chen Y. 2005. The cellular and molecular etiology of the cleft secondary palate in Fgf10 mutant mice. Dev Biol. 277(1):102–113. [DOI] [PubMed] [Google Scholar]
- Almaidhan A, Cesario J, Malt AL, Zhao Y, Sharma N, Choi V, Jeong J. 2014. Neural crest–specific deletion of Ldb1 leads to cleft secondary palate with impaired palatal shelf elevation. BMC Dev Biol. 14:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkley LL, Morris-Wiman J. 1987. Computer-assisted analysis of hyaluronate distribution during morphogenesis of the mouse secondary palate. Development. 100(4):629–635. [DOI] [PubMed] [Google Scholar]
- Brock LJ, Economou AD, Cobourne MT, Green JB. 2016. Mapping cellular processes in the mesenchyme during palatal development in the absence of Tbx1 reveals complex proliferation changes and perturbed cell packing and polarity. J Anat. 228(3):464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush JO, Jiang R. 2012. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 139(2):231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey LM, Lan Y, Cho ES, Maltby KM, Gridley T, Jiang R. 2006. Jag2-Notch1 signaling regulates oral epithelial differentiation and palate development. Dev Dyn. 235(7):1830–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesario JM, Malt AL, Deacon LJ, Sandberg M, Vogt D, Tang Z, Zhao Y, Brown S, Rubenstein JL, Jeong J. 2015. Lhx6 and Lhx8 promote palate development through negative regulation of a cell cycle inhibitor gene, p57Kip2. Hum Mol Gen. 24(17):5024–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiquet M, Blumer S, Angelini M, Mitsiadis TA, Katsaros C. 2016. Mesenchymal remodeling during palatal shelf elevation revealed by extracellular matrix and F-actin expression patterns. Front Physiol. 7:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Herreros AG, Baulida J. 2012. Cooperation, amplification, and feed-back in epithelial-mesenchymal transition. Biochim Biophys Acta. 1825(2):223–228. [DOI] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. 2011. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 12(3):167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson MW. 1988. Palate development. Development. 103(Suppl):41–61. [DOI] [PubMed] [Google Scholar]
- Han J, Mayo J, Xu X, Li J, Bringas P, Maas RL, Rubenstein JL, Chai Y. 2009. Indirect modulation of Shh signaling by Dlx5 affects the oral-nasal patterning of palate and rescues cleft palate in Msx1-null mice. Development. 136(24):4225–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Xiong W, Yu X, Espinoza-Lewis R, Liu C, Gu S, Nishita M, Suzuki K, Yamada G, Minami Y, et al. 2008. Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development. 135(23):3871–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Liu J, Li Z, Ozturk F, Gurumurthy C, Romano RA, Sinha S, Nawshad A. 2015. TGFβ3 regulates periderm removal through ΔNp63 in the developing palate. J Cell Physiol. 230(6):1212–1225. [DOI] [PubMed] [Google Scholar]
- Ingraham CR, Kinoshita A, Kondo S, Yang B, Sajan S, Trout KJ, Malik MI, Dunnwald M, Goudy SL, Lovett M, et al. 2006. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 ( Irf6). Nat Genet. 38(11):1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J, Suzuki A, Pelikan RC, Ho TV, Sanchez-Lara PA, Chai Y. 2014. Modulation of lipid metabolic defects rescues cleft palate in Tgfbr2 mutant mice. Hum Mol Genet. 23(1):182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J, Suzuki A, Pelikan RC, Ho TV, Sanchez-Lara PA, Urata M, Dixon MJ, Chai Y. 2013. Smad4-Irf6 genetic interaction and TGFβ-mediated IRF6 signaling cascade are crucial for palatal fusion in mice. Development. 140(6):1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalali A, Zhu X, Liu C, Nawshad A. 2012. Induction of palate epithelial mesenchymal transition by transforming growth factor β3 signaling. Dev Growth Differ. 54(6):633–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. 2004. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 18(8):937–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JZ, Tan M, Warner DR, Darling DS, Higashi Y, Gridley T, Ding J. 2010. Mesenchymal cell remodeling during mouse secondary palate reorientation. Dev Dyn. 239(7):2110–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JZ, Warner DR, Lu Q, Pisano MM, Greene RM, Ding J. 2014. Deciphering TGF-β3 function in medial edge epithelium specification and fusion during mouse secondary palate development. Dev Dyn. 243(12):1536–1543. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Cui XM, Heisterkamp N, Groffen J, Shuler CF. 1997. Transforming growth factor-β3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev Dyn. 209(3):255–260. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. 1995. Abnormal lung development and cleft palate in mice lacking TGF-β3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 11(4):415–421. [DOI] [PubMed] [Google Scholar]
- Ke CY, Xiao WL, Chen CM, Lo LJ, Wong FH. 2015. IRF6 is the mediator of TGFβ3 during regulation of the epithelial mesenchymal transition and palatal fusion. Sci Rep. 5:12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lewis AE, Singh V, Ma X, Adelstein R, Bush JO. 2015. Convergence and extrusion are required for normal fusion of the mammalian secondary palate. PLoS Biol. 13(4):e1002122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousa YA, Schutte BC. 2016. Toward an orofacial gene regulatory network. Dev Dyn. 245(3):220–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Jiang R. 2009. Sonic hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal outgrowth. Development. 136(8):1387–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Ovitt CE, Cho ES, Maltby KM, Wang Q, Jiang R. 2004. Odd-skipped related 2 ( Osr2) encodes a key intrinsic regulator of secondary palate growth and morphogenesis. Development. 131(13):3207–3216. [DOI] [PubMed] [Google Scholar]
- Lan Y, Xu J, Jiang R. 2015. Cellular and molecular mechanisms of palatogenesis. Curr Top Dev Biol. 115:59–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Zhang N, Liu H, Xu J, Jiang R. 2016. Golgb1 regulates protein glycosylation and is crucial for mammalian palate development. Development. 143(13):2344–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J, Kaartinen V. 2014. Signaling networks in palate development. Wiley Interdiscip Rev Syst Biol Med. 6(3):271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J, Yumoto K, Azhar M, Ninomiya-Tsuji J, Inagaki M, Hu Y, Deng CX, Kim J, Mishina Y, Kaartinen V. 2015. Tak1, Smad4 and Trim33 redundantly mediate TGF-β3 signaling during palate development. Dev Biol. 398(2):231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J, Yumoto K, Pisano J, Azhar M, Thomas PS, Kaartinen V. 2014. Control elements targeting Tgfb3 expression to the palatal epithelium are located intergenically and in introns of the upstream Ift43 gene. Front Physiol. 5:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Ding J. 2007. Gene expression analysis reveals that formation of the mouse anterior secondary palate involves recruitment of cells from the posterior side. Int J Dev Biol. 51(2):167–172. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang M, Zhao W, Yuan X, Yang X, Li Y, Qiu M, Zhu XJ, Zhang Z. 2015. Gpr177-mediated Wnt signaling is required for secondary palate development. J Dent Res. 94(7):961–967. [DOI] [PubMed] [Google Scholar]
- Mima J, Koshino A, Oka K, Uchida H, Hieda Y, Nohara K, Kogo M, Chai Y, Sakai T. 2013. Regulation of the epithelial adhesion molecule CEACAM1 is important for palate formation. PLoS One. 8(4):e61653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti F, Marinari B, Lo Iacono N, Botti E, Giunta A, Spallone G, Garaffo G, Vernersson-Lindahl E, Merlo G, Mills AA, et al. 2010. A regulatory feedback loop involving p63 and IRF6 links the pathogenesis of 2 genetically different human ectodermal dysplasias. J Clin Invest. 120(5):1570–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray SA, Oram KF, Gridley T. 2007. Multiple functions of Snail family genes during palate development in mice. Development. 134(9):1789–1797. [DOI] [PubMed] [Google Scholar]
- Nawshad A. 2008. Palatal seam disintegration: to die or not to die? that is no longer the question. Dev Dyn. 237(10):2643–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik AM, Johansson JA, Ghiami M, Reyahi A, Carlsson P. 2016. Foxf2 is required for secondary palate development and Tgfβ signaling in palatal shelf mesenchyme. Dev Biol. 415(1):14-23. [DOI] [PubMed] [Google Scholar]
- Nishihara H, Kobayashi N, Kimura-Yoshida C, Yan K, Bormuth O, Ding Q, Nakanishi A, Sasaki T, Hirakawa M, Sumiyama K, et al. 2016. Coordinately co-opted multiple transposable elements constitute an enhancer for wnt5a expression in the mammalian secondary palate. PLoS Genet. 12(10):e1006380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda K, Mishina Y, Komatsu Y. 2016. Constitutively active mutation of ACVR1 in oral epithelium causes submucous cleft palate in mice. Dev Biol. 415(2):306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauws E, Hoshino A, Bentley L, Prajapati S, Keller C, Hammond P, Martinez-Barbera JP, Moore GE, Stanier P. 2009. Tbx22null mice have a submucous cleft palate due to reduced palatal bone formation and also display ankyloglossia and choanal atresia phenotypes. Hum Mol Gen. 18(21):4171–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrard-Janvid M, Leslie EJ, Kousa YA, Smith TL, Dunnwald M, Magnusson M, Lentz BA, Unneberg P, Fransson I, Koillinen HK, et al. 2014. Dominant mutations in GRHL3 cause Van der Woude syndrome and disrupt oral periderm development. Am J Hum Genet. 94(1):23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. 1995. Transforming growth factor-β3 is required for secondary palate fusion. Nature Genet. 11(4):409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP. 2004. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 113(12):1692–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Dixon J, Jiang R, Dixon MJ. 2009. Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum Mol Gen. 18(14):2632–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Dixon J, Malhotra S, Hardman MJ, Knowles L, Boot-Handford RP, Shore P, Whitmarsh A, Dixon MJ. 2006. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nature Genet. 38(11):1329–1334. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Hammond NL, Coulombe PA, Saloranta C, Nousiainen HO, Salonen R, Berry A, Hanley N, Headon D, Karikoski R, et al. 2014. Periderm prevents pathological epithelial adhesions during embryogenesis. J Clin Invest. 124(9):3891–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick AE, D’Souza-Schorey C. 2016. Wnt signaling in cell motility and invasion: drawing parallels between development and cancer. Cancers (Basel). 8(9):E80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelan RS, Mukhopadhyay P, Pisano MM, Greene RM. 2012. Developmental epigenetics of the murine secondary palate. ILAR J. 53(3–4): 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano MJ, Liu J, Svoboda KK, Nawshad A, Benson MD. 2015. Ephrin reverse signaling mediates palatal fusion and epithelial-to-mesenchymal transition independently of Tgfβ3. J Cell Physiol. 230(12):2961–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Li L, Jin C, Lee JM, Jung HS. 2015. Role of region-distinctive expression of Rac1 in regulating fibronectin arrangement during palatal shelf elevation. Cell Tissue Res. 361(3):857–868. [DOI] [PubMed] [Google Scholar]
- Xu J, Liu H, Lan Y, Aronow BJ, Kalinichenko VV, Jiang R. 2016. A Shh-Foxf-Fgf18-Shh molecular circuit regulating palate development. PLoS Genet. 12(1):e1005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Han J, Ito Y, Bringas P, Deng C, Chai Y. 2008. Ectodermal Smad4 and p38 MAPK are functionally redundant in mediating TGF-β/BMP signaling during tooth and palate development. Dev Cell. 15(2):322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Jia Z, Bryant-Pike W, Chandrasekhar A, Murray JC, Fritzsch B, Bassuk AG. 2014. Analysis of PRICKLE1 in human cleft palate and mouse development demonstrates rare and common variants involved in human malformations. Mol Genet Genomic Med. 2(2):138–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Smallwood PM, Wang Y, Vidaltamayo R, Reed R, Nathans J. 2010. Frizzled 1 and frizzled 2 genes function in palate, ventricular septum and neural tube closure: general implications for tissue fusion processes. Development. 137(21):3707–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Ornitz DM. 2011. Histomorphological study of palatal shelf elevation during murine secondary palate formation. Dev Dyn. 240(7):1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Gu S, Alappat S, Song Y, Yan M, Zhang X, Zhang G, Jiang Y, Zhang Z, Zhang Y, et al. 2005. Shox2-deficient mice exhibit a rare type of incomplete clefting of the secondary palate. Development. 132(19):4397–4406. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. 2002. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 129(17):4135–4146. [DOI] [PubMed] [Google Scholar]
- Zhou J, Gao Y, Lan Y, Jia S, Jiang R. 2013. Pax9 regulates a molecular network involving Bmp4, Fgf10, Shh signaling and the Osr2 transcription factor to control palate morphogenesis. Development. 140(23):4709–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]