

Abstract
Here we describe the genotype-phenotype correlations of diseases caused by variants in Fibroblast Growth Factor Receptor 1 (FGFR1) and report a novel, de novo variant in FGFR1 in an individual with multiple congenital anomalies. The proband presented with bilateral cleft lip and palate, malformed auricles, and bilateral ectrodactyly of his hands and feet at birth. He was later diagnosed with diabetes insipidus, spastic quadriplegia, developmental delay, agenesis of the corpus callosum, and enlargement of the third cerebral ventricle. We noted the substantial phenotypic overlap with individuals with Hartsfield syndrome, the rare combination of holoprosencephaly and ectrodactyly. Sequencing of FGFR1 identified a previously unreported de novo variant in exon 11 (p.Gly487Cys), which we modeled to determine its predicted effect on the protein structure. Although it was not predicted to significantly alter protein folding stability, it is possible this variant leads to the formation of nonnative intra- or intermolecular disulfide bonds. We then mapped this and other disease-associated variants to a 3-dimensional model of FGFR1 to assess which protein domains harbored the highest number of pathogenic changes. We observed the greatest number of variants within the domains involved in FGF binding and FGFR activation. To further explore the contribution of each variant to disease, we recorded the phenotype resulting from each FGFR1 variant to generate a series of phenotype-specific protein maps and compared our results to benign variants appearing in control databases. It is our hope that the use of phenotypic maps such as these will further the understanding of genetic disease in general and diseases caused by variation in FGFR1 specifically.
Keywords: craniofacial anomalies, craniofacial biology/genetics, genetics, protein modeling, Hartsfield syndrome, clinical phenotypes
Introduction
Hartsfield syndrome (MIM#615465), the co-occurrence of holoprosencephaly (MIM#236100) and ectrodactyly (MIM# 183600), is a rare condition reported in 32 individuals to date (Hartsfield 1984; Appendix Table 1). It is most commonly diagnosed in individuals with lobar or alobar holoprosencephaly and bilateral ectrodactyly but more broadly includes midline defects (such as a single central incisor, cleft lip and/or palate, microcephaly, or midface hypoplasia; Muenke and Beachy 2014)) in the presence of limb anomalies (Hong et al. 2016). Clinical presentation of Hartsfield syndrome often includes hypernatremia secondary to central diabetes insipidus, mild to severe intellectual disability, developmental delay, and pituitary insufficiency. Hartsfield syndrome results from heterozygous or biallelic variants in Fibroblast Growth Factor Receptor 1 (FGFR1) (Simonis et al. 2013).
Here we report a 33-y-old man with Hartsfield syndrome manifesting as agenesis of the corpus callosum, cleft lip and palate, ectrodactyly, and additional clinical features (Appendix Table 2). Using Sanger sequencing, we detected a novel, de novo FGFR1 variant in exon 11 replacing the glycine residue at amino acid position 487 with a cysteine (p.Gly487Cys). Notably, a missense variant changing this glycine residue to an aspartic acid (p.Gly487Asp) was previously reported in an individual with Hartsfield syndrome and an individual with olfactogenital (Kallmann; MIM#147950) syndrome, the co-occurrence of congenital hypogonadotropic hypogonadism and anosmia (Hong et al. 2016; Nair et al. 2016). We modeled this variant in silico to assess its effect on the protein structure. In addition, because variants in FGFR1 lead to a variety of disorders including normosomic congenital hypogonadotropic hypogonadism, Kallmann syndrome (Dodé et al. 2003), osteoglophonic dysplasia (MIM#166250; White et al. 2005), Pfeiffer syndrome (MIM#101600; Muenke et al. 1994), and trigonocephaly 1 (MIM#190440; Kress et al. 2000) and were detected in somatic tissues of individuals with encephalocraniocutaneous lipomatosis (MIM#613001; Bennett et al. 2016) and other types of cancer, we performed a comprehensive assessment of published FGFR1 mutations by domain and disease phenotype.
Clinical Report
The proband, the first child of nonconsanguineous parents, was born vaginally at 41 wk gestation. Apgar scores were 5 and 6 (1 and 5 min). Birth weight and head circumference were normal (2820 g and 33 cm). Family history was unremarkable. He was intubated immediately after birth but removed from respiratory support 3 h later. He was diagnosed with bilateral cleft lip and clefting of the hard and soft palates, malformed auricles (small, cup shaped, low set), an abnormally small penis, and bilateral ectrodactyly of hands and feet. Hand X-rays showed 2 normal ulnar digits with a possible third metacarpal without any phalanges bilaterally. At the time of birth, his clinical features were not ascribable to a specific disorder.
At 1 y of age, he presented with a coarse head tremor. Head computerized tomography (CT) scans revealed agenesis of the corpus callosum and enlargement of the third cerebral ventricle. Chest X-rays were negative for cardiac and lung abnormalities, and renal ultrasound was negative for size and shape abnormalities or hydronephrosis. At age 2, hypogonadism was noted, but the patient responded to human chorionic gonadotropin, suggesting functional testes. Notably, no teeth had erupted by 17 mo of age, with 6 teeth present at age 2. By age 11, several permanent teeth were missing with eruption of teeth 3 and 14, and by age 16 he was diagnosed with late/mixed dentition, deformities of teeth 8 and 9, clinically significant gingivitis, a class II malocclusion, class II molar occlusion, 5 mm palatal fistula, and an anterior and inferior premaxilla. From birth until age 4 he received 4 myringotomies with the placement of pressure-equalizer tubes, and audiograms were conducted throughout early development. A pure-tone audiogram at age 11 detected mild to borderline normal hearing in his right ear and moderate hearing loss in his left ear. Assessment for hyposmia or anosmia was not conducted, and olfactory bulbs were not assessed on his CT scan. At 1 y of age, his T4 level was noted as borderline low (5.8 and 6.0), and he was diagnosed with mild hypothyroidism. His sleep growth hormone level was 0.99 but deemed inaccurate because of waking during a difficult stick and was not repeated. He underwent multiple surgeries including repairs of the cleft lip, repair of the cleft palate, orchiopexy, otoplasty, and procedures to treat his lower extremity spasticity and hip dysplasia. He was diagnosed with hypernatremia secondary to central diabetes insipidus, which is treated by taking 3 × 3, 0.2-mg pills of desmopressin daily. He is intellectually delayed, nonverbal, and nonambulatory but communicates using gestures and a DynaVox. He currently resides in a home with 24-h community living support staff.
Materials and Methods
Samples
All participants in this study (proband, clinically normal sister and parents) were recruited after obtaining signed informed consent in compliance with the Institutional Review Board (IRB) at the University of Iowa (IRB No. 199804080). DNA was extracted from whole-blood samples using the Quickgene 610L (Autogen). DNA quantification was performed using the Qubit 2.0 Fluorometer (Life Technologies), with concentrations ranging from 0 to 550 ng/uL.
Sanger Sequencing
Sanger sequencing was used to genotype this family for exonic regions encoding isoform 1 of FGFR1 (exons 1 to 18; alternative exons were not sequenced). We designed primers flanking the exons using Primer3 (http://biotools.umassmed.edu/bioapps/primer3_www.cgi) and amplified these regions using polymerase chain reaction (PCR). Sequencing was completed on the resulting PCR products using an ABI 3730XL DNA Sequencer (Functional Biosciences, Inc.). Chromatograms were base called with PHRED (v.0.961028), assembled with PHRAP (v.0.960731), scanned by POLYPHRED (v.0.970312), and viewed with the CONSED program (v.4.0). The sequences were compared with the UCSC Genome Browser (HG19 build) context sequence. All variants were noted, and genotypes were determined.
Protein Model Building and Testing
Methods used for building a protein model for isoform 1 of FGFR1 (ENST00000447712) and testing the mutated amino acid residue within the tyrosine kinase (TK) domain can be found in the Appendix.
Combined Annotation Dependent Depletion Mapping
To generate a map of FGFR1 domains intolerant to variation, maximum Combined Annotation Dependent Depletion (CADD) Phred scores (a measure of the deleteriousness of single nucleotide variants compared with all possible substitutions in the genome; Kircher et al. 2014)) for each residue of FGFR1 were obtained from dbNSFP (Liu et al. 2011) and applied as a colored gradient to an FGFR1 model using Pymol (Schrodinger 2015). Exonic variants associated with disease phenotypes in PubMed were similarly mapped to an FGFR1 model by all diseases, individual disease, and subphenotype. Variants occurring in control databases were obtained from ExAC (ENST00000447712; Lek et al. 2016), 1000 Genomes (1000 Genomes Project Consortium et al. 2012), dbSNP (Sherry et al. 2001), ClinVar (Landrum et al. 2016), and EVS (http://evs.gs.washington.edu/EVS/) and classified as benign or pathogenic using the Deafness Variation Database (2010) approach followed by mapping to a FGFR1 model.
Statistical Analysis
Fisher’s exact test with Bonferroni correction for multiple testing (P = 0.0003) was used to compare variants occurring within specific protein domains of FGFR1. Pathogenic variants were assessed if they were exonic and the affected individual did not harbor variants in a second gene, since the effects of these variants as phenotypic modifiers could not be ruled out. Variants listed as benign, likely benign, or as a variant of unknown significance (VUS) were considered nonpathogenic. Comparisons were made by protein domain between all pathogenic versus nonpathogenic variants, as well as between associated disease phenotypes with greater than or equal to 5 pathogenic variants. The phenotypes assessed were Hartsfield syndrome, Pfeiffer syndrome/osteoglophonic dysplasia, congenital hypogonadotropic hypogonadism/Kallmann syndrome, dental anomalies, cleft lip and/or palate, ear malformations, hearing loss, limb defects, craniosynostosis, intellectual disability/developmental delay, corpus callosum agenesis, holoprosencephaly, craniofacial anomalies (dental anomalies, cleft lip and/or palate, ear malformations, craniosynostosis, holoprosencephaly), and craniofacial anomalies with limb defects. Congenital hypogonadotropic hypogonadism and Kallmann syndrome were analyzed together since they are often not reported as phenotypically separate diseases. Similarly, both Pfeiffer syndrome and osteoglophonic dysplasia were assessed together because they result from gain-of-function FGFR1 variants. The protein regions considered were the signal peptide, Immunoglobulin-like (Ig) I, acidic box, IgII, IgII-IgIII linker, IgIII, juxtamembrane, transmembrane, intramembrane, TK domains, and c-terminus.
Results
Sequencing
The proband was previously sequenced for approximately 500 Kb of genomic DNA around 13 loci associated with clefting (8q24.21 and coding exons of ADAMTS20, TFAP2A, ARHGAP29, LHX8, MSX1, TP63, PVRL1, PVRL2, RFC1, SKI1, TGFA, and TGFB3; Leslie et al. 2013), and no variants were detected. Upon reevaluating the patient’s phenotype and noticing the close resemblance to Hartsfield syndrome, we performed Sanger sequencing for all coding exons of isoform 1 of FGFR1 (NM_023110.2). He harbored a missense mutation in exon 11 (HG19; chr8:38275481; c.1459G>T; p.Gly487Cys), which was confirmed as a novel, de novo variant upon sequencing his unaffected mother, father, and sister (see Appendix Fig. 1 for pedigree and chromatograms). Polyphen 2 predicted the variant was probably damaging with a score of 1.00, and SIFT predicted the variant was damaging. Our variant was absent in ExAC, EVS, dbSNP, or 1000 Genomes.
Protein Model Testing
The change in protein folding stability (ΔΔG) for p.Gly487Cys was estimated using molecular simulations (see Appendix, Protein Model Building and Testing). From a Bennet’s acceptance ratio analysis of the simulation results, the difference in stability between the variant and wild-type states was 0.7 ± 0.1 kcal/mol. This modest destabilization is consistent with the variant causing a negligible reduction in folded protein, and therefore, the hypothesis that p.Gly487Cys causes unfolding of FGFR1’s TK domain was rejected. However, we note that this thermodynamic analysis does not account for the possibility that the introduction of cysteine at position 487 may result in nonnative disulfide bond formation and concomitant change or loss in function, although this would rely on FGFR1 encountering an oxidizing environment likely forming a disulfide intermediate (Cumming et al. 2004).
CADD Mapping
We obtained the max CADD Phred scores of each amino acid of FGFR1 and mapped them to a 3-dimensional protein model to assess which regions are predicted to be pathogenic when altered. The highest (most deleterious) scores occurred in the TK (which houses the ATP binding site and leads to downstream signaling), acidic box (which autoinhibits FGFR1), and immunoglobulin-like (Ig) II, IgII-IgIII linker, and IgIII domains (which allow for FGF binding; Eswarakumar et al. 2005; Fig. 1). We compared this prediction to the location of pathogenic FGFR1 variants reported in PubMed (Appendix Table 3) by CADD Phred score, protein domain, and disease phenotype (Fig. 2). As predicted, the domains harboring the median highest CADD Phred (scores) were the acidic box (31), IgII (27), IgII-IgIII linker (31), IgIII (28), and TK domains (29). Next, we annotated the CADD Phred scores of all exonic, nonpathogenic variants (see the Materials and Methods section) in the ExAC, dbSNP, 1000 Genomes, and EVS databases (Appendix Table 4) and mapped them to our model (Fig. 3). Of the 429 nonpathogenic variants, 11 were reported as likely disease causing in PubMed (Appendix Table 3). The median CADD Phred score of the nonpathogenic variants was 18, while the median CADD Phred score of pathogenic variants was 27. Finally, we assessed the location of pathogenic versus nonpathogenic variants within the protein domains as well if there were domain-specific disease associations within the pathogenic variants (Table). Interestingly, pathogenic variants across all diseases were less likely to occur in the C-terminal region compared with VUS and benign variants (P = 0.0001), while the presence of disease-associated variants in the IgIII domain neared significance (P = 0.0004) but did not withstand Bonferroni correction for multiple comparisons when compared with nonpathogenic variants (P = 0.0003). Assessment of all other diseases and disease phenotypes by protein domain of FGFR1 approached but did not reach significance after Bonferroni correction. These include variants occurring within the TK domain of individuals with Hartsfield syndrome (P = 0.004), cleft lip and/or palate (P = 0.005), limb anomalies (P = 0.003), holoprosencephaly (P = 0.001), and the presence of craniofacial dysmorphologies with limb defects (P = 0.006), as well as within the juxtamembrane domain in Pfeiffer syndrome/osteoglophonic dysplasia (P = 0.0005) or craniosynostosis (P = 0.0009).
Figure 1.
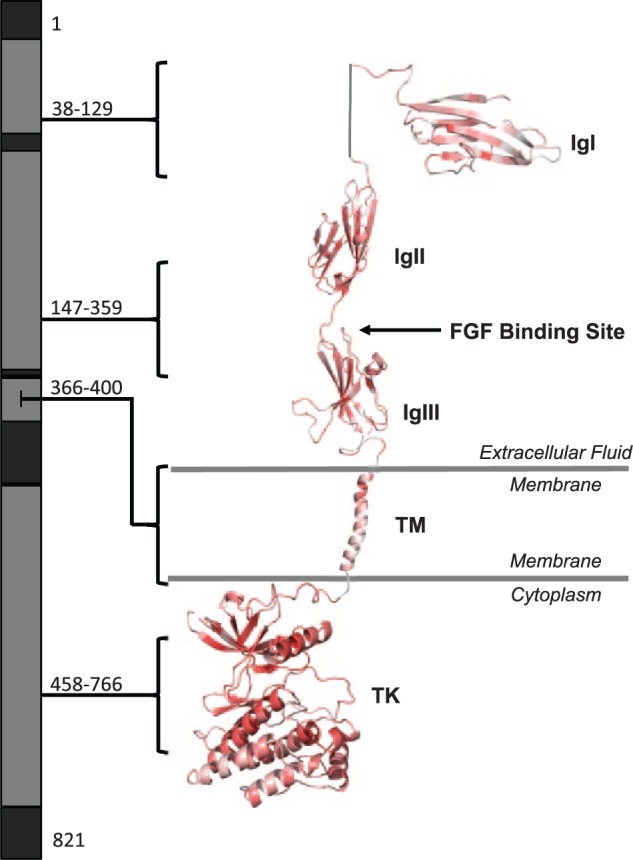
Map of Combined Annotation Dependent Depletion (CADD) scores across domains of FGFR1. Maximum CADD Phred scores were mapped back to the protein structure of FGFR1 to determine if specific domains are predicted to be more pathogenic when altered. CADD scores were color coded by predicted deleteriousness (vibrant red, more deleterious; dull red, less deleterious). The amino acid residues contributing to each domain are highlighted (gray), and residue numbers are listed, while those for which the protein structure is unknown (black) were not included in the model. Note particularly deleterious changes are predicted to occur in the TK domain and near the FGF binding site. IgI, immunoglobulin-like 1 domain; IgII, immunoglobulin-like 2 domain; IgIII, immunoglobulin-like 3 domain; TM, transmembrane domain; TK, tyrosine kinase domain.
Figure 2.
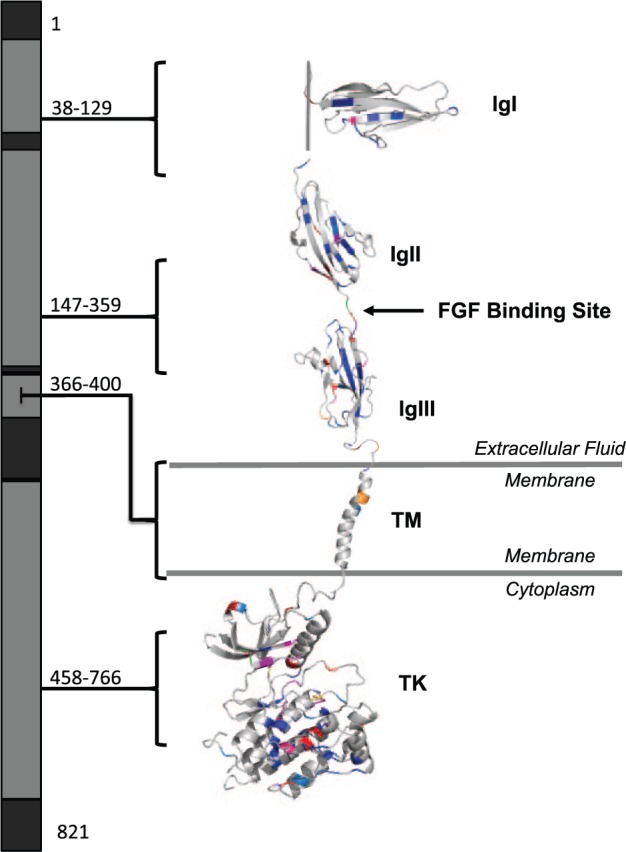
Map of pathogenic FGFR1 variants by protein domain. All reported variants of FGFR1 and their clinical phenotypes were obtained from PubMed. Altered residues were color coded by disease (Hartsfield syndrome, purple; normosomic congenital hypogonadotropic hypogonadism/Kallmann syndrome, blue; Pfeiffer syndrome/osteoglophonic dysplasia, orange; septo-optic dysplasia, green; encephalocraniocutaneous lipomatosis, yellow; cleft lip and/or palate, red) and Combined Annotation Dependent Depletion Phred score of predicted deleteriousness (vibrant colors, more deleterious). IgI, immunoglobulin-like 1 domain; IgII, immunoglobulin-like 2 domain; IgIII, immunoglobulin-like 3 domain; TM, transmembrane domain; TK, tyrosine kinase domain.
Figure 3.
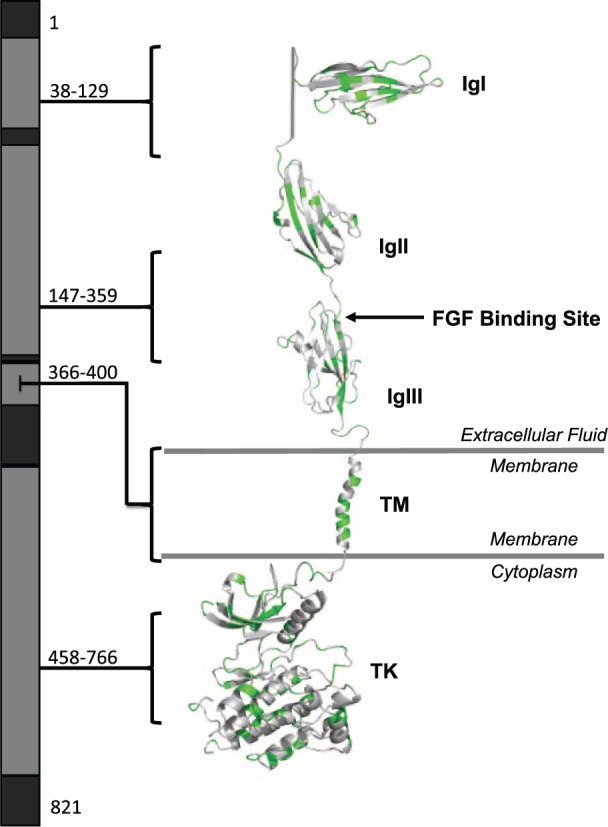
Map of benign, likely benign, and variants of unknown significance of FGFR1. Nonpathogenic variants of FGFR1 identified in publicly available databases (ExAC, dbSNP, 1000 Genomes, and EVS) were mapped to the protein structure. Vibrancy of green corresponds with predicted deleteriousness per Combined Annotation Dependent Depletion Phred score (more vibrant, more deleterious; less vibrant, less deleterious). IgI, immunoglobulin-like 1 domain; IgII, immunoglobulin-like 2 domain; IgIII, immunoglobulin-like 3 domain; TM, transmembrane domain; TK, tyrosine kinase domain.
Table.
Location of Disease or Phenotype by Protein Domain of FGFR1.
| Domain vs. Disease | SP | IgI | AB | IgII | IgII to IgIII | IgIII | JM | TM | ID | TK | CT |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Hartsfield syndrome | x | x | x | x | |||||||
| Pfeiffer syndrome/osteoglophonic dysplasia | x | x | x | ||||||||
| Encephalocraniocutaneous lipomatosis | x | ||||||||||
| Septo-optic dysplasia | x | x | x | x | |||||||
| CHH/Kallmann syndrome | x | x | x | x | x | x | x | x | x | x | x |
| Dental agenesis | x | x | x | x | x | x | x | x | x | ||
| Cleft lip and palate | x | x | x | x | x | x | x | x | x | ||
| Malformed ears | x | x | |||||||||
| Hearing loss | x | x | x | x | x | ||||||
| Limb defects | x | x | x | x | x | x | x | ||||
| Craniosynostosis | x | x | x | x | x | ||||||
| Intellectual disability/developmental delay | x | x | x | x | x | ||||||
| Corpus callosum agenesis | x | x | x | x | x | x | |||||
| Holoprosencephaly | x | x | x | x | x | x | |||||
| Craniofacial anomalies | x | x | x | x | x | x | x | x | x | ||
| Craniofacial anomalies + limb defects | x | x | x | x | x |
AB, acidic box; CHH, normosomic congenital hypogonadotropic hypogonadism; CT, C terminus; ID, interdomain; IgI, immunoglobulin-like 1 domain; IgII, immunoglobulin-like 2 domain; IgII-IgIII, IgII/IgIII linker domain; IgIII, immunoglobulin-like 3 domain; JM, juxtamembrane domain; SP, signal peptide; TK, tyrosine kinase domain; TM, transmembrane domain. Limb defects include ectrodactyly, syndactyly, polydactyly, and oligodactyly.
Discussion
Holoprosencephaly is the most common structural malformation of the human forebrain, occurring 1 in every 10,000 births (Muenke and Beachy 2000; Orioli and Castilla 2010). It ranges in severity from cyclopia, to alobar holoprosencephaly (1 cerebral ventricle and no interhemispheric fissure) or lobar holoprosencephaly (separate cerebral ventricles present with incomplete frontal cortical separation), to a single central incisor or hypotelorism (Muenke and Beachy 2014). Ectrodactyly is a limb abnormality with syndactyly and median clefts of the hands and feet, occurring in 1 in every 18,000 births (Elliott et al. 2005). Although each of these conditions occurs as an isolated event or as a separate phenotype in multiple syndromes, their co-occurrence is the hallmark of Hartsfield syndrome (Hartsfield 1984).
Hartsfield syndrome is an exceedingly rare condition caused by de novo, monoallelic or biallelic mutations in FGFR1 (see Appendix Table 1 for a complete list of Hartsfield syndrome–associated variants; Simonis et al. 2013). Here, we report a male with Hartsfield syndrome who harbors a novel, de novo glycine to cysteine change (p.Gly487Cys) in FGFR1. A missense change from this glycine to aspartic acid has been reported in 2 individuals. The first was diagnosed with Kallmann syndrome (Nair et al. 2016). The second has Hartsfield syndrome presenting as lobar holoprosencephaly, an absent septum pellucidum and anterior corpus callosum, bilateral ectrodactyly of the feet, bilateral cleft lip and palate, and developmental delay but does not exhibit the diabetes insipidus, total corpus callosum agenesis, and hypothyroidism detected in our proband (Hong et al. 2016; Appendix Table 2). Eight studies exploring genetic causes of congenital hypogonadotropic hypogonadism and Kallmann syndrome (both caused by variants in FGFR1; Pitteloud et al. 2007; Raivio et al. 2009; Sykiotis et al. 2010; Shaw et al. 2011; Abel et al. 2013; Tommiska et al. 2014; Dubourg et al. 2016; Quaynor et al. 2016) and 1 exploring Hartsfield-related phenotypes (Hong et al. 2016) demonstrated oligogenicity. This suggests these variable phenotypes may be due to modifier genes or the effects of the amino acid change on protein structure or function.
To assess if p.Gly487Cys affected protein folding stability, we modeled it using molecular dynamics simulations (see Appendix, Protein Model Building and Testing). The change in protein folding free energy was not large enough to cause a major destabilization, indicating p.Gly487Cys does not disrupt folding of FGFR1’s TK domain. Glycine 487 is found on a beta strand and is the most flexible amino acid because of its small side chain. Thus, it can adopt phi-psi angles that are not observed experimentally for other proteinogenic amino acids. The native glycine’s phi-psi angles were –167.0 and 97.5, respectively, and within the range that cysteine (and other amino acids) can also adopt. This observation and the simulation results support the conclusion that replacement of glycine with cysteine does not strain the polypeptide backbone. However, the thermodynamic analysis does not account for the potential formation of nonnative disulfide intermediates between the introduced cysteine and native cysteine residues in FGFR1 (or other proteins) in the presence of intracellular oxidative stress, which may disrupt protein folding (Cumming et al. 2004).
In addition to Hartfield syndrome, variants in FGFR1 are associated with at least 5 clinical diseases (McKusick 2007) including normosomic congenital hypogonadotropic hypogonadism and olfactogenital (Kallmann) syndrome, osteoglophonic dysplasia, Pfeiffer syndrome, trigonocephaly 1, and encephalocraniocutaneous lipomatosis. Osteoglophonic dysplasia and the craniosynostosis syndromes (Pfeiffer and trigonocephaly 1) result from gain-of-function variants thought to cause increased cell proliferation and early osteoblast differentiation (Johnson and Wilkie 2011), whereas normosomic hypogonadotropic hypogonadism and Kallmann syndrome are likely caused by loss-of-function variants. Recently, several variants associated with Hartsfield syndrome showed a dominant-negative effect when modeled in zebrafish, possibly explaining the phenotypic severity of the disease (Hong et al. 2016). Although mosaic activating mutations were identified in encephalocraniocutaneous lipomatosis (Bennett et al. 2016), similar variants are reported in a variety of cancers; therefore, the causal role of FGFR1 in this disease needs further exploration. Finally, FGFR1 variants were identified in individuals with nonsyndromic cleft lip and/or palate (Riley et al. 2007). A summary of published FGFR1 variants and their associated diseases can be found in Appendix Table 3.
Intriguingly, Hartsfield syndrome encompasses the phenotypic spectrum of many of these conditions. Cleft lip and/or palate often co-occurs with Kallmann syndrome arising from FGFR1 mutations (in about 30% of the cases; Dodé et al. 2003; Pitteloud et al. 2007), the presence of a micropenis and undescended testes is a hallmark of congenital hypogonadotropic hypogonadism in male infants, and agenesis of the corpus callosum and intellectual disability have been reported in cases of encephalocraniocutaneous lipomatosis and Kallmann syndrome (Chandravanshi 2014; Dodé et al. 2003). To better understand the phenotypic variability of these diseases, we investigated the location of their corresponding variants within the 3-dimensional context of FGFR1.
FGFR1 is 1 of 4 structurally similar fibroblast growth factor (FGF) receptors consisting of 3 extracellular immunoglobulin-like (Ig) domains, a helical transmembrane domain, and an intracellular TK domain (Ornitz and Itoh 2015). FGFRs are activated when 1 of at least 22 structurally related FGFs bind the extracellular domain (Eswarakumar et al. 2005). Proper binding relies on the presence of heparin, which induces oligomerization of FGF (Eswarakumar et al. 2005; Ornitz and Itoh 2015), allowing for receptor dimerization and transautophosphorylation of the cytoplasmic TK domains followed by the activation of downstream signaling (Ornitz and Itoh 2015). Changes in the extracellular domain can alter FGF-FGFR1 binding affinity, while variation in the TK domain can increase dimer stabilization and activation or cause reduced protein activation (Neilson and Friesel 1996).
When assessing the overall CADD Phred scores of pathogenic variants, domains critical for FGF binding (acidic box, IgII, IgII-IgIII linker, IgIII) and FGFR1 activation (TK) contained the highest scoring residues, suggesting these domains are most likely pathogenic when altered. However, after comparing the localization of variants within the protein domains of FGFR1 by disease and phenotype, only the association of Pfeiffer syndrome and osteoglophonic dysplasia (P = 0.0005) or craniosynostosis (P = 0.0009) with the juxtamembrane domain neared statistical significance (P = 0.0003). While this demonstrates gain-of-function mutations may localize near the cellular membrane of FGFR1, correlations between loss-of-function or dominant negative variants and a specific domain were not observed. Notably, a major limitation of this study is the reliance on accurate and comprehensive phenotyping in the published literature. The acquisition of additional cases combined with thorough subphenotyping is necessary to draw additional genotypic-phenotypic conclusions.
In this report, we identify a novel residue change in an individual with Hartsfield syndrome, and we categorize published disease variants of FGFR1 by their clinical manifestation and location within the protein. Our study supports the need for systematic reporting of the sequence variants and corresponding abnormal phenotypes in the literature to maximize the clinical utility of these findings (Richards et al. 2015) and demonstrates the current lack of understanding of the functional implications of identified variants within the protein. In addition, work exploring the spatiotemporal, isoform-specific, and species-specific effects of these variants is still needed. We hope the genotype-phenotype maps described here will be useful in interpreting the clinical significance of these variants in the future and encourage more critical assessments of their impact within the protein structure.
Author Contributions
L.A. Lansdon, contributed to conception, design, data acquisition, analysis, and interpretation and drafted and critically revised the manuscript; H.V. Bernabe, contributed to data analysis and drafted the manuscript; N. Nidey, contributed to data acquisition and drafted the manuscript; J. Standley, contributed to data analysis and interpretation and drafted the manuscript; M.J. Schnieders, contributed to design, data analysis, and interpretation and critically revised the manuscript; J.C. Murray, contributed to conception, design, and data interpretation and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplementary Material
Acknowledgments
We are especially grateful to the proband and his family for their participation in this study.
Footnotes
A supplemental appendix to this article is available online.
This work was funded by the National Institutes of Health (grants R37DE-08559, R01DC002842, and T32GM008629) and the National Science Foundation (grant CHE-1404147).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
Authors’ Note: Data underlying the results here are available upon request from the authors.
References
- 1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature. 491(7422):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel BS, Shaw ND, Brown JM, Adams JM, Alati T, Martin KA, Pitteloud N, Seminara SB, Plummer L, Pignatelli D, et al. 2013. Responsiveness to a physiological regimen of gnrh therapy and relation to genotype in women with isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 98(2):E206–E216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett JT, Tan TY, Alcantara D, Tetrault M, Timms AE, Jensen D, Collins S, Nowaczyk MJ, Lindhurst MJ, Christensen KM, et al. 2016. Mosaic activating mutations in fgfr1 cause encephalocraniocutaneous lipomatosis. Am J Hum Genet. 98(3):579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandravanshi SL. 2014. Encephalocraniocutaneous lipomatosis: a case report and review of the literature. Indian J Ophthalmol. 62(5):622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming RC, Andon NL, Haynes PA, Park M, Fischer WH, Schubert D. 2004. Protein disulfide bond formation in the cytoplasm during oxidative stress. J Biol Chem. 279(21):21749–21758. [DOI] [PubMed] [Google Scholar]
- Deafness Variation Database. 2010. The molecular otolaryngology and renal research laboratories. Iowa City: The University of Iowa; [accessed Jul 25, 2017]. http://deafnessvariationdatabase.org/. [Google Scholar]
- Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Dû N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, et al. 2003. Loss-of-function mutations in fgfr1 cause autosomal dominant kallmann syndrome. Nat Genet. 33(4):463–465. [DOI] [PubMed] [Google Scholar]
- Dubourg C, Carré W, Hamdi-Rozé H, Mouden C, Roume J, Abdelmajid B, Amram D, Baumann C, Chassaing N, Coubes C, et al. 2016. Mutational spectrum in holoprosencephaly shows that fgf is a new major signaling pathway. Hum Mutat. 37(12):1329–1339. [DOI] [PubMed] [Google Scholar]
- Elliott AM, Evans JA, Chudley AE. 2005. Split hand foot malformation (shfm). Clin Genet. 68(6):501–505. [DOI] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. 2005. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 16(2):139–149. [DOI] [PubMed] [Google Scholar]
- Exome variant server, nhlbi go exome sequencing project (esp). Seattle (WA); [accessed Jul 25, 2017]. https://esp.gs.washington.edu/drupal/. [Google Scholar]
- Hartsfield J. 1984. Syndrome identification case report 119. Hypertelorism associated with holoprocencephaly and ectrodactyly. Clin Dysmorphol. 2(0962–8827):27–31. [Google Scholar]
- Hong S, Hu P, Marino J, Hufnagel SB, Hopkin RJ, Toromanovic A, Richieri-Costa A, Ribeiro-Bicudo LA, Kruszka P, Roessler E, et al. 2016. Dominant-negative kinase domain mutations in fgfr1 can explain the clinical severity of hartsfield syndrome. Hum Mol Genet. 25(10):1912–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D, Wilkie AO. 2011. Craniosynostosis. Eur J Hum Genet. 19(4):369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 46(3):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress W, Petersen B, Collmann H, Grimm T. 2000. An unusual fgfr1 mutation (fibroblast growth factor receptor 1 mutation) in a girl with non-syndromic trigonocephaly. Cytogenet Cell Genet. 91(1–4):138–140. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. 2016. Clinvar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44(D1):D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie EJ, Mancuso JL, Schutte BC, Cooper ME, Durda KM, L’Heureux J, Zucchero TM, Marazita ML, Murray JC. 2013. Search for genetic modifiers of irf6 and genotype-phenotype correlations in van der woude and popliteal pterygium syndromes. Am J Med Genet A. 161A(10):2535–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jian X, Boerwinkle E. 2011. Dbnsfp: a lightweight database of human nonsynonymous snps and their functional predictions. Hum Mutat. 32(8):894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKusick VA. 2007. Mendelian inheritance in man and its online version, omim. Am J Hum Genet. 80(4):588–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenke M, Beachy PA. 2000. Genetics of ventral forebrain development and holoprosencephaly. Curr Opin Genet Dev. 10(3):262–269. [DOI] [PubMed] [Google Scholar]
- Muenke M, Beachy PA. 2014. Holoprosencephaly. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G. editors. The online metabolic and molecular bases of inherited disease. New York (NY): McGraw-Hill; http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62664261. [Google Scholar]
- Muenke M, Schell U, Hehr A, Robin NH, Losken HW, Schinzel A, Pulleyn LJ, Rutland P, Reardon W, Malcolm S, et al. 1994. A common mutation in the fibroblast growth factor receptor 1 gene in pfeiffer syndrome. Nat Genet. 8(3):269–274. [DOI] [PubMed] [Google Scholar]
- Nair S, Jadhav S, Lila A, Jagtap V, Bukan A, Pandit R, Ekbote A, Dharmalingam M, Kumar P, Kalra P, et al. 2016. Spectrum of phenotype and genotype of congenital isolated hypogonadotropic hypogonadism in asian indians. Clin Endocrinol (Oxf). 85(1):100–109. [DOI] [PubMed] [Google Scholar]
- Neilson KM, Friesel R. 1996. Ligand-independent activation of fibroblast growth factor receptors by point mutations in the extracellular, transmembrane, and kinase domains. J Biol Chem. 271(40):25049–25057. [DOI] [PubMed] [Google Scholar]
- Orioli IM, Castilla EE. 2010. Epidemiology of holoprosencephaly: prevalence and risk factors. Am J Med Genet C Semin Med Genet. 154C(1):13–21. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. 2015. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 4(3):215–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, Dwyer A, Plummer L, Hughes V, Seminara S, Cheng YZ, et al. 2007. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest. 117(2):457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaynor SD, Bosley ME, Duckworth CG, Porter KR, Kim SH, Kim HG, Chorich LP, Sullivan ME, Choi JH, Cameron RS, et al. 2016. Targeted next generation sequencing approach identifies eighteen new candidate genes in normosmic hypogonadotropic hypogonadism and kallmann syndrome. Mol Cell Endocrinol. 437:86–96. [DOI] [PubMed] [Google Scholar]
- Raivio T, Sidis Y, Plummer L, Chen H, Ma J, Mukherjee A, Jacobson-Dickman E, Quinton R, Van Vliet G, Lavoie H, et al. 2009. Frequency of impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. Endocr Rev. 30(7):934. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dode C, Mohammadi M, et al. 2007. Impaired fgf signaling contributes to cleft lip and palate. Proc Natl Acad Sci U S A. 104(11):4512–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger LLC. 2015. The PyMOL molecular graphics system, version 1.8. New York (NY) Schrödinger, LLC. [Google Scholar]
- Shaw ND, Seminara SB, Welt CK, Au MG, Plummer L, Hughes VA, Dwyer AA, Martin KA, Quinton R, Mericq V, et al. 2011. Expanding the phenotype and genotype of female gnrh deficiency. J Clin Endocrinol Metab. 96(3):E566–E576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. Dbsnp: the NCBI database of genetic variation. Nucleic Acids Res. 29(1):308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonis N, Migeotte I, Lambert N, Perazzolo C, de Silva DC, Dimitrov B, Heinrichs C, Janssens S, Kerr B, Mortier G, et al. 2013. Fgfr1 mutations cause hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly. J Med Genet. 50(9):585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, Dwyer AA, Quinton R, Hall JE, Gusella JF, et al. 2010. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U S A. 107(34):15140–15144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tommiska J, Kansakoski J, Christiansen P, Jorgensen N, Lawaetz JG, Juul A, Raivio T. 2014. Genetics of congenital hypogonadotropic hypogonadism in denmark. Eur J Med Genet. 57(7):345–348. [DOI] [PubMed] [Google Scholar]
- White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu X, Shaw NJ, McLellan NJ, et al. 2005. Mutations that cause osteoglophonic dysplasia define novel roles for fgfr1 in bone elongation. Am J Hum Genet. 76(2):361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.