

Abstract
Background & Aims
Non-alcoholic fatty liver disease (NAFLD) is the most common form of liver disease. Activation of hedgehog (Hh) signaling has been implicated in the progression of NAFLD and proposed as a therapeutic target; however, the effects of Hh signaling inhibition have not been studied in humans with germline mutations affecting this pathway.
Methods
Patients with holoprosencephaly (HPE), a disorder associated with germline mutations disrupting Sonic hedgehog (SHH) signaling, were clinically evaluated for NAFLD. A combined mouse model of Hh signaling attenuation (Gli2 heterozygous null: Gli2+/−) and diet-induced NAFLD was used to examine aspects of NAFLD and hepatic gene expression profiles, including molecular markers of hepatic fibrosis and inflammation.
Results
Patients with HPE had a higher prevalence of liver steatosis compared to the general population, independent of obesity. Exposure of Gli2+/− mice to fatty liver-inducing diets resulted in increased liver steatosis compared to wild-type mice. Similar to humans, this effect was independent of obesity in the mutant mice and was associated with decreased expression of pro-fibrotic and pro-inflammatory genes, and increased expression of PPARγ, a potent anti-fibrogenic and anti-inflammatory regulator. Interestingly, tumor suppressors p53 and p16INK4 were found to be downregulated in the Gli2+/− mice exposed to high-fat diet.
Conclusions
Our results indicate that germline mutations disrupting Hh signaling promote liver steatosis, independent of obesity, with reduced fibrosis. While Hh signaling inhibition has been associated with a better NAFLD prognosis, further studies are required to evaluate the long-term effects of mutations affecting this pathway.
Keywords: non-alcoholic fatty liver disease, NAFLD, metabolic disease, hedgehog, SHH, GLI2, NASH, liver fibrosis, cirrhosis, hepatocellular carcinoma, holoprosencephaly, HPE
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common form of liver disease in the United States, affecting 20–30% of adults and 7.6% of children [1]. The severity of NAFLD ranges from simple steatosis (fat deposition without inflammation) to non-alcoholic steatohepatitis (NASH), which can progress to cirrhosis and hepatocellular carcinoma (HCC) [2, 3]. Although NAFLD is usually associated with obesity, type-2 diabetes, and hyperlipidemia [4]—primarily as a result of high intake of foods rich in sugar and saturated fats (e.g., Western-style diet) and inactive lifestyles—epidemiological studies, differences in prevalence among ethnicities, and evidence of familial aggregation suggest a genetic component [5–7].
Hedgehog (Hh) ligands are critical developmental morphogens necessary for embryonic patterning, growth and cell differentiation [8]. Beyond embryogenesis, several types of cells that reside in healthy adult livers are capable of producing and/or responding to Hh ligands and this pathway is activated in many types of acute and chronic liver injury, with tissue responses including expansion of liver progenitor populations, myofibroblast accumulation and fibrogenesis, repair-associated inflammation, and vascular remodeling [9]. In addition to its role in liver repair, several studies have also suggested Hh signaling involvement in the regulation of lipid metabolism in adipocytes [10, 11] and more recently, in hepatocytes [12], This cumulative evidence suggests an important role for Hh signaling in liver function, but the effect of human germline Hh signaling mutations on the development and progression of NAFLD has not been investigated.
Patients and Methods
Human subjects
Patients were recruited in accordance with the ethical guidelines and recommendations of the National Human Genome Research Institute (NHGRI) Institutional Review Board. An informed consent was obtained for all participants. Approximately 1,645 HPE cases and their biological relatives were enrolled under research protocol 98-HG-0249 (clinicaltrials.gov: NCT00341978) until June 2015. Each individual was tested for mutations in the four most common genes involved in nonchromosomal, nonsyndromic holoprosencephaly (HPE) (SHH, ZIC2, SIX3, and TGIF1) using previously described methods [13]. In addition to the standard evaluation described elsewhere [14], a subset of patients was examined at the NIH Clinical Center (NIH-CC) under NHGRI protocol 04-HG-00923 (clinicaltrials.gov: NCT00088426). Clinical testing included an abdominal ultrasound, fasting lipid profiles, and hepatic function tests. All individuals with a clinical diagnosis of HPE seen at the NIH-CC, including those with microform HPE, were included in the analysis. Additionally, 61 autopsy reports from individuals who succumbed to severe HPE were reviewed.
Mouse model
The animal protocol was approved by the NHGRI Animal Care and Use Committee (protocol G-13-2) in compliance with the ethical regulations stated in the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. The results are reported according to the ARRIVE guidelines [15]. Gli2 heterozygous null (Gli2+/−) mice were used as a model of attenuated Hh signaling. GLI2 is a downstream target involved in Hh signaling [16], which allows us to investigate the convergence of multiple factors affecting this key pathway. Unlike humans, Gli2+/− mice have not been previously reported to show any clinical phenotype [17], although a functional haploinsufficiency resulting in severe HPE has been demonstrated in gene-environment interaction studies [18, 19]. Additional information on this mouse model and specific experimental procedures can be found in the Supplementary Material.
Dietary interventions
At 4 weeks of age, initial weight was recorded and Gli2+/− and wild-type (WT) mice were switched from standard breeder chow to: group 1) high-fat (HF) diet (60% kcal from fat) or a control diet (10% kcal from fat); or group 2) methionine- and choline-deficient (MCD) diet or a control diet. Mice in group 1 were exposed to diets for 12 weeks and mice in group 2 for 4 weeks. Animals were fed ad libitum. Weight was recorded again at the end of the intervention period, body fat composition determined and mice euthanized for blood and tissue collection (Supplementary Methods). The presence and severity of liver steatosis was assessed by histological evaluation and triglyceride quantification. A subset of livers was selected for RNA expression profiling, and a subset of pituitary glands was sent for histological evaluation. Because sex differences exist in the response of C57BL/6J mice to dietary models [20], we compared same-sex animals in experimental groups.
Details about histological, biochemical, gene expression, bioinformatics and statistical analyses can be found in the Supplementary Material.
Results
Fatty liver is more prevalent in patients with HPE, a disease caused by SHH signaling disruption
HPE is a disorder of forebrain development associated with genetic and teratogenic lesions disrupting Sonic hedgehog (SHH) signaling [18, 19, 21]. Clinical evaluation of 29 patients with HPE (18 children and 11 adults) at the NIH-CC showed sonographic evidence of fatty liver in 12 individuals (33.33% of children [P = 0.002], and 54.50% of adults [P < 0.001]) (Table 1). The mean age of individuals with NAFLD was 30.3 years (P = 0.03) for adults and 5.5 years (P = 0.04) for children. Three of the 6 adults (50%), but only 1 of the 6 children (16.67%) with fatty liver were obese. Hyperlipidemia, the most common morbidity associated with NAFLD, was significantly lower in patients with HPE than in the general population (16.67% versus 69.16%, P = 0.005) (Table 1). Overall, there was no statistically significant correlation between the presence of fatty liver and the severity of brain malformation, age, diabetes, hypertension, obesity, dyslipidemia, or hepatic function tests (Tables S1 and S2). Example pedigrees of individuals with HPE and NAFLD are shown in Figure 1.
Table 1.
Comparison of individuals with NAFLD in the HPE cohort compared to the general population
| Population estimates | HPE cohort | P value | |
|---|---|---|---|
| Adults with NAFLD (n=6) | |||
| Percentage | 24.13 [4] | 54.50 | <0.001 |
| Age (y) | 40.10 [57] | 30.30 | 0.026 |
| Obesity | 51.34 [4] | 50.00 | 1 |
| BMI* (Kg/m2) | 28.20 [57] | 27.98 | 0.960 |
| Diabetes | 22.51 [4] | 16.67 | 1 |
| Dyslipidemia | 69.16 [4] | 16.67 | 0.005 |
| Hypertension | 39.34 [4] | 25.00 | 1 |
| Children with NAFLD (n=6) | |||
| Percentage | 7.60 [1] | 33.33 | 0.002 |
| Age | 11 [58] | 5.50 | 0.037 |
Body mass index was calculated as weight (kg)/height2 (m2). P compared to population estimates using two-sided t tests.
Figure 1.
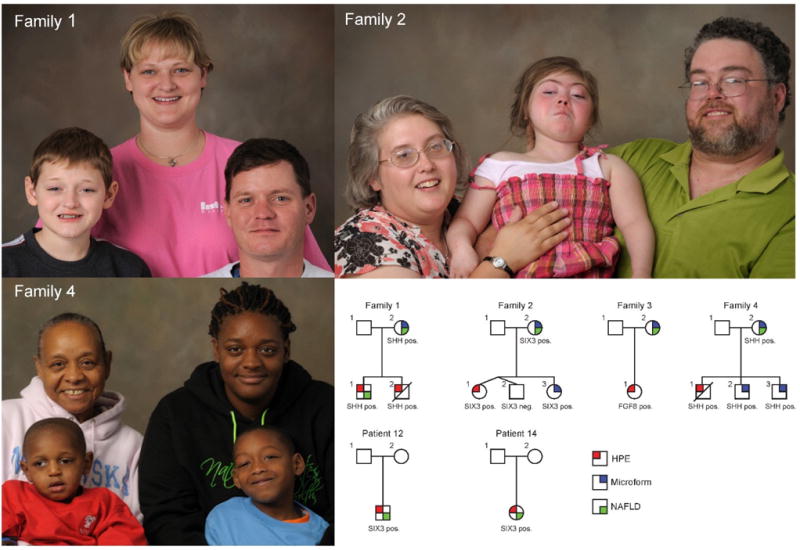
Example of families segregating mutations that perturb SHH signaling. Families 1 and 4 correspond to kindreds segregating heterozygous SHH mutations. The majority of affected individuals in these two families have microform HPE (mutation positive individuals with subtle midline facial differences), though both families have deceased individuals who succumbed to severe HPE. The child with HPE and the mutation-positive mother in family 1 had fatty liver at the time of examination. The grandmother in family 4 also carried the mutation and had a history of fatty liver, but was not available for examination. In family 2, both the proband and her mother carry a SIX3 nonsense mutation; the proband has frank HPE, while the mother (as well as other family members who are not shown), demonstrate microform features only.
In addition, we evaluated 67 autopsy reports from individuals with severe HPE. Sixty-two reports were from patients who had fetal or immediate postnatal demise, but only 42 included a histological evaluation of the liver. Of those, 3 (7.32%) had fatty liver. Two of 6 (33%) individuals who survived the newborn period also had some degree of fatty liver (Table S3). These results suggest earlier onset and higher prevalence of fatty liver in our HPE cohort compared to the general population.
Hh signaling attenuation increases liver steatosis in mice
We used a Gli2 heterozygous null (Gli2+/−) mouse as a model of attenuated Hh signaling. In order to at least partly mimic Western-style dietary influences in humans, Gli2+/− mice and their age-matched WT littermates were fed HF or control diet. Mice fed the HF diet showed a significant increase in body weight; body fat composition; serum levels of triglycerides, nonesterified fatty acids, total and LDL cholesterol; and increased hepatic steatosis (Figure S1, Table S4). These results are in agreement with previous animal studies showing similar metabolic effects of HF diets [20, 22], which validates our dietary model.
We next compared Gli2+/− and WT on HF diet to determine the effect of Hh signaling attenuation on diet-induced fatty liver disease. While we were not able to detect any significant differences between strains in body weight, body fat composition, serum lipid profiles or liver triglyceride content (Figure S2 and S3A, Table S5), Gli2+/− mice showed increased liver steatosis scores upon histological evaluation compared to their WT littermates (Figure 2A). This disagreement between liver triglyceride quantification and steatosis scores highlights the importance of histological evaluation in the diagnosis of NAFLD, as the pattern of fat deposition can vary from one individual to another. In general, our mice exposed to HF diet for 12 weeks revealed a multifocal deposition phenotype, which limits our ability to accurately quantify triglycerides from liver hunks that may not be representative of the entire organ’s lipid content.
Figure 2.
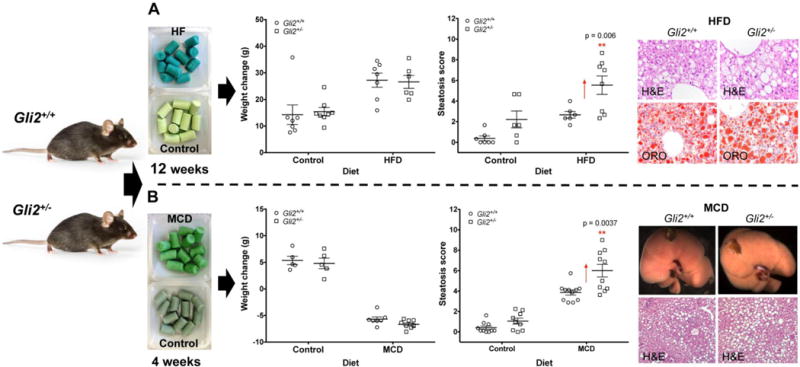
Dietary interventions in C57BL/6J-Gli2+/− and C57BL/6J-WT littermates. Animals were exposed to (A) a high-fat (HF) diet for 12 weeks (females) or (B) a methionine-and choline-deficient (MCD) diet for 4 weeks (males). While there were no differences in body mass and body fat between strains fed the HF diet (see also Table S5), Gli2+/− mutants showed an increase in hepatic steatosis compared to WTs. Gli2+/− mice on the MCD diet also showed an increase in macrovesicular liver steatosis but in the presence of weight loss. Results are expressed as the mean ± SEM. **P<0.01. Student t test with Holm-Sidak correction for multiple comparisons was used for statistical analysis. Magnification: top panel, 400×; bottom panel, 100×. H&E, hematoxylin and eosin staining. ORO, Oil Red O staining.
The increased hepatic steatosis without body fat increase suggested that Hh signaling disruption might affect hepatic lipid metabolism independent of systemic adiposity. To test this hypothesis, we exposed Gli2+/− and WT mice to an MCD diet, which is known to induce severe NASH in the presence of weight loss and does not induce features of metabolic syndrome, an important risk factor for NAFLD [2, 23]. Interestingly, Gli2+/− mice still exhibited increased liver steatosis compared to WT animals (Figure 2B). Different from the HF diet, the livers of mutant mice exposed to MCD diet exhibited increased triglyceride content (Figure S3B) consistent with a more diffuse pattern of hepatic steatosis induced by this diet [24, 25]. These results indicate that Hh signaling defects can result in hepatic lipid accumulation in the absence of obesity.
Because GLI2 mutations have been associated with pituitary anomalies in humans [16, 26], and this could independently influence energy metabolism, we examined basal pituitary, adrenal and thyroid secretory function in adult mice exposed to breeder chow. We did not find any differences between Gli2+/− and WT mice in pituitary histology or serum levels of adrenocorticotrophic hormone (ACTH), growth hormone (GH), cortisone, triiodothyronine (T3) or thyroxine (T4) (Figures S4 and S5).
Hh signaling attenuation causes dysregulation of genes involved in hepatic energy metabolism
The increased liver steatosis seen in the Gli2+/− mice suggests an intrinsic hepatic metabolic defect that predisposes to diet-induced NAFLD. To examine the molecular effects of Hh signaling attenuation alone, we compared the hepatic gene expression profiles of Gli2+/− vs. WT mice exposed to control chow. Dysregulated genes are shown in Table S6. Gene network analysis identified key pathways in lipid and xenobiotic metabolism, lipid mobilization/transport and cellular migration (Figure S6A). To analyze and interpret the biological meaning of differential gene expression profiles we used Ingenuity Pathway Analysis (IPA) (see Supplementary Material). Interestingly, IPA Downstream Effects analysis identified fatty acid oxidation and immunological function as the most significantly decreased categories in the Gli2+/− mice, suggesting that Hh attenuation predisposes to hepatic fat accumulation while decreasing inflammatory response (Figure 3). Upstream Regulator analysis predicted several regulators to be inhibited, such as peroxisome proliferator- activated receptor alpha (PPARα), nuclear receptor subfamily 1, group I, member 2 (NR1I2, also knows as pregnane X receptor) and tumor suppressor p53 (TP53), while others were predicted to be activated, such as proto-oncogene c-Myc (Figure 4).
Figure 3.
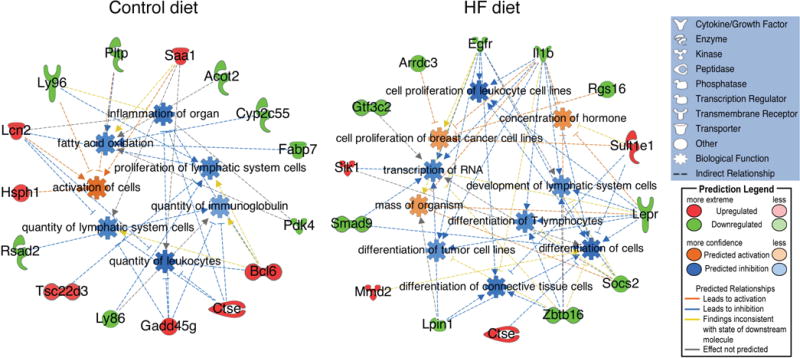
IPA Downstream Effects analysis of genes differentially expressed in the livers of C57BL/6J-Gli2+/− mice (n=4) compared to C57BL/6J-WT (n=4) fed control or HF diet (females). Molecules in green and red correspond to down- and upregulated genes from our dataset, respectively.
Figure 4.
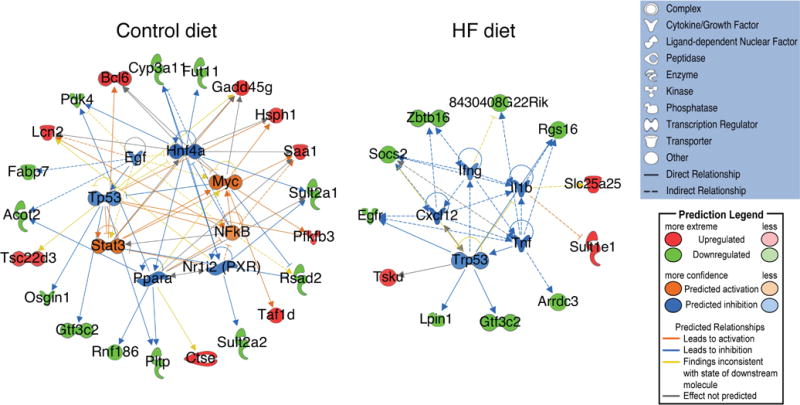
IPA network analysis of upstream regulators identified in the C57BL/6J-Gli2+/− (n=4) compared to C57BL/6J-WT (n=4) mice fed control or HF diet (females). Molecules in green and red correspond to down- and upregulated genes from our dataset, respectively.
We next compared the hepatic gene expression profiles of Gli2+/− vs. WT mice exposed to HF diet, as Western-style dietary influences impact liver metabolism and the development and progression of NAFLD in humans. Dysregulated genes are shown in Table S7. Gene network analysis identified pathways in cell signaling, nucleic acid metabolism, small molecule metabolism (in particular lipid and xenobiotic metabolism), amino acid metabolism and molecular transport (Figure S6B). Compared to control diet, the HF diet showed statistically significant dysregulation of pathways related to liver proliferation, liver fibrosis and liver steatosis. Similar to control diet, IPA Downstream Effects analysis also predicted an inhibition of inflammatory response, but new functions included inhibited differentiation of connective tissue cells and inhibited differentiation of tumor cells (Figure 3). Upstream Regulator Analysis predicted inhibition of important regulators of inflammation (IL1β, TNFα, INFγ and CXCL12) and cell cycle progression and apoptosis (p53) (Figure 4). These are interesting observations in view of the normal progression of NAFLD, which conversely involves increased inflammation and increased activation of hepatic stellate cells (HSC) leading to liver fibrosis and NASH cirrhosis [27]. Another interesting finding was the predicted inhibition of p53 and decreased tumor cell differentiation.
Hh signaling attenuation decreases liver inflammation and fibrogenesis
Gene expression analyses suggested that Hh signaling attenuation has a negative effect on liver reparative functions. Normally, liver damage as seen in steatosis and NASH induces the local production of Hh ligands that stimulate the proliferation and activation of cells involved in liver repair, including HSCs [9, 28]. Sustained hepatic damage induces the expression by HSCs of genes involved in extracellular matrix (ECM) remodeling, such as matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) [29]. Pro-inflammatory cytokines also play a major role in liver regeneration and the pathogenesis of NAFLD, in particular the IL1β inflammasome [30], as well as PPARs, which are master regulators of energy metabolism and overall liver function [31].
When exposed to control diet, we did not detect any differences between Gli2+/− and WT mice in mRNA levels of fibrosis markers collagen type I, alpha-smooth muscle actin (α-SMA), vimentin, MMP-2 or TIMP-2 (Figure 5A). We did not detect any expression of TIMP-1. Likewise, mRNAs levels of pro-fibrogenic and pro-inflammatory cytokines TGFβ1, IL1β and CCL2 remained unchanged. There was a slight increase in F4/80 mRNA expression, a macrophage surface marker. IL6 and TNFα mRNAs were not detectable in either strain. Contrary to IPA Upstream Regulator prediction (Figure 4), PPARα remained unchanged. PPARγ was also not detectable.
Figure 5.
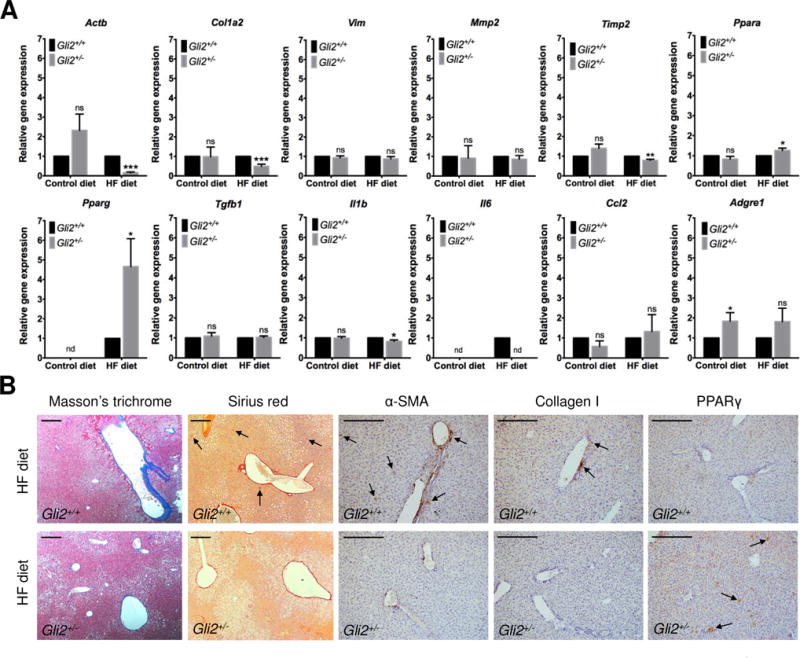
Expression of hepatic fibrosis and inflammation genes in the C57BL/6J-Gli2+/− (n=8) and C57BL/6J-WT (n=11) mice fed HF or control diet for 12 weeks (females). (A) Relative mRNA expression for α-SMA (Actb), collagen type I (Col1a2), vimentin (Vim), MMP-2 (Mmp2), TIMP-2 (Timp2), PPARγ (Pparg), PPARα (Ppara), TGFβ1 (Tgfb1), IL1β (Il1b), IL6 (Il6), CCL2 (Ccl2), and F4/80 (Adgre1). (B) Masson’s trichrome and Sirius red staining of connective tissue, and immunohistochemistry of α-SMA, collagen type I and PPARγ proteins in mice fed the HF diet. Results are expressed as the mean ± SEM. *P<0.05, **P<0.01 and ***P<0.001. Student t test with Holm-Sidak correction for multiple comparisons was used for statistical analysis. Scale bar: 100 μm. ns: not significant. nd: not detected.
When exposed to HF diet, however, the Gli2+/− mice showed reduced hepatic mRNA expression of collagen type I α-SMA, (Figure 5A) that was confirmed by immunohistochemistry and connective tissue staining (Figure 5B). No increase was observed in vimentin, MMP-2 or TIMP-2 mRNA levels, and TIMP-1 was still not detected, all of which are markers of HSC activation usually upregulated in NAFLD. IL1β and IL6 mRNA expression was reduced but no difference was detected in TGFβ1, CCL or F4/80 levels (also usually upregulated in NAFLD). TNFα was still not detected. These results suggest an attenuated pro-fibrogenic and inflammatory response in the Gli2+/− compared to WT mice. Interestingly, PPARγ and PPARα were upregulated in the Gli2+/− mice, although PPARγ expression was more pronouncedly increased. These results are in agreement with our IPA prediction of decreased inflammation and decreased differentiation of connective tissue (Figure 3), suggesting a new mechanism of NAFLD manifestation with increased steatosis but reduced or absent fibrosis and inflammation.
Hh signaling attenuation alters the expression of cell cycle regulators
Gene network analyses predicted inhibition of p53 activity and tumor cell differentiation function in the Gli2+/− mice. p53 is a master regulator of cell cycle progression and has been associated with the development of many cancer types including HCC [32, 33]. We confirmed that hepatic p53 mRNA was in fact reduced in the Gli2+/− mice on HF diet compared to WT controls (P = 5.3 × 10−11) (Figure 6). HCC markers alpha-fetoprotein (Afp), heat-shock protein 70-1b (Hspa1b) and glypican 3 (Gpc3) were not detected in any group, indicating absence of carcinoma. No differences were detected in expression of mitochondrial pro-apoptotic genes Bax and Bad. Interestingly, mRNA expression of tumor suppressor p16INK4 was also reduced in the livers of Gli2+/− mice fed the HF diet (Figure 6).
Figure 6.
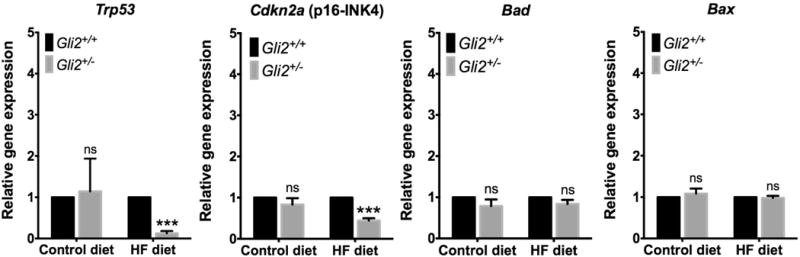
mRNA expression of cell cycle regulation, apoptosis, and carcinogenesis markers in the C57BL/6J-Gli2+/− (n=8) and C57BL/6J-WT (n=11) mice (females). No differences were observed between strains when fed control chow. Expression of p53 and p16INK4 mRNAs was significantly downregulated in Gli2+/− mice fed HF diet. HCC markers alpha-fetoprotein (Afp), heat-shock protein 70 (Hspa1b) and glypican 3 (Gpc3) were not detectable. Results are expressed as the mean ± SEM. ***P<0.001. Student t test with Holm-Sidak correction for multiple comparisons was used for statistical analysis. ns: not significant.
Evaluation of our HPE probands and their families did not reveal any evidence of HCC. However, given the young age of these individuals (oldest adults are now in their early 40’s), follow-up and collection of additional family history related to liver disease outcomes are required.
Discussion
The genetic factors that contribute to the pathology of NAFLD remain largely unidentified. The research presented here shows for the first time that germline disruption of SHH signaling in humans promotes hepatic lipid accumulation. This observation was further supported by a combined mouse model of attenuated Hh signaling and diet-induced NAFLD. While normally indistinguishable from their wild type littermates, previous reports have shown that Gli2+/− mice are sensitized to teratogen-induced HPE [18, 19, 21], demonstrating a functional GLI2 haploinsufficiency and providing an excellent model for the study of gene-environment interactions.
We found that germline Hh signaling attenuation predisposes mice to diet-induced liver steatosis independent of obesity. This is in agreement with our observation in HPE patients— most of whom are not overweight—and suggests a new mechanism of NAFLD manifestation. These findings are in concert with the recent work of Matz-Soja et al. [12] showing that conditional hepatocyte-specific deletion of Smoothened (a Patched-inhibited Hh co-receptor) in adult mice produces liver steatosis independent of insulin resistance. Previous studies have indicated that Hh signaling defects stimulate systemic adipogenesis [11, 34, 35], which can lead to metabolic complications such as dyslipidemia, type 2 diabetes and NAFLD. For instance, the study by Lee et al. [36] showed that mice lacking Hh co-receptor BOC display increased white adipose tissue. However, we did not see this effect in our Gli2+/− mice. One possible explanation is that BOC-deficient mice are fundamentally different from our Gli2+/− mice at least in terms of gene dosage (null vs. haploinsufficient, respectively), which requires additional experiments to examine the long-term effects of GLI2 haploinsufficiency and determine the gene expression threshold required for Hh-mediated dysregulation of adipogenesis.
Altered lipid homeostasis is considered the pathological hallmark of NAFLD [37]. Metabolic conditions leading to an imbalance between lipid acquisition and removal in hepatocytes will result in liver steatosis. To investigate the molecular mechanism underlying the susceptibility to hepatic steatosis in the context of attenuated Hh signaling, we analyzed hepatic gene expression profiles of Gli2+/− and WT mice exposed to low-fat (control) or HF caloric intakes. Interestingly, the livers of Gli2+/− mice showed dysregulation of genes participating in key cellular processes implicated in the pathology of NAFLD. Gene network analyses revealed dysregulation of genes involved in fatty acid β- and ω-oxidation and sterol oxidation (Cyp3a5, Cyp2c9, Sult2a1, Sult2a2, Acot2) [38–41]; lipid mobilization (Pltp and Fabp7) [42, 43]; and glucose metabolism (Pdk4, Pfkfb3) [44, 45], indicating that Hh signaling disruption affects hepatic energy metabolism.
Injured hepatocytes produce Hh ligands to initiate progenitor-cell mediated liver regeneration. In this process, Hh signaling activates quiescent HSC resulting in ECM deposition [28, 46]. However, in order to maintain hepatic tissue homeostasis, a healthy balance between matrix breakdown mediated by MMPs and matrix protein synthesis (mainly collagen deposition) is required. In NAFLD, this balance is disrupted resulting in excess ECM. Contrary to classical NAFLD molecular pathogenesis, the Gli2+/− mice showed reduced expression of pro-fibrogenic markers when exposed to HF diet. Based on our results, this could be mechanistically explained by a combination of reduced collagen deposition mediated by PPARγ, and increased ECM degradation by MMPs due to decreased expression of TIMPs [29]. Counter-intuitively, MMP-2 is considered a profibrogenic MMP, as it regulates the degradation of basal lamina allowing replacement by fibrillar collagens during fibrogenesis [47].
In addition to its widely recognized requirement for normal adipocyte function and lipid metabolism [48], PPARγ plays a central role in liver repair and the development and progression of NAFLD [31]. Some studies have proposed that increased PPARγ expression promotes liver steatosis and have attributed a causal role to PPARγ through mechanisms involving activation of lipogenic genes and de novo lipogenesis [12, 31]. Furthermore, it has been demonstrated that PPARγ inhibits monocyte and macrophage inflammatory responses and prevents myofibroblastic transdifferentiation of HSC [31], which explains the reduced expression or lack of induction of fibrosis markers and pro-inflammatory cytokines. Our results are supported by recent reports showing decreased inflammatory response upon inhibition of Hh signaling [49].
Another interesting finding was the dysregulation of molecules involved in cell cycle control and apoptosis. p53 and p16INK4 were downregulated in our Gli2+/− mice exposed to HF diet. About 50% of human tumors carry mutant p53, and many p53 mutants facilitate oncogenic functions such as increased proliferation, survival, and metastasis [33]. Mutations in p53 are found in 12–48% of HCC cases [50, 51] and are particularly frequent in hepatitis B-related HCC [50, 52], where tumorigenesis may arise in non-cirrhotic livers and display overexpression of hepatic progenitor cell genes [32]. Similarly, p16INK4 inactivation has been associated with the development of many human cancers, including HCC [53, 54], frequently associated with reduced or absent expression of the gene (34–48% of cases) [55]. However, these findings are counterintuitive in view of the established paradigm of otherwise activated Hh signaling during NAFLD progression and hepatocarcinogenesis [56].
Based on the results of this study, we suggest a novel mechanism of NAFLD manifestation in the context of Hh signaling attenuation and exposure to Western-style diets (Figure 7). Additional studies are required to evaluate the long-term effects of Hh signaling inhibition on liver phenotype and determine the physiological consequences of increased liver steatosis. More immediately, our results have important clinical implications for the management and care of patients with HPE and their mutation-positive relatives, as this disorder has traditionally been considered as an “above the neck” diagnosis. NAFLD is associated with other health risks that might have historically been neglected in families affected by HPE.
Figure 7.
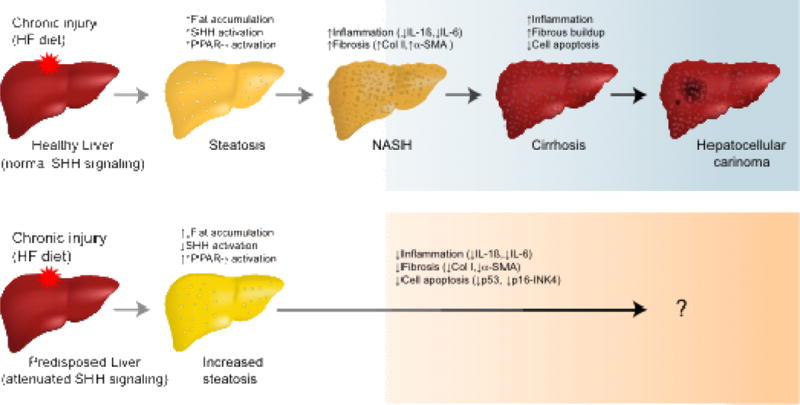
Proposed mechanism of NAFLD progression in the context of SHH signaling and exposure to Western-style diets. In a normal liver (top panel), prolonged exposure to high-fat (HF) diet results in fatty liver with a concurrent activation of SHH and PPARγ pathways. The liver becomes chronically inflamed and the SHH ligand produced by ballooned hepatocytes activates quiescent hepatic stellate cells to differentiate into myofibroblasts with increased production of pro-fibrogenic markers (e.g., collagen type I and α-SMA) and pro-inflammatory cytokines (e.g., IL1β, IL6). About 20% of patients with NASH eventually develop cirrhosis, which prevents the liver from functioning properly. They may also develop hepatocellular carcinoma. In the presence of HF diet and attenuated SHH signaling (bottom panel), there is increased susceptibility to fat accumulation but decreased deposition of fibrous tissue and inflammation due to overactivation of PPARγ, which reduces the risk of cirrhosis. The course of NAFLD in this scenario remains to be elucidated.
Supplementary Material
Lay summary.
Non-alcoholic fatty liver disease (NAFLD) is characterized by excess fat deposition in the liver predominantly due to high-calorie intake and a sedentary life style. NAFLD progression is usually accompanied by activation of the Sonic hedgehog (SHH) pathway leading to fibrous buildup (scar tissue) and inflammation of the liver tissue. In this research, the authors show for the first time that patients with holoprosencephaly, a disease caused by SHH signaling mutations, have increased liver steatosis independent of obesity. This observation was recapitulated in a mouse model of attenuated SHH signaling that also showed increased liver steatosis but with decreased fibrosis and inflammation. While SHH inhibition is associated with a good NAFLD prognosis, this increase in liver fat accumulation in the context of SHH signaling inhibition must be studied prospectively to evaluate its long-term effects, especially in individuals with Western-type dietary habits.
Highlights.
Humans with holoprosencephaly, a disorder of Sonic hedgehog signaling disruption, have a higher incidence of fatty liver
Mutant mice with attenuated Hedgehog signaling are predisposed to develop fatty liver when exposed to fatty liver-inducing diets.
Contrary to the normal progression of non-alcoholic fatty liver disease, mutant mice with attenuated Hedgehog signaling have reduced expression of pro-fibrogenic and pro-inflammatory markers.
Mutant mice with attenuated Hedgehog signaling have decreased expression of cell cycle regulators p53 and p16.
Acknowledgments
We are grateful to the NHGRI Animal Core Facility and the NIH Mouse Imaging Facility for their help with mouse care and husbandry and their technical assistance in mouse procedures. This research was supported by National Human Genome Research Institute intramural funds, National Institute on Alcohol Abuse and Alcoholism grant P60-AA011605 (to KKS), and National Institute of Dental and Craniofacial Research grant K99DE022101 (to RJL).
Financial support: This research was supported by the Division of Intramural Research, National Human Genome Research Institute (to MM), National Institute on Alcohol Abuse and Alcoholism grant P60-AA011605 (to KKS), and National Institute of Dental and Craniofacial Research grant K99DE022101 (to RJL).
Abbreviations
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- HCC
hepatocellular carcinoma
- Hh
hedgehog
- HPE
holoprosencephaly
- SHH
sonic hedgehog
- GLI2
GLI family zinc finger 2
- ZIC2
Zic family member 2
- SIX3
SIX homeobox 3
- TGIF1
TGFB induced factor homeobox 1
- NIH-CC
National Institutes of Health Clinical Center
- HF
high fat
- MCD
methionine and choline deficient
- WT
wild type
- ACTH
adrenocorticotropic hormone
- GH
growth hormone
- T3
triiodothyronine
- T4
thyroxine
- IPA
Ingenuity Pathway Analysis
- PPARα
peroxisome proliferator-activated receptor alpha
- TP53
tumor protein p53
- IL1β
interleukin 1 beta
- TNFα
tumor necrosis factor alpha
- INFγ
interferon gamma
- CXCL12
C-X-C motif chemokine ligand 12
- CCL2
C-C motif chemokine ligand 2
- HSC
hepatic stellate cells
- PPARγ
peroxisome proliferator-activated receptor gamma
- mRNA
messenger RNA
- α-SMA
alpha-smooth muscle actin
- TGFβ1
transforming growth factor beta 1
- IL6
interleukin 6
- PCR
polymerase chain reaction
- p16INK4
cyclin dependent kinase inhibitor 2A (CDKN2A)
- MMP-2
matrix metalloproteinase 2
- TIMP-1
inhibitor of matrix metalloproteinase-1
- TIMP-2
inhibitor of matrix metalloproteinase-2
- ECM
extracellular matrix
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors do not have any conflict of interest to declare.
Author contributions: BDS, PK, MJGS, KW and MM evaluated and recruited the patients at the NIH Clinical Center. RB, BDS and MM participated in study design, interpretation of the data and writing of the manuscript. RJL, MJGS and AFM participated in study design, mouse husbandry, data collection and interpretation, and writing of the manuscript. JLE participated in animal experiments. DK participated in the evaluation of human pathology samples. KKS and JMW participated in the evaluation of liver pathology samples.
References
- 1.Anderson EL, Howe LD, Jones HE, Higgins JP, Lawlor DA, Fraser A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS One. 2015;10:e0140908. doi: 10.1371/journal.pone.0140908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Machado MV, Cortez-Pinto H. Non-alcoholic fatty liver disease: what the clinician needs to know. World J Gastroenterol. 2014;20:12956–12980. doi: 10.3748/wjg.v20.i36.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McPherson S, Hardy T, Henderson E, Burt AD, Day CP, Anstee QM. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol. 2015;62:1148–1155. doi: 10.1016/j.jhep.2014.11.034. [DOI] [PubMed] [Google Scholar]
- 4.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global Epidemiology of Non-Alcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence and Outcomes. Hepatology. 2015;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 5.Anstee QM, Day CP. The genetics of NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10:645–655. doi: 10.1038/nrgastro.2013.182. [DOI] [PubMed] [Google Scholar]
- 6.Macaluso FS, Maida M, Petta S. Genetic background in nonalcoholic fatty liver disease: A comprehensive review. World J Gastroenterol. 2015;21:11088–11111. doi: 10.3748/wjg.v21.i39.11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anstee QM, Day CP. The Genetics of Nonalcoholic Fatty Liver Disease: Spotlight on PNPLA3 and TM6SF2. Semin Liver Dis. 2015;35:270–290. doi: 10.1055/s-0035-1562947. [DOI] [PubMed] [Google Scholar]
- 8.Ryan KE, Chiang C. Hedgehog secretion and signal transduction in vertebrates. J Biol Chem. 2012;287:17905–17913. doi: 10.1074/jbc.R112.356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Omenetti A, Choi S, Michelotti G, Diehl AM. Hedgehog signaling in the liver. J Hepatol. 2011;54:366–373. doi: 10.1016/j.jhep.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moisan A, Lee YK, Zhang JD, Hudak CS, Meyer CA, Prummer M, et al. White-to-brown metabolic conversion of human adipocytes by JAK inhibition. Nat Cell Biol. 2015;17:57–67. doi: 10.1038/ncb3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suh JM, Gao X, McKay J, McKay R, Salo Z, Graff JM. Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metab. 2006;3:25–34. doi: 10.1016/j.cmet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 12.Matz-Soja M, Rennert C, Schonefeld K, Aleithe S, Boettger J, Schmidt-Heck W, et al. Hedgehog signaling is a potent regulator of liver lipid metabolism and reveals a GLI-code associated with steatosis. Elife. 2016;5 doi: 10.7554/eLife.13308. pii: e13308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roessler E, El-Jaick KB, Dubourg C, Vélez JI, Solomon BD, Pineda-Álvarez DE, et al. The mutational spectrum of holoprosencephaly-associated changes within the SHH gene in humans predicts loss-of-function through either key structural alterations of the ligand or its altered synthesis. Hum Mutat. 2009;30:E921–E935. doi: 10.1002/humu.21090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Solomon BD, Bear KA, Wyllie A, Keaton AA, Dubourg C, David V, et al. Genotypic and phenotypic analysis of 396 individuals with mutations in Sonic Hedgehog. J Med Genet. 2012;49:473–479. doi: 10.1136/jmedgenet-2012-101008. [DOI] [PubMed] [Google Scholar]
- 15.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, et al. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci U S A. 2003;100:13424–13429. doi: 10.1073/pnas.2235734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124:113–123. doi: 10.1242/dev.124.1.113. [DOI] [PubMed] [Google Scholar]
- 18.Heyne GW, Everson JL, Ansen-Wilson LJ, Melberg CG, Fink DM, Parins KF, et al. Gli2 gene-environment interactions contribute to the etiological complexity of holoprosencephaly: evidence from a mouse model. Dis Model Mech. 2016;9:1307–1315. doi: 10.1242/dmm.026328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kietzman HW, Everson JL, Sulik KK, Lipinski RJ. The Teratogenic Effects of Prenatal Ethanol Exposure Are Exacerbated by Sonic Hedgehog or Gli2 Haploinsufficiency in the Mouse. PLoS ONE. 2014;9:e89448. doi: 10.1371/journal.pone.0089448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, Smith DL, Jr, Keating KD, Allison DB, Nagy TR. Variations in body weight, food intake and body composition after long-term high-fat diet feeding in C57BL/6J mice. Obesity. 2014;22:2147–2155. doi: 10.1002/oby.20811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lipinski RJ, Godin EA, O’Leary-Moore SK, Parnell SE, Sulik KK. Genesis of teratogen-induced holoprosencephaly in mice. Am J Med Genet C Semin Med Genet. 2010;154C:29–42. doi: 10.1002/ajmg.c.30239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakamura A, Terauchi Y. Lessons from mouse models of high-fat diet-induced NAFLD. Int J Mol Sci. 2013;14:21240–21257. doi: 10.3390/ijms141121240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itagaki H, Shimizu K, Morikawa S, Ogawa K, Ezaki T. Morphological and functional characterization of non-alcoholic fatty liver disease induced by a methionine-choline-deficient diet in C57BL/6 mice. Int J Clin Exp Pathol. 2013;6:2683–2696. [PMC free article] [PubMed] [Google Scholar]
- 24.Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Fujii H, et al. Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine choline-deficient diet-fed mice. Lab Invest. 2010;90:1169–1178. doi: 10.1038/labinvest.2010.75. [DOI] [PubMed] [Google Scholar]
- 25.Salamone F, Galvano F, Cappello F, Mangiameli A, Barbagallo I, Li Volti G. Silibinin modulates lipid homeostasis and inhibits nuclear factor kappa B activation in experimental nonalcoholic steatohepatitis. Transl Res. 2012;159:477–486. doi: 10.1016/j.trsl.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 26.Arnhold IJP, Franca MM, Carvalho LR, Mendonca BB, Jorge AAL. Role of GLI2 in hypopituitarism phenotype. J Mol Endocrinol. 2015;54:R141–R150. doi: 10.1530/JME-15-0009. [DOI] [PubMed] [Google Scholar]
- 27.De Minicis S, Day C, Svegliati-Baroni G. From NAFLD to NASH and HCC: pathogenetic mechanisms and therapeutic insights. Curr Pharm Des. 2013;19:5239–5249. [PubMed] [Google Scholar]
- 28.Guy CD, Suzuki A, Zdanowicz M, Abdelmalek MF, Burchette J, Unalp A, et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology. 2012;55:1711–1721. doi: 10.1002/hep.25559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barbero-Becerra VJ, Giraudi PJ, Chavez-Tapia NC, Uribe M, Tiribelli C, Rosso N. The interplay between hepatic stellate cells and hepatocytes in an in vitro model of NASH. Toxicol In Vitro. 2015;29:1753–1758. doi: 10.1016/j.tiv.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 30.Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol. 2015;12:387–400. doi: 10.1038/nrgastro.2015.94. [DOI] [PubMed] [Google Scholar]
- 31.Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta. 2012;1821:809–818. doi: 10.1016/j.bbalip.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 32.Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 2016;64:S84–S101. doi: 10.1016/j.jhep.2016.02.021. [DOI] [PubMed] [Google Scholar]
- 33.Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 34.Teperino R, Amann S, Bayer M, McGee Sean L, Loipetzberger A, Connor T, et al. Hedgehog Partial Agonism Drives Warburg-like Metabolism in Muscle and Brown Fat. Cell. 2012;151:414–426. doi: 10.1016/j.cell.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 35.Nosavanh L, Yu DH, Jaehnig EJ, Tong Q, Shen L, Chen MH. Cell-autonomous activation of Hedgehog signaling inhibits brown adipose tissue development. Proc Natl Acad Sci U S A. 2015;112:5069–5074. doi: 10.1073/pnas.1420978112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee HJ, Jo SB, Romer AI, Lim HJ, Kim MJ, Koo SH, et al. Overweight in mice and enhanced adipogenesis in vitro are associated with lack of the hedgehog coreceptor boc. Diabetes. 2015;64:2092–2103. doi: 10.2337/db14-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanni E, Bugianesi E, Kotronen A, De Minicis S, Yki-Jarvinen H, Svegliati-Baroni G. From the metabolic syndrome to NAFLD or vice versa? Dig Liver Dis. 2010;42:320–330. doi: 10.1016/j.dld.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 38.Krauser JA, Guengerich FP. Cytochrome P450 3A4-catalyzed testosterone 6beta-hydroxylation stereochemistry, kinetic deuterium isotope effects, and rate-limiting steps. J Biol Chem. 2005;280:19496–19506. doi: 10.1074/jbc.M501854200. [DOI] [PubMed] [Google Scholar]
- 39.Gu J, Weng Y, Zhang QY, Cui H, Behr M, Wu L, et al. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J Biol Chem. 2003;278:25895–25901. doi: 10.1074/jbc.M303125200. [DOI] [PubMed] [Google Scholar]
- 40.Gamage N, Barnett A, Hempel N, Duggleby RG, Windmill KF, Martin JL, et al. Human sulfotransferases and their role in chemical metabolism. Toxicol Sci. 2006;90:5–22. doi: 10.1093/toxsci/kfj061. [DOI] [PubMed] [Google Scholar]
- 41.Moffat C, Bhatia L, Nguyen T, Lynch P, Wang M, Wang D, et al. Acyl-CoA thioesterase-2 facilitates mitochondrial fatty acid oxidation in the liver. J Lipid Res. 2014;55:2458–70. doi: 10.1194/jlr.M046961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thumser AE, Moore JB, Plant NJ. Fatty acid binding proteins: tissue-specific functions in health and disease. Curr Opin Clin Nutr Metab Care. 2014;17:124–129. doi: 10.1097/MCO.0000000000000031. [DOI] [PubMed] [Google Scholar]
- 43.Qin S, Song G, Yu Y. Phospholipid transfer protein in diabetes, metabolic syndrome and obesity. Cardiovasc Hematol Disord Drug Targets. 2014;14:149–153. doi: 10.2174/1871529x1402140807144435. [DOI] [PubMed] [Google Scholar]
- 44.Sugden MC, Bulmer K, Gibbons GF, Knight BL, Holness MJ. Peroxisome-proliferator-activated receptor-alpha (PPARalpha) deficiency leads to dysregulation of hepatic lipid and carbohydrate metabolism by fatty acids and insulin. Biochem J. 2002;364:361–368. doi: 10.1042/BJ20011699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu P, Blair PV, Sato J, Jaskiewicz J, Popov KM, Harris RA. Starvation increases the amount of pyruvate dehydrogenase kinase in several mammalian tissues. Arch Biochem Biophys. 2000;381:1–7. doi: 10.1006/abbi.2000.1946. [DOI] [PubMed] [Google Scholar]
- 46.Syn WK, Jung Y, Omenetti A, Abdelmalek M, Guy CD, Yang L, et al. Hedgehog-Mediated Epithelial-to-Mesenchymal Transition and Fibrogenic Repair in Nonalcoholic Fatty Liver Disease. Gastroenterology. 2009;137:1478–1488.e1478. doi: 10.1053/j.gastro.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedman SL. Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol. 2004;1:98–105. doi: 10.1038/ncpgasthep0055. [DOI] [PubMed] [Google Scholar]
- 48.Ferre P. The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53(Suppl 1):S43–50. doi: 10.2337/diabetes.53.2007.s43. [DOI] [PubMed] [Google Scholar]
- 49.Kwon H, Song K, Han C, Chen W, Wang Y, Dash S, et al. Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease. Hepatology. 2016;63:1155–1169. doi: 10.1002/hep.28289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amaddeo G, Cao Q, Ladeiro Y, Imbeaud S, Nault JC, Jaoui D, et al. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut. 2015;64:820–829. doi: 10.1136/gutjnl-2013-306228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chaubert P, Gayer R, Zimmermann A, Fontolliet C, Stamm B, Bosman F, et al. Germ-line mutations of the p16INK4(MTS1) gene occur in a subset of patients with hepatocellular carcinoma. Hepatology. 1997;25:1376–1381. doi: 10.1002/hep.510250613. [DOI] [PubMed] [Google Scholar]
- 54.Hui AM, Sakamoto M, Kanai Y, Ino Y, Gotoh M, Yokota J, et al. Inactivation of p16INK4 in hepatocellular carcinoma. Hepatology. 1996;24:575–579. doi: 10.1002/hep.510240319. [DOI] [PubMed] [Google Scholar]
- 55.Matsuda Y, Ichida T, Matsuzawa J, Sugimura K, Asakura H. p16(INK4) is inactivated by extensive CpG methylation in human hepatocellular carcinoma. Gastroenterology. 1999;116:394–400. doi: 10.1016/s0016-5085(99)70137-x. [DOI] [PubMed] [Google Scholar]
- 56.Cai H, Li H, Li J, Li X, Li Y, Shi Y, et al. Sonic hedgehog signaling pathway mediates development of hepatocellular carcinoma. Tumour Biol. 2016;37:16199. doi: 10.1007/s13277-016-5463-6. [DOI] [PubMed] [Google Scholar]
- 57.Minervini MI, Ruppert K, Fontes P, Volpes R, Vizzini G, de Vera ME, et al. Liver biopsy findings from healthy potential living liver donors: Reasons for disqualification, silent diseases and correlation with liver injury tests. J Hepatol. 2009;50:501–510. doi: 10.1016/j.jhep.2008.10.030. [DOI] [PubMed] [Google Scholar]
- 58.Schwimmer J. Definitive Diagnosis and Assessment of Risk for Nonalcoholic Fatty Liver Disease in Children and Adolescents. Semin Liver Dis. 2007;27:312–318. doi: 10.1055/s-2007-985075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.