

Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) infections are a global public health problem. MRSA strains have acquired a non-native penicillin-binding protein called PBP2a that crosslinks peptidoglycan when the native S. aureus PBPs are inhibited by β-lactams. If assembly of the pentaglycine branch on the cell wall precursor Lipid II is genetically blocked, MRSA strains become susceptible to β-lactams. Therefore, it has been proposed that PBP2a can only crosslink peptidoglycan strands bearing a complete pentaglycine branch. This hypothesis has never been tested because the necessary substrates have not been available. Here, we obtained S. aureus Lipid II variants having shorter glycine branches and have tested whether PBP2a and two other S. aureus transpeptidases, PBP2 and PBP4, can crosslink peptidoglycan strands made from the variants. There are striking differences in enzymatic activity among these enzymes depending on the length of the glycine branch, but we find that PBP2a can, in fact, crosslink glycan strands bearing triglycine. We report experiments in cells that are consistent with our in vitro findings about the crosslinking preferences of these PBPs. In addition to providing insights into the cell wall physiology of a major pathogen, our studies identify the best target for β-lactam potentiators to treat MRSA.
Graphical Abstract
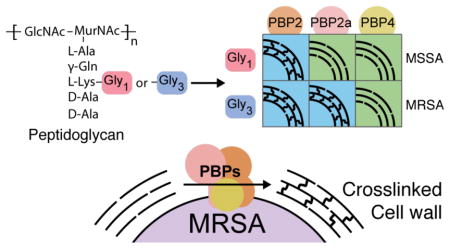
Bacterial cells are encased in a macromolecular mesh known as peptidoglycan (PG), which is required for survival. Peptidoglycan biosynthesis is a target for many clinically used antibiotics, including β-lactams. β-lactams remain widely used due to their broad spectrum and excellent safety profile. They covalently inactivate the transpeptidase (TP) domains of penicillin-binding proteins (PBPs), thereby preventing crosslinking of peptidoglycan strands (SI Figure 1).1 In most organisms, β-lactam resistance is due to expression of β-lactamases that destroy β-lactams before they can inactivate their lethal targets. A major strategy for overcoming this form of resistance is to use a β-lactamase inhibitor in combination with a β-lactam.1 In Staphylococcus aureus, resistance arises through acquisition of a gene cassette containing mecA, which encodes the intrinsically resistant transpeptidase, PBP2a.2 This enzyme crosslinks peptidoglycan when other transpeptidases are inhibited by β-lactams. These methicillin-resistant S. aureus strains (MRSA) are responsible for more than half of deaths due to antibiotic resistant infections in the United States.3 Strategies to overcome β-lactam resistance have focused on developing new β-lactams that inhibit PBP2a, such as ceftaroline and ceftobiprole, and on identifying β-lactam potentiators for use in combination with β-lactams.2a,4
β-lactam potentiators increase the susceptibility of MRSA strains to β-lactams. Genetic studies have identified over two dozen potentiator targets, including FemA and FemB, which are required for synthesis of the pentaglycine branch on S. aureus Lipid II, the peptidoglycan precursor (Figure 1).5 S. aureus Lipid II consists of a lipid-linked disaccharide containing a pentapeptide stem with a pentaglycine branch attached to the lysine of the stem peptide. FemA installs the second and third glycines of this branch, making Gly3-Lipid II, and FemB installs the fourth and fifth, making Gly5-Lipid II. Once assembled, Gly5-Lipid II is flipped to the extracellular surface of the membrane where it is polymerized and crosslinked (Figure 1). S. aureus strains lacking the femA or femB genes are viable, implying that one or more of the native PBPs can crosslink peptidoglycan containing Gly1- and Gly3-peptidoglycan.5a,5b However, deletion of these genes increases susceptibility to β-lactams even when PBP2a is expressed. Therefore, it has been proposed PBP2a can only crosslink Gly5-peptidoglycan.6
Figure 1.
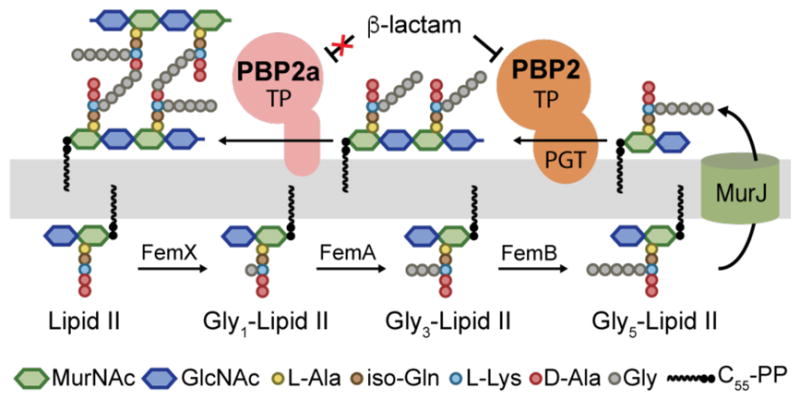
Schematic of peptidoglycan biosynthesis in methicillin-resistant S. aureus. Three enzymes, FemXAB, attach five glycines to the lysine side chain of the peptidoglycan precursor Lipid II. FemX is essential but cells can survive without FemA or FemB.5 After export to the cell surface, Gly5-Lipid II is polymerized by peptidoglycan glycosyltransferases (PGTs) and the glycan strands are crosslinked by transpeptidases (TPs). PGT domains are found in bifunctional enzymes such as PBP2 (shown), which also contain transpeptidase domains, or in monofunctional enzymes. PBP2a crosslinks glycan strands when native TPs, e.g., as in PBP2, are inhibited by β-lactams.
The crosslinking preferences of S. aureus PBPs have not been characterized due to the major challenges involved in obtaining uncrosslinked peptidoglycan substrates bearing different glycine branches. We recently developed a method to obtain Gly5-Lipid II in large amounts from S. aureus, enabling access to Gly5-peptidoglycan.7,8 Here we report that it is possible to obtain substantial quantities of Gly1- and Gly3-Lipid II from appropriate S. aureus mutant strains, which has enabled us to examine the crosslinking preferences of native S. aureus PBPs as well as PBP2a.
The transpeptidase activity of PBP2a has not been previously reconstituted. Therefore, we first developed conditions to monitor crosslinking of uncrosslinked glycan strands produced from the native cell wall precursor, Gly5-Lipid II. Gly5-Lipid II was incubated with PBP2a and a mutant of PBP2 (PBP2-TP*) that contains an active peptidoglycan glycosyl-transferase (PGT) domain but a catalytically inactive trans-peptidase domain. Reactions were quenched after 3 h and the polymeric products were treated with mutanolysin and NaBH4 to produce muropeptide fragments for LC/MS analysis (Figure 2a).9 We detected three major products: monomer (pink peak), hydrolyzed monomer (monomer minus D-Ala, blue peak), and dimer (orange peak). The hydrolyzed monomer and dimer peaks disappeared when PBP2a was genetically inactivated by mutating the active site serine to alanine (Figure 2a, PBP2a S403A), confirming that they result from PBP2a activity.10 Therefore, PBP2a can activate the terminal amide bond in the pentapeptide, resulting in cross-linking if the pentaglycine attacks the covalent intermediate or hydrolysis if water attacks (see SI Figure 1).
Figure 2.

Reconstitution of PBP2a activity and inhibition. (a) Demonstration of PBP2a activity by LC/MS. Extracted ion chromatogram (EIC) for the wild type PBP2a (PBP2a wt) shows both hydrolysis and crosslinked products, but the catalytically inactive mutant (PBP2a S403A) or ceftaroline-treated PBP2a do not. (b) Western blot showing inhibition of PBP2a traspeptidation in the presence of different concentrations of ceftaroline and meropenem. Peptidoglycan fragments incorporate biotin-D-lysine unless PBP2a transpeptidase activity is inhibited (see Methods).
We next examined the β-lactam susceptibility profile of PBP2a. Previous studies showed that PBP2a can be inactivated by the fifth-generation cephalosporins, ceftaroline and ceftobiprole, and is weakly inhibited by meropenem, but not other β-lactams.2a,4b,11 We found that incubation of PBP2a with ceftaroline prior to adding PBP2-TP* and Gly5-Lipid II abolished formation of the dimer and hydrolyzed monomer peaks in the LC/MS assay (Fig. 2a). We also used a PAGE-immunoblot assay to compare inhibitions of transpeptidase activity by these β-lactams (Figure 2b, SI Figure 2).7 Meropenem inhibited PBP2a, but was about twenty-fold less potent than ceftaroline, consistent with results obtained from competition binding assays for these β-lactams.11–12
We next examined the substrate preferences of PBP2a and two other S. aureus PBPs, PBP2 and PBP4. PBP2 is the only PBP in S. aureus that contains both a peptidoglycan glycosyltransferase domain and a transpeptidase domain, and it is essential for survival. PBP4, which contains only a transpeptidase domain, is not essential for survival, but is known for installing additional crosslinks in peptidoglycan and has been implicated in β-lactam resistance in some MRSA strains.13 Using the method previously developed to obtain Gly5-Lipid II, we isolated Gly1- and Gly3-Lipid II from ΔfemA and ΔfemB S. aureus strains, respectively (Figure 3a–3c).7 To investigate crosslinking preferences, we incubated each substrate with either (1) PBP2 TP* and PBP2a, (2) PBP2 wt, or (3) PBP2 TP* and PBP4. For each condition, we quantified the ratio of crosslinked muropeptide and hydrolyzed monomer (minus D-Ala) using LC/MS (Figure 3d). The combined crosslinking and hydrolysis activities were approximately constant across different substrates for each enzyme, showing that formation of the acyl-enzyme intermediate was not sensitive to the length of the glycine branch; however, the proportion of activity due to crosslinking versus hydrolysis varied greatly. PBP2a preferred Gly5-peptidoglycan, but also crosslinked Gly3-peptidoglycan; however, it hydrolyzed Gly1-peptidoglycan. PBP2 crosslinked all three substrates. PBP4, the low-molecular weight PBP, showed the most dramatic switch in activity as the glycine branch was truncated. For Gly5-peptidoglycan, PBP4 performed almost exclusively crosslinking, whereas 90% of products were due to hydrolysis with Gly3-peptidoglycan. With Gly1-peptidoglycan, PBP4 acted only as a carboxypeptidase.
Figure 3.

The ratio of crosslinking to hydrolysis for S. aureus PBPs depends on both the enzyme and the substrate. (a) Schematic of the Lipid II isolation procedure with removal of the lipid tail for LC/MS analysis. S. aureus wt. or mutant strains are treated with moenomycin A, which inhibits PGT activity, to accumulate Lipid II, which is then isolated by a two-step extraction procedure (see Methods). Acidic hydrolysis of the isolated Lipid II removes the undecaprenyl-phosphate chain for LC/MS analysis. (b) Chemical structure of Lipid II monophosphates after acid hydrolysis. (c) LC/MS analysis of Gly1- and Gly3-Lipid II monophosphates isolated from the ΔfemA and ΔfemB mutants, respectively. (d) A bar graph showing ratios of crosslinking and hydrolysis activities for PBP2a, PBP2 and PBP4 with Gly5-, Gly3-, and Gly1-peptidoglycan (see Methods).
To assess the relevance of the in vitro findings on PBP4, we quantified the percentage of hydrolyzed monomer compared to other muropeptides in cell wall isolated from ΔfemB strains with or without pbp4 (Figure 4a).13a In wildtype S. aureus, hydrolyzed muropeptide makes up about 10% of the total muropeptide products and this increases to 20% in the ΔfemB mutant. When pbp4 is deleted in the ΔfemB background, hydrolyzed muropeptide decreases to wildtype levels. Complementation of Δpbp4ΔfemB with pbp4 doubles the amount of hydrolyzed muropeptide. We concluded that PBP4 has substantial carboxypeptidase activity in cells producing Gly3-peptidoglycan, consistent with in vitro findings.
Figure 4.
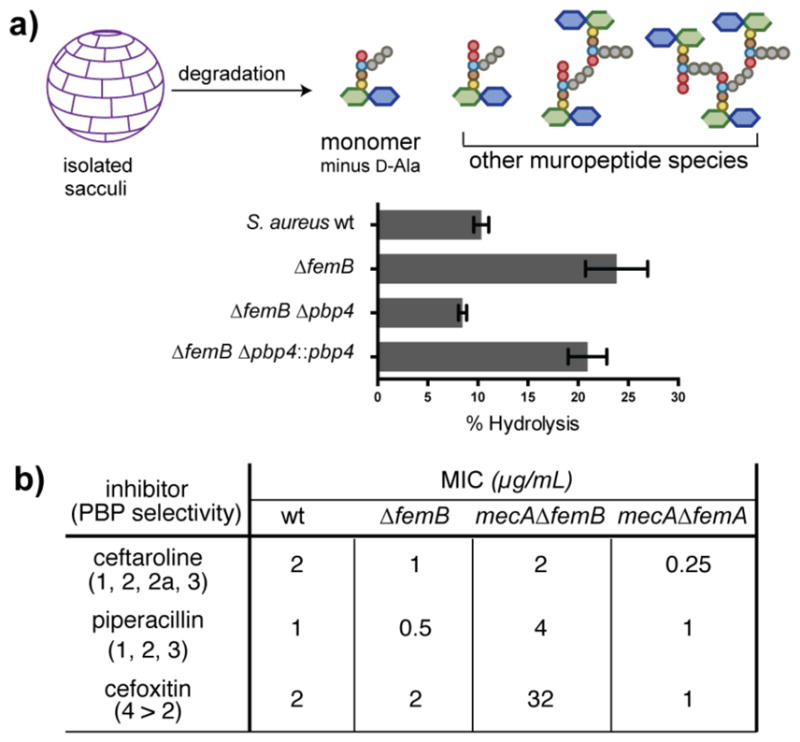
In cells, Gly3-peptidoglycan undergoes hydrolysis by PBP4 yet can be crosslinked by PBP2a. (a) S. aureus sacculi were degraded and analyzed by LC/MS to determine the extent of D-Ala hydrolysis. The bar graph shows the percentage of hydrolyzed muropeptide compared to other depicted species. Deleting PBP4 reduces hydrolysis. (b) Minimum inhibitory concentrations (MICs) for three different β-lactams against four S. aureus strains, with the targeted PBPs for each β-lactam shown in parentheses. The presence of mecA, encoding PBP2a, in the ΔfemB background increases the MIC of piperacillin and cefoxitin, but not ceftaroline, consistent with the in vitro data that PBP2a can crosslink Gly3-peptidoglycan. In the ΔfemA background, mecA does not confer resistance.
Determining whether PBP2a can crosslink Gly3-peptidoglycan in vivo was more challenging than probing PBP4’s role. We compared the minimum inhibitory concentrations (MIC) of three β-lactams, ceftaroline, piperacillin, and cefoxitin, against four S. aureus strains, wildtype, ΔfemB, mecAΔfemB, and mecAΔfemA (Figure 4b).14 The mecA strains contain a cassette that expresses PBP2a. Deleting femB had only a modest effect on the MICs of the three drugs. Introducing mecA into the ΔfemB background, however, increased the MICs of piperacillin and cefoxitin by 8-fold and 16-fold, respectively, but did not significantly affect the MIC of ceftaroline. These findings are consistent with the in vitro data showing that PBP2a can crosslink Gly3-peptidoglycan when the native PBPs are inhibited. In contrast, the mecAΔfemA strain remained susceptible to all three β-lactams, confirming the in vitro results that PBP2a preferentially hydrolyzes Gly1-peptidoglycan.
We have found that three S. aureus PBPs in MRSA show substantial differences in crosslinking versus hydrolysis behavior as the glycine branch on Lipid II is shortened. Based on our results, we draw the following conclusions. First, because the transpeptidase activity of S. aureus PBP2 is relatively insensitive to glycine branch length, this PBP must play a major role in crosslinking Gly1-peptidoglycan to allow survival of strains lacking femA. Second, PBP4, although noted for its ability to make highly crosslinked peptidoglycan, primarily acts as a carboxypeptidase to remove D-Ala from the stem peptide when there are fewer than five glycines in the branch peptide. The sensitivity of PBP4 to substrate structure indicates a delicate balance between crosslinking and hydrolytic activities, and as most β-lactams do not inhibit PBP4, its hydrolytic activity may contribute to lethality in ΔfemA or ΔfemB strains treated with β-lactams. Finally, although PBP2a does not crosslink Gly1-peptidoglycan, it unexpectedly crosslinks Gly3-peptidoglycan. These findings have important implications for strategies to treat MRSA infections because they imply that FemA is a better target than FemB for potentiators designed to restore susceptibility to β-lactams.15
Supplementary Material
Acknowledgments
We thank Prof. S. J. Kim for providing the UT34-2 and UK17 strains and Prof. A. Cheung for the MW2 Δpbp4, MW2 Δpbp4::pbp4 strains. We also thank J. X. Wang at the Harvard Small Molecule Mass Spectrometry Facility for help with MS.
Funding Sources
This work was funded by NIH grants GM076710 (S.W.) GM066174 (D.K.) and a Singapore A*STAR NSS (Y.Q).
Footnotes
Notes
The authors declare no competing financial interest.
Experimental procedures, protein purification protocol, LC/MS analysis, and Western blot analysis. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Walsh CTWT. Antibiotics: Challenges, Mechanisms, Opportunities. ASM press; Washington, DC: 2016. [Google Scholar]
- 2.(a) Fishovitz J, Rojas-Altuve A, Otero LH, Dawley M, Carrasco-López C, Chang M, Hermoso JA, Mobashery S. J Am Chem Soc. 2014;136:9814. doi: 10.1021/ja5030657. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fishovitz J, Taghizadeh N, Fisher JF, Chang M, Mobashery S. J Am Chem Soc. 2015;137:6500. doi: 10.1021/jacs.5b01374. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) de Lencastre H, Oliveira D, Tomasz A. Curr Opin Microbiol. 2007;10:428. doi: 10.1016/j.mib.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Center for Disease Control and Prevention Office of Infectious Disease. [accessed May 1, 2017]; https://www.cdc.gov/drugresistance/threat-report-2013.
- 4.(a) Villegas-Estrada A, Lee M, Hesek D, Vakulenko SB, Mobashery S. J Am Chem Soc. 2008;130:9212. doi: 10.1021/ja8029448. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lovering AL, Gretes MC, Safadi SS, Danel F, De Castro L, Page MGP, Strynadka NCJ. J Biol Chem. 2012 doi: 10.1074/jbc.M112.355644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Maidhof H, Reinicke B, Blumel P, Berger-Bachi B, Labischinski H. J Bacteriol. 1991;173:3507. doi: 10.1128/jb.173.11.3507-3513.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Henze U, Sidow T, Wecke J, Labischinski H, Berger-Bächi B. J Bacteriol. 1993;175:1612. doi: 10.1128/jb.175.6.1612-1620.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rohrer S, Berger-Bächi B. Antimicrob Agents Chemother. 2003;47:837. doi: 10.1128/AAC.47.3.837-846.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Strandén AM, Ehlert K, Labischinski H, Berger-Bächi B. J Bacteriol. 1997;179:9. doi: 10.1128/jb.179.1.9-16.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tschierske M, Ehlert K, Stranden AM, Berger-Bachi B. FEMS Microbiol Lett. 1997;153:261. doi: 10.1016/s0378-1097(97)00234-6. [DOI] [PubMed] [Google Scholar]
- 7.Qiao Y, Srisuknimit V, Rubino F, Schaefer K, Ruiz N, Walker S, Kahne D. Nat Chem Biol. 2017 doi: 10.1038/nchembio.2388. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Ye XY, Lo MC, Brunner L, Walker D, Kahne D, Walker S. J Am Chem Soc. 2001;123:3155. doi: 10.1021/ja010028q. [DOI] [PubMed] [Google Scholar]; (b) Schwartz B, Markwalder JA, Wang Y. J Am Chem Soc. 2001;123:11638. doi: 10.1021/ja0166848. [DOI] [PubMed] [Google Scholar]; (c) VanNieuwenhze MS, Mauldin SC, Zia-Ebrahimi M, Winger BE, Hornback WJ, Saha SL, Aikins JA, Blaszczak LC. J Am Chem Soc. 2002;124:3656. doi: 10.1021/ja017386d. [DOI] [PubMed] [Google Scholar]; (d) Breukink E, van Heusden HE, Vollmerhaus PJ, Swiezewska E, Brunner L, Walker S, Heck AJR, de Kruijff B. J Biol Chem. 2003;278:19898. doi: 10.1074/jbc.M301463200. [DOI] [PubMed] [Google Scholar]; (e) Lloyd AJ, Gilbey AM, Blewett AM, De Pascale G, El Zoeiby A, Levesque RC, Catherwood AC, Tomasz A, Bugg TDH, Roper DI, Dowson CG. J Biol Chem. 2008;283:6402. doi: 10.1074/jbc.M708105200. [DOI] [PubMed] [Google Scholar]; (f) Huang LY, Huang SH, Chang YC, Cheng WC, Cheng TJR, Wong CH. Angew Chem Int Ed. 2014;53:8060. doi: 10.1002/anie.201402313. [DOI] [PubMed] [Google Scholar]
- 9.(a) Lebar MD, Lupoli TJ, Tsukamoto H, May JM, Walker S, Kahne D. J Am Chem Soc. 2013;135:4632. doi: 10.1021/ja312510m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lebar MD, May JM, Meeske AJ, Leiman SA, Lupoli TJ, Tsukamoto H, Losick R, Rudner DZ, Walker S, Kahne D. J Am Chem Soc. 2014;136:10874. doi: 10.1021/ja505668f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lupoli TJ, Lebar MD, Markovski M, Bernhardt T, Kahne D, Walker S. J Am Chem Soc. 2014;136:52. doi: 10.1021/ja410813j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Qiao Y, Lebar MD, Schirner K, Schaefer K, Tsukamoto H, Kahne D, Walker S. J Am Chem Soc. 2014;136:14678. doi: 10.1021/ja508147s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Lim D, Strynadka NCJ. Nat Struct Mol Biol. 2002;9:870. doi: 10.1038/nsb858. [DOI] [PubMed] [Google Scholar]; (b) Fuda C, Suvorov M, Vakulenko SB, Mobashery S. J Biol Chem. 2004;279:40802. doi: 10.1074/jbc.M403589200. [DOI] [PubMed] [Google Scholar]
- 11.Bobba S, Ponnaluri VKC, Mukherji M, Gutheil WG. Antimicrob Agents Chemother. 2011;55:2783. doi: 10.1128/AAC.01327-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Davies TA, Page MGP, Shang W, Andrew T, Kania M, Bush K. Antimicrob Agents Chemother. 2007;51:2621. doi: 10.1128/AAC.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moisan H, Pruneau M, Malouin F. J Antimicrob Chemother. 2010;65:713. doi: 10.1093/jac/dkp503. [DOI] [PubMed] [Google Scholar]
- 13.(a) Memmi G, Filipe SR, Pinho MG, Fu Z, Cheung A. Antimicrob Agents Chemother. 2008;52:3955. doi: 10.1128/AAC.00049-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chan LC, Basuino L, Diep B, Hamilton S, Chatterjee SS, Chambers HF. Antimicrob Agents Chemother. 2015;59:2960. doi: 10.1128/AAC.05004-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharif S, Kim SJ, Labischinski H, Chen J, Schaefer J. J Bacteriol. 2013;195:1421. doi: 10.1128/JB.01471-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukumoto A, Kim YP, Hanaki H, Shiomi K, Tomoda H, Ōmura S. J Antibiot. 2008;61:7. doi: 10.1038/ja.2008.102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.