

Abstract
Rare mutations in IRF6 and GRHL3 cause Van der Woude syndrome, an autosomal dominant orofacial clefting disorder. Common variants in IRF6 and GRHL3 also contribute risk for isolated orofacial clefting. Similarly, variants within genes that encode receptor tyrosine kinase (RTK) signaling components, including members of the FGF pathway, EPHA3 and SPRY2, also contribute risk for isolated orofacial clefting. In the mouse, loss of Irf6 or perturbation of Fgf signaling leads to abnormal oral epithelial adhesions and cleft palate. Oral adhesions can result from a disruption of periderm formation. Here, we find that IRF6 and SPRY4 signaling interact in periderm function. We crossed Irf6 heterozygous (Irf6+/–) mice with transgenic mice that express Spry4 in the basal epithelial layer (TgKRT14::Spry4). While embryos with either of these mutations can have abnormal oral adhesions, using a new quantitative assay, we observed a nonadditive effect of abnormal oral epithelial adhesions in the most severely affected double mutant embryos (Irf6+/–;TgKRT14::Spry4). At the molecular level, the sites of abnormal oral adhesions maintained periderm-like cells that express keratin 6, but we observed abnormal expression of GRHL3. Together, these data suggest that Irf6 and RTK signaling interact in regulating periderm differentiation and function, as well as provide a rationale to screen for epistatic interactions between variants in IRF6 and RTK signaling pathway genes in human orofacial clefting populations.
Keywords: GRHL3 protein, cleft lip with or without cleft palate nonsyndromic, receptor protein-tyrosine kinases, Van der Woude syndrome, popliteal pterygium syndrome, oral adhesions
Introduction
Interferon regulatory factor 6 (IRF6) regulates epidermal proliferation and differentiation (Ingraham et al. 2006; Richardson et al. 2006). Rare mutations in IRF6 cause Van der Woude syndrome (VWS; Online Mendelian Inheritance in Man [OMIM] 119300) or popliteal pterygium syndrome (PPS; OMIM 263650) (Kondo et al. 2002), which can include cleft lip and palate, lip pits, vertebral anomalies, and cutaneous and limb defects. Common variants in IRF6 can contribute risk toward isolated orofacial clefting (iCLP; OMIM 119530) (Zucchero et al. 2004; Rahimov et al. 2008). Recent work shows that IRF6 is required for periderm formation (Richardson et al. 2009; Richardson et al. 2014). Failure of periderm formation leads to pathological epithelial adhesions (Richardson et al. 2014), and it is thought that these adhesions hinder development of the palatal shelves and can lead to cleft palate.
Palatal development is a fluid and continuous process that in the mouse starts at E12.5 and ends between E15.5 and E16.5 (Bush and Jiang 2012). The progressive stages include formation of the palatal shelves, proliferation, apposition, adhesion, and fusion (Kousa and Schutte 2016). The palatal shelves are lined by an epithelium that might contact many adjacent structures in the oral cavity, including the mandible and tongue. The oral epithelium is composed of 2 layers: the superficial periderm and the basal cells. Oral periderm is a flat, squamous epithelial layer that covers basal cells. Oral periderm has many critical roles in palatal development, including preventing pathological oral adhesions between the palatal shelves and adjacent structures and then dissolving to allow strong adhesions between the apposing palatal shelves. Basal epithelium is a cuboidal epithelial layer that sits between periderm and the basement membrane, and beneath the basement membrane is mesenchyme. Basal cells proliferate to support the expanding and underlying palatal mesenchyme and then also dissolve to allow for the formation of the palate as a confluent mesenchymal bridge.
IRF6 has diverse roles in palatal development mediated through interactions with different transcription factors. IRF6 has been shown to regulate GRHL3 (de la Garza et al. 2013; Peyrard-Janvid et al. 2014), and loss of GRHL3 can lead to abnormal periderm development and cleft palate (Peyrard-Janvid et al. 2014). Furthermore, mutations in GRHL3 can also lead to VWS2 (OMIM 606713). A missense variant in GRHL3 also contributes risk toward iCP (OMIM 119540) (Leslie et al. 2016). In addition, IRF6 and TP63 are required for proper periderm development and palatogenesis (Thomason et al. 2010).
Receptor tyrosine kinase (RTK) signaling pathways are both evolutionarily conserved and diverse, regulating networks in embryonic development, cancer, and inflammation through cellular processes such as proliferation, differentiation, cell migration, and metabolism (Citri and Yarden 2006; Lemmon and Schlessinger 2010). RTK signaling pathways are composed of 20 subfamilies, including fibroblast growth factors (FGFs). RTK signaling can drive expression of Sprouty (Spry) genes, whose protein products in turn modulate RTK signaling as part of a negative feedback loop (Minowada et al. 1999; Mason et al. 2006; Dorey and Amaya 2010). This signaling cascade and feedback loop are relevant to palatal development, as common and rare variants in genes that encode for FGF ligands and receptors and SPRY2 have been implicated in isolated orofacial clefting (Vieira et al. 2005; Riley et al. 2007; Ludwig et al. 2012). Spry2 knockout mice have higher levels of FGF signaling and a cleft palate (Matsumura et al. 2011). Fgf10 and Fgfr2 null mice have palatal-lingual oral adhesions and cleft palate (Rice et al. 2004), suggesting a role in periderm development (Alappat et al. 2005).
In this work, we used mice carrying a null Irf6 allele (Ingraham et al. 2006) and a transgenic allele expressing Spry4 under the control of the KRT14 promoter (TgKRT14::Spry4) (Charles et al. 2011) to test for a genetic interaction between IRF6 and RTK signaling in palatal development. While embryos with either mutation can have abnormal oral epithelial adhesions, we find, using a new quantitative assay, that double heterozygous embryos (Irf6+/–;TgKRT14Spry4) have more extensive pathological oral epithelial adhesions. Consistently, the expression of GRHL3 was altered in the oral periderm of double heterozygous embryos. These data suggest that proper periderm function, in addition to formation, is required to prevent abnormal oral epithelial adhesions. These data also suggest that IRF6 and SPRY4 genetically interact in regulating periderm function. This is important, since common and rare FGF and IRF6 variants are known to contribute risk in human orofacial clefting, providing justification for testing for genetic interactions in orofacial clefting populations.
Methods
Murine Crosses
All murine experiments were approved by the Michigan State University Institutional Animal Care and Use Committee (AUF 05/12-093-00). Presence of a copulation plug was denoted as E0.5. We used harem matings, defined as 1 breeder male with 4 females, to enhance pregnancy rates. The Irf6 heterozygous allele is a genetrap, which functions as a null allele (Ingraham et al. 2006). We intercrossed Irf6 heterozgyous (Irf6+/gt, referred to as Irf6+/–) mice with those containing the TgKRT14::Spry4 allele, which is the KRT14 promoter upstream of Spry4 complementary DNA (cDNA) (Charles et al. 2011). TgKRT14::Spry4 were created in a CBA/J × C57BL/10J 1-cell zygote and have been maintained for nearly 10 y on a hybridized CBA/J × C57BL/10J background. The Irf6+/– genetrap allele has been maintained on an inbred C57BL/6J background for over 10 y. We examined littermate embryos at 2 developmental periods, including E13.5 to E15.5 and E16.5 to E17.5. Genotyping for the Irf6 heterozygous allele was performed as previously described (Ingraham et al. 2006). Genotyping for embryos containing the TgKRT14::Spry4 allele was performed with primers for the KRT14 promoter.
Morphological and Histological Analyses
After gross inspection, embryos were fixed in paraformaldehyde (245 to 684, per protocol). After fixing for 16 to 24 h, embryos were placed in 50% to 80% ethanol until paraffin embedding. Coronal sections of the head were obtained at 7-µm intervals. At E13.5, the tooth germ was used as a landmark to determine locations of interest in the oral cavity. Slides were selected for hematoxylin (GHS332; Sigma-Aldrich) and eosin (E511-25; Fisher Chemical) staining (Ingraham et al. 2006).
Molecular Analyses of the Murine Oral Cavity
We immunostained wild-type (n = 2), Irf6 heterozygous (Irf6+/–) (n = 3), KRT14::Spry4 transgenic (TgKRT14::Spry4) (n = 4), and double heterozygous embryos (Irf6+/–;TgKRT14::Spry4) (n = 5) for every molecular target in this manuscript. Antigen retrieval was completed in sodium citrate (pH 6.0). After antigen retrieval, permeabilization was performed in Triton X-100 (VWR). The slides were then washed to remove detergent. Two 1-h-long blocking steps were performed. The 2 incubations were bovine serum albumin (BSA) in phosphate-buffered saline (PBS) (10%) and goat anti-mouse Fab fragment in PBS (40 µg/mL) (115-007-003; Jackson ImmunoResearch Laboratories). After blocking, primary antibodies were incubated for 18 to 24 h at 4°C. Primary antibodies included TP63 (4A4, sc-8431; Santa Cruz), keratin 6 (PRB-169P; Covance), keratin 14 (NCL-L-LL002; Novocastra), IRF6 (SAB2102995; Sigma-Aldrich), GRHL3 (a kind gift from Dr. B. Andersen, University of California, Irvine), and activated caspase 3 (Ab13847; Abcam). Secondary antibodies, including goat anti-mouse and goat anti-rabbit (Molecular Probes), were incubated for 1 to 2 h at room temperature. Nuclei were labeled with DAPI (Invitrogen).
Bioimaging Using the Stereomicroscope and the Upright/Fluorescent Microscope
Images of whole-mount embryos were obtained using the SMZ1000 Nikon microscope and the NIS Elements Software v4.11. Coronel sections were visualized using a Nikon Eclipse 90i upright microscope, and images were obtained with NIS Elements Advanced Research v3.10 software. Images were enhanced uniformly using standard program algorithms, including deconvolution and sharpening. Adobe Photoshop Elements v9.0 was used to assemble and produce the figures.
Oral Adhesion Assay
NIS Elements Advanced Research v3.10 software was used to obtain measurements from hematoxylin and eosin (H&E) histology stains of oral cavity tissue at E13.5. The total oral cavity surface spanned from the medial aspect of the palatal shelves to the lateral edges of the oral cavity. The interactive measurement feature was used to measure the arc length of oral adhesions and the total oral cavity surface. These measures were exported to Microsoft Excel. Total arc length of oral adhesions and oral cavity were obtained and percent oral adhesion for each embryo was calculated based on these numbers. In immunostained images, this approach was used to measure the arc length of epithelium expressing GRHL3 relative to the total imaged epithelial surface as marked by DAPI.
Statistics
Data analysis, tables, and histograms were obtained using Excel (version 2010) and GraphPad Prism Software (version 5). A 1-way analysis of variance (ANOVA) assuming a nonparametric distribution (Kruskal-Wallis test) was performed on the percent oral adhesions and the percent oral epithelium expressing GRHL3 in both epithelial surfaces of palatal-lingual and maxillary-mandibular oral adhesions. In addition, we performed a post hoc analysis to compare all pairs of genotypes in each assay (Dunn’s multiple comparison test). We used a significance level of 0.05 (95% confidence interval).
Results
To explore potential genetic interactions between IRF6 and SPRY4 signaling, we crossed Irf6 heterozygous mice (Irf6+/gt, herein referred to as Irf6+/–) (Ingraham et al. 2006) with mice carrying the TgKRT14::Spry4 allele (Charles et al. 2011). The TgKRT14::Spry4 allele expresses Spry4 under the control of the KRT14 promoter. The distribution of embryonic genotypes was at the expected Mendelian ratios (Appendix Table 1), and we did not detect an increase in the number of resorptions. We observed no gross anatomical defects at E17.5 (82 embryos) and E15.5 (37 embryos). Embryos carrying the KRT14::Spry4 transgene appeared to have a vascular defect (Fig. 1B) and an eyelid closure defect in single transgenic (TgKRT14::Spry4) and double heterozygous embryos (Irf6+/–;TgKRT14::Spry4) (Fig. 1B, C). We did not detect orofacial clefting in E17.5 embryos based on histological staining of coronal sections.
Figure 1.
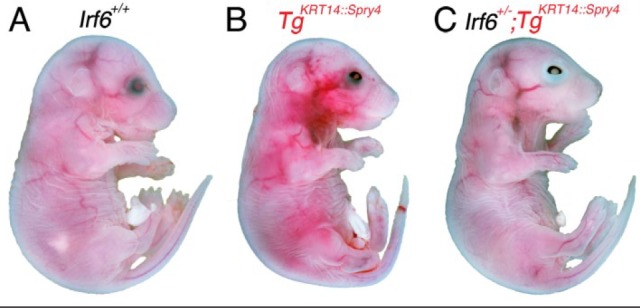
Eye closure defect in TgKRT14::Spry4 embryos at E17.5. Analysis and whole-mount imaging of wild-type (A), TgKRT14::Spry4 (B), and Irf6+/–;TgKRT14::Spry4 embryos (C) show an eye closure defect in embryos containing KRT14::Spry4 transgene but not wild-type littermates. Irf6 heterozygous embryos did not have an eye closure defect (data not shown). The eye closure defects were similar between TgKRT14::Spry4 and experimental embryos (B, C). A vascular defect was also observed in TgKRT14::Spry4 embryos (B). No other anatomic defects were detected on gross evaluation.
However, we identified abnormal oral epithelial adhesions in the oral cavity at E13.5 in embryos that were single heterozygous for Irf6 or the transgene and in embryos that were double heterozygous (Irf6+/–;TgKRT14::Spry4) (Fig. 2). In the singly heterozygous littermates, the abnormal oral adhesions occurred more frequently between the maxilla and mandible (maxillary-mandibular) and were superficial to the tooth germ (Fig. 2B, C). However in the double heterozygous embryos, the abnormal oral adhesions appeared more extensive, because they also occurred between the palate and tongue (palatal-lingual) (Fig. 2D).
Figure 2.

Irf6 and Spry4 regulate oral epithelium development at E13.5 at the tooth germ. Insert in (A) shows plane and approximate location of sections (A–X). Images and analysis of oral epithelium using hematoxylin and eosin (H&E) staining (A–D) to identify oral adhesions and immunostaining (E–X) to examine expression of KRT6/TP63 (E–H), KRT14 (I–L), IRF6 (M–P), GRHL3 (Q–T), and activated caspase 3 (CASP3, U–X) at the tooth germ. Genotypes separated by columns, including wild-type (A, E, I, M, Q, U), Irf6 heterozygous (Irf6+/–) (B, F, J, N, R, V), TgKRT14::Spry4 (C, G, K, O, S, W), and Irf6+/–;TgKRT14::Spry4 embryos (D, H, L, P, T, X). Relative to wild-type embryos (A), Irf6 heterozygous embryos had minor oral adhesions between the mandibular epithelium and either the palatal or maxillary epithelium (B, black arrowheads). TgKRT14::Spry4 embryos appeared to have similar maxillary-mandibular oral adhesions (C, black arrowhead), but other transgenic embryos TgKRT14::Spry4 also had oral adhesions between the palate and tongue (Fig. 4). Irf6+/–;TgKRT14::Spry4 embryos had more extensive maxillary-mandibular and palatal-lingual oral adhesions (D, black arrowheads). (E, F) The morphology of periderm cells was more cuboidal than squamous in TgKRT14::Spry4 (G) and Irf6+/–;TgKRT14::Spry4 (H) embryos (white arrowheads). Immunostaining shows irregular KRT6 staining and an expanded basal layer (TP63) in TgKRT14::Spry4 (G) and Irf6+/–;TgKRT14::Spry4 (H) embryos. (I–L) KRT14 immunostaining was slightly reduced in TgKRT14::Spry4 embryos (K) but highlighted the extent and severity of oral adhesions in Irf6+/–;TgKRT14::Spry4 (L) embryos (white arrow). (M–P) IRF6 staining showed expression in epithelium (white arrowheads), including sites of oral adhesions (P). (Q–T) GRHL3 expression was detected in periderm (Q–S, white arrowheads) but not in sites of oral adhesions in Irf6+/–;TgKRT14::Spry4 embryos (T, white arrowhead). (U–X) Activated caspase 3 staining in epithelium was similar for all genotypes. Scale bars: (A–D) 250 µm; (E–X) 40 µm. Mn, mandible; Mx, maxilla, P, palate; T, tongue; Tg, tooth germ.
To further examine these tissues, we performed immunostaining and examined both maxillary-mandibular and palatal-lingual oral adhesions. Maxillary-mandibular oral adhesions were most frequently detected at the tooth germ (Fig. 2). At the molecular level, sites of oral adhesions above the tooth germs in the transgenic embryos (TgKRT14::Spry4) and the double heterozygous embryos appeared to have an expanded basal cell layer, as marked by TP63 (Fig. 2, compare E, F with G, H). Furthermore, while we detected periderm, as marked by KRT6 expression, the cells appeared more cuboidal than squamous in morphology at the site of oral adhesions (Fig. 2E–H). KRT14 expression was less prominent in TgKRT14::Spry4 embryos compared with littermates (Fig. 2K). IRF6 appeared to be evenly expressed in the epithelium, including sites of oral adhesions (Fig. 2P). Importantly, GRHL3 expression was not uniformly detected at sites of oral adhesions in the tooth germ. Furthermore, cells expressing GRHL3 appeared to be more squamous than cuboidal in morphology (Fig. 2T). We did not detect differences in activated caspase 3 expression, including at sites of oral adhesions (Fig. 2U–X).
For most markers, expression at oral adhesions between the palate and tongue was different compared with maxillary-mandibular adhesions. For TP63, we did not observe an expanded basal cell layer in either lingual or palatal epithelium at adhesion sites (Fig. 3A–D). KRT6 expression was absent along the lingual epithelium (Fig. 3D). KRT14 staining in double heterozygous embryos (Irf6+/–;TgKRT14::Spry4) revealed loss or detachment of the lingual epithelium (Fig. 3H). This degree of oral adhesion severity was not detected in wild-type, Irf6 heterozygous, or transgenic (TgKRT14::Spry4) littermates. Interestingly, compared with wild-type and Irf6 heterozygous embryos, IRF6 expression appeared more prominent in lingual mesenchyme of transgenic littermates (TgKRT14::Spry4) (Fig. 3K). Similar to maxillary-mandibular oral adhesions, GRHL3 expression was not detected at sites of oral adhesion between the tongue and palate in double heterozygous embryos (Irf6+/–;TgKRT14::Spry4) (Fig. 3P). Furthermore, the GRHL3 signal appeared to be discontinuous in mutant embryos (Fig. 3N–P) compared with wild-type littermates (Fig. 3M). Finally, in contrast to maxillary-mandibular oral adhesions, sites of palatal-lingual oral adhesion appeared to express activated caspase 3 in double heterozygous embryos (Irf6+/–;TgKRT14::Spry4) (Fig. 3T).
Figure 3.

Irf6 and Spry4 interaction leads to loss of periderm and oral adhesions between the tongue and palate at E13.5. (A–T) Images and analysis of oral adhesions and immunostaining to examine expression of KRT6/TP63 (A–D), KRT14 (E–H), IRF6 (I–L), GRHL3 (M–P), and activated caspase 3 (CASP3, Q–T) between the tongue and palate. Genotypes separated by columns, including wild-type (A, E, I, M, Q), Irf6 heterozygous (Irf6+/–) (B, F, J, N, R), TgKRT14::Spry4 (C, G, K, O, S), and Irf6+/–;TgKRT14::Spry4 embryos (D, H, L, P, T). (A–D) Immunostaining showed an abnormal expression pattern of both KRT6 and TP63 (compare white arrowheads in A–D), with loss of periderm and basal cells (white arrow) in Irf6+/–;TgKRT14::Spry4 embryos (D). (E–H) KRT14 immunostaining highlighted adhesions in TgKRT14::Spry4 embryos (G) and loss of lingual oral epithelium in Irf6+/–;TgKRT14::Spry4 embryos (H). (I–L) IRF6 stained both the periderm and basal epithelial cells in wild-type embryos (I) and Irf6 heterozygous (J). There appeared to be ectopic IRF6 expression in the lingual mesenchyme of both TgKRT14::Spry4 (K) and Irf6+/–;TgKRT14::Spry4 (L) embryos. Epithelial IRF6 expression did not appear different (I–L, white arrowheads). (M–P) GRHL3 expression was found in both the periderm and basal epithelial cells of tongue and, to a lesser extent, the palatal shelves (M). There was reduced GRHL3 expression in the periderm of Irf6 heterozygous (N) and TgKRT14::Spry4 embryos (O). Loss of periderm contributed to the apparent reduction GRHL3 expression in Irf6+/–;TgKRT14::Spry4 (P) embryos (white arrowheads). (Q–T) Activated caspase 3 expression appeared more prominent in sites of oral adhesion in Irf6+/–;TgKRT14::Spry4 (T) embryos (white arrows). Scale bar: (A–T) 40 µm. P, palate; T, tongue.
This qualitative analysis suggested that oral adhesions were more common and extensive in Irf6+/–;TgKRT14::Spry4 embryos. To quantify this observation, we developed an assay to more precisely measure and compare oral adhesions among the different genotypes (Fig. 4). We measured the visual field by using the interactive measurement feature in Nikon Elements Software. To account for the curvilinear structures within the oral cavity, we measured the arc length of oral adhesions as a fraction of the total oral cavity surface (Fig. 4). The nasal cavity epithelium was excluded from our measurements (black line above tongue in Fig. 4B, D, F) because even complete oral adhesions spare this surface (Ingraham et al. 2006; Richardson et al. 2006). We separately measured maxillary-mandibular and palatal-lingual oral adhesions, which allowed us to calculate the percentage of the oral cavity that was adhered in total and at each of these sites.
Figure 4.

Quantitative oral adhesion assay. (A–F) Coronal sections of E13.5 oral cavities. (A) Inset in right lower corner of this images shows cartoon with a black line indicating the plan of section and approximate location of sections. Two genotypes are shown to illustrate different types of oral adhesions and how they were measured: wild-type (A, B) and Irf6 heterozygous, transgenic embryos (Irf6+/–;TgKRT14::Spry4) (CF). We used standard microscope software to measure the distance around the curved surface of the oral cavity using multiple linear segments (arc length). Representative images and lines before (left column: A, C, E) and after (right column: B, D, F) surface measurements indicated with colored lines. We measured oral epithelium from the lateral edges of the oral cavity to the top of the tongue (black horizontal line bisecting oral epithelium) (B, D, F). We obtained the arc length for the whole oral cavity (blue line), maxilla-mandible oral adhesion (yellow-dotted line), and palatal-lingual oral adhesions (green-dotted line) (B, D, F). Mn, mandible; Mx, maxilla; PS, palatal shelf; T, tongue; Tg, tooth germ.
We did not detect oral adhesion in wild-type embryos (Fig. 5A). Irf6 heterozygous embryos had mean oral adhesions involving 5.6% of the oral cavity, with a maximum of 16% (Fig. 5A). All of these adhesions were between the maxilla and mandible. TgKRT14::Spry4 embryos had mean total oral adhesions of 6.5% with a maximum of 13%. The majority of these also occurred between the maxilla and mandible (Fig. 5B). In contrast, double heterozygous embryos (Irf6+/–;TgKRT14::Spry4) had mean oral adhesions of 11.2% and a maximum of 35.8% (Fig. 5A). Mean oral adhesions between the maxilla and mandible were 6.7% of the oral cavity with a maximum of 35.8%. Between palate and tongue, an average of 5% of oral adhesions were between the tongue and palate with a maximum of 22% (Fig. 5C). Assuming a nonparametric distribution (Kruskal-Wallis test), we did not identify a significant difference among the different genotypes in total oral (P = 0.09), palatal-lingual (P = 0.24), or maxillary-mandibular (P = 0.17) oral adhesions.
Figure 5.

A quantitative adhesion assay shows that Irf6+/–;TgKRT14::Spry4 embryos have more extensive oral adhesions at E13.5. (A–C) We obtained a percent arc length of total oral adhesion, relative to the entire arc length of the oral cavity (A). We then calculated the individual contributions of palatal-lingual (B) and maxillary-mandibular (C) oral adhesions as a percentage of the total arc length. (D–G) Quantifying percent oral epithelium expressing GRHL3. We calculated the percent oral epithelium where GRHL3 was detected on each epithelial surface (D–G) for both palatal-lingual (D, E) and maxillary-mandibular (F, G) oral adhesions. The y-axis shows the percent epithelium where GRHL3 was detected. Three images were quantified for each epithelial surface and each genotype using the arc length tool in NIS Elements Advanced Research v3.10. Four genotypes were tested, including wild-type (white triangle), Irf6 heterozygous (Irf6+/–) (black square), TgKRT14::Spry4 (white circle), and Irf6+/–;TgKRT14::Spry4 (black diamond) embryos.
Our qualitative analysis suggested that GRHL3 expression was reduced in double heterozygous embryos (Irf6+/–; TgKRT14::Spry4). Similar to the oral adhesion assay, we answered this question directly by measuring epithelial arc length to calculate the percent oral epithelium with GRHL3 expression. We examined both epithelial surfaces involved in palatal-lingual and maxillary-mandibular oral adhesions. For each type of oral adhesion, we measured the total length of the epithelium and the length of epithelium expressing GRHL3. We used these measurements to calculate the percent oral epithelium with GRHL3 expression. In palatal and lingual epithelium, statistical analysis showed a significant difference in the percent epithelium with GRHL3 expression among the 4 genotypes examined (ANOVA, P < 0.05) (Fig. 5D, E). Dunn’s multiple comparison test showed a significant difference in the percent lingual and palatal epithelium with GRHL3 expression when comparing wild-type and double heterozygous embryos (P < 0.05). In mandibular and maxillary oral epithelium, we detected a significant difference in epithelial GRHL3 expression (ANOVA, P < 0.05). However, no significant differences were found between the 4 genotypes using Dunn’s multiple comparison (Fig. 5F, G).
Discussion
Common and rare variants in IRF6 and RTK signaling genes have been implicated in orofacial clefting. Here, we report a genetic interaction between IRF6 and SPRY4, a regulator of RTK signaling. These results imply that IRF6 and RTK signaling might also interact in contributing to human orofacial development. This conclusion is supported by both the qualitative and quantitative data obtained from in vivo epistasis experiments. Using a quantitative oral adhesion assay developed here, we observed a nonadditive effect in double heterozygous embryos (Irf6+/–;TgKRT14::Spry4). This quantitative assay may be useful in future work to detect epistatic relationships between other genes necessary for the development of oral epithelium.
Previous work showed that loss of periderm leads to oral adhesions (Richardson et al. 2014). Similarly, we observed a loss in periderm cells at palatal-lingual adhesions. However, KRT6 staining was detected at maxillary-mandibular oral adhesions. However, the morphology of these superficial cells was more cuboidal than squamous, suggesting abnormalities in periderm function. Maxillary-mandibular epithelium at the tooth germ also seemed to be the most common or sensitive site to screen for oral adhesions. It is not clear why the site of the tooth germ was most susceptible to oral adhesions. Finally, oral adhesions were less severe and extensive compared with the previously described Irf6 null embryos. Based on these observations, we hypothesize that the abnormal periderm cell morphology represents an intermediate phenotype resulting from periderm dysfunction.
Importantly, of all the markers examined, GRHL3 expression appeared to be the best correlative molecular marker of both periderm cell morphology and function. Interestingly, loss of IRF6 (de la Garza et al. 2013; Peyrard-Janvid et al. 2014) and overexpression of SPRY4 seemed to converge at GRHL3 expression, and loss of GRHL3 can lead to orofacial clefting. Periderm cells involved in oral adhesions did not express GRHL3, while periderm cells with squamous morphology expressing GRHL3 did not appear to participate in adhesion. Loss of GRHL3 expression, abnormal cellular morphology, and presence of oral adhesions implicates abnormal cellular function, in addition to development, in pathologic oral adhesions. Therefore, we have identified GRHL3 as a common molecular target upon which these signaling pathways could interact to alter periderm formation and palatal development. Future experiments testing the role of FGF10 and FGFR2 in IRF6 and SPRY4 or SPRY2 signaling might identify how these elements interact.
While Irf6 heterozygous mice were only found to have maxillary-mandibular oral adhesions, transgenic embryos (TgKRT14::Spry4) had both maxillary-mandibular and palatal-lingual oral adhesions. These data suggest that there are different mechanisms associated with the function and maintenance of periderm along the surface of the oral cavity. However, these pathways are likely to converge because in both locations of oral adhesions, GRHL3 served as a specific marker of functional periderm, while KRT6 was useful in detecting periderm cells. In addition, activated caspase 3 expression seemed slightly more prominent at sites of oral adhesions between the lingual and palatal epithelium.
Prior work shows that “rounded” periderm cells stretch across an open eye between E15 and E16 to mediate eyelid closure (Harris and Juriloff 1986; Juriloff and Harris 1989). Loss of Sprouty 1 and 2 resulted in an eyelid closure defect by reducing periderm migration (ERK and c-Jun signaling) and altering epithelial proliferation (FGF-ERK signaling) (Kuracha et al. 2013). Interestingly, our data show that overexpressing SPRY4 in epithelium resulted in an eye closure defect, which supports the role of SPRY4 and FGF signaling in periderm development and function. However, the eyelid closure defect in transgenic embryos (TgKRT14::Spry4) was not rescued in double heterozygous embryos (Irf6+/–;TgKRT14::Spry4), suggesting that an interaction between IRF6 and RTK signaling is specific to oral periderm.
Sprouty genes inhibit several pathways, including those downstream of FGFR signaling (Dorey and Amaya 2010). Therefore, our results can be interpreted as a genetic interaction with SPRY4 and as a means of screening for a genetic interaction with RTK signaling pathways, including FGF genes. Future work may uncover which components of the FGF signaling network could interact with IRF6. Downstream, IRF6 interacts with TP63 (Thomason et al. 2010), and TP63 regulates BMP and FGF signaling in orofacial clefting (Thomason et al. 2008). Our data suggested that double heterozygous embryos (Irf6+/–;TgKRT14::Spry4) have an expanded basal epithelial layer, as marked by TP63 expression. This relationship has also been observed in Irf6 knockout embryos (Ingraham et al. 2006). Importantly, our expression analysis also suggests the presence of increased IRF6 expression in the lingual mesenchyme of TgKRT14::Spry4 embryos (Goudy et al. 2013). These data suggest that RTK signaling might regulate IRF6 expression in the mesoderm. It will be interesting in the future to delineate how SPRY4 and RTK signaling interact with IRF6 to regulate GRHL3 and TP63 expression in oral epithelium and possibly the mesenchyme.
Author Contributions
Y.A. Kousa, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; R. Roushangar, contributed to design, data acquisition, and analysis and critically revised the manuscript; N. Patel, A. Walter, P. Marangoni, contributed to data acquisition and critically revised the manuscript; R. Krumlauf, O.D. Klein, contributed to design and critically revised the manuscript; B.C. Schutte, contributed to conception, data analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplementary Material
Footnotes
A supplemental appendix to this article is available online.
Financial support to YAK (F31DE022696-01) came from the National Institutes of Health (NIH), National Institute of Dental and Craniofacial Research (NIDCR). Additional support to BCS was provided from NIH (DE13513) and Michigan State University grants.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Alappat SR, Zhang Z, Suzuki K, Zhang X, Liu H, Jiang R, Yamada G, Chen Y. 2005. The cellular and molecular etiology of the cleft secondary palate in fgf10 mutant mice. Dev Biol. 277(1):102–113. [DOI] [PubMed] [Google Scholar]
- Bush JO, Jiang R. 2012. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 139(2):231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles C, Hovorakova M, Ahn Y, Lyons DB, Marangoni P, Churava S, Biehs B, Jheon A, Lesot H, Balooch G, et al. 2011. Regulation of tooth number by fine-tuning levels of receptor-tyrosine kinase signaling. Development. 138(18):4063–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Yarden Y. 2006. Egf-erbb signalling: towards the systems level. Nat Rev Mol Cell Biol. 7(7):505–516. [DOI] [PubMed] [Google Scholar]
- de la Garza G, Schleiffarth JR, Dunnwald M, Mankad A, Weirather JL, Bonde G, Butcher S, Mansour TA, Kousa YA, Fukazawa CF, et al. 2013. Interferon regulatory factor 6 promotes differentiation of the periderm by activating expression of grainyhead-like 3. J Invest Dermatol. 133(1):68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorey K, Amaya E. 2010. Fgf signalling: diverse roles during early vertebrate embryogenesis. Development. 137(22):3731–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudy S, Angel P, Jacobs B, Hill C, Mainini V, Smith AL, Kousa YA, Caprioli R, Prince LS, Baldwin S, et al. 2013. Cell-autonomous and non-cell-autonomous roles for Irf6 during development of the tongue. PLoS One. 8(2):e56270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MJ, Juriloff DM. 1986. Eyelid development and fusion induced by cortisone treatment in mutant, lidgap-Miller, foetal mice: a scanning electron microscope study. J Embryol Exp Morphol. 91:1–18. [PubMed] [Google Scholar]
- Ingraham CR, Kinoshita A, Kondo S, Yang B, Sajan S, Trout KJ, Malik MI, Dunnwald M, Goudy SL, Lovett M, et al. 2006. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat Genet. 38(11):1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ. 1989. A scanning electron microscope study of fetal eyelid closure accelerated by cortisone in SWV/Bc mice. Teratology. 40(1):59–66. [DOI] [PubMed] [Google Scholar]
- Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S, Sander A, et al. 2002. Mutations in Irf6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 32(2):285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousa YA, Schutte BC. 2016. Toward an orofacial gene regulatory network. Dev Dyn. 245(3):220–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuracha MR, Siefker E, Licht JD, Govindarajan V. 2013. Spry1 and spry2 are necessary for eyelid closure. Dev Biol. 383(2):227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. 2010. Cell signaling by receptor tyrosine kinases. Cell. 141(7):1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EJ, Liu H, Carlson JC, Shaffer JR, Feingold E, Wehby G, Laurie CA, Jain D, Laurie CC, Doheny KF, et al. 2016. A genome-wide association study of nonsyndromic cleft palate identifies an etiologic missense variant in GRHL3. Am J Hum Genet. 98(4):744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig KU, Mangold E, Herms S, Nowak S, Reutter H, Paul A, Becker J, Herberz R, AlChawa T, Nasser E, et al. 2012. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat Genet. 44(9):968–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JM, Morrison DJ, Basson MA, Licht JD. 2006. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 16(1):45–54. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Taketomi T, Yoshizaki K, Arai S, Sanui T, Yoshiga D, Yoshimura A, Nakamura S. 2011. Sprouty2 controls proliferation of palate mesenchymal cells via fibroblast growth factor signaling. Biochem Biophys Res Commun. 404(4):1076–1082. [DOI] [PubMed] [Google Scholar]
- Minowada G, Jarvis LA, Chi CL, Neubuser A, Sun X, Hacohen N, Krasnow MA, Martin GR. 1999. Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development. 126(20):4465–4475. [DOI] [PubMed] [Google Scholar]
- Peyrard-Janvid M, Leslie EJ, Kousa YA, Smith TL, Dunnwald M, Magnusson M, Lentz BA, Unneberg P, Fransson I, Koillinen HK, et al. 2014. Dominant mutations in GRHL3 cause Van der Woude syndrome and disrupt oral periderm development. Am J Hum Genet. 94(1):23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimov F, Marazita ML, Visel A, Cooper ME, Hitchler MJ, Rubini M, Domann FE, Govil M, Christensen K, Bille C, et al. 2008. Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat Genet. 40(11):1341–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP. 2004. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 113(12):1692–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Dixon J, Jiang R, Dixon MJ. 2009. Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum Mol Genet. 18(14):2632–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Dixon J, Malhotra S, Hardman MJ, Knowles L, Boot-Handford RP, Shore P, Whitmarsh A, Dixon MJ. 2006. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat Genet. 38(11):1329–1334. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Hammond NL, Coulombe PA, Saloranta C, Nousiainen HO, Salonen R, Berry A, Hanley N, Headon D, Karikoski R, et al. 2014. Periderm prevents pathological epithelial adhesions during embryogenesis. J Clin Invest. 124(9):3891–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dode C, Mohammadi M, et al. 2007. Impaired FGF signaling contributes to cleft lip and palate. Proc Natl Acad Sci USA. 104(11):4512–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason HA, Dixon MJ, Dixon J. 2008. Facial clefting in Tp63 deficient mice results from altered Bmp4, Fgf8 and Shh signaling. Dev Biol. 321(1):273–282. [DOI] [PubMed] [Google Scholar]
- Thomason HA, Zhou H, Kouwenhoven EN, Dotto GP, Restivo G, Nguyen BC, Little H, Dixon MJ, van Bokhoven H, Dixon J. 2010. Cooperation between the transcription factors P63 and IRF6 is essential to prevent cleft palate in mice. J Clin Invest. 120(5):1561–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira AR, Avila JR, Daack-Hirsch S, Dragan E, Felix TM, Rahimov F, Harrington J, Schultz RR, Watanabe Y, Johnson M, et al. 2005. Medical sequencing of candidate genes for nonsyndromic cleft lip and palate. PLoS Genet. 1(6):e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchero TM, Cooper ME, Maher BS, Daack-Hirsch S, Nepomuceno B, Ribeiro L, Caprau D, Christensen K, Suzuki Y, Machida J, et al. 2004. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 351(8):769–778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.