

Abstract
Processing a tissue of interest to generate a microscopic image that supports a scientific argument can be challenging. The acquisition of high-quality microscopic images is not entirely dependent upon the quality of the microscope, but also upon the methods of tissue processing, which often involve multiple critical actions or steps. Furthermore, mesenchymal cell types in the skin and other tissues represent a new challenge for tissue preparation and imaging. Here, we present a complete process, from tissue harvest to microscopy. Our technique, called "horizontal whole mount," is one that novices can quickly become proficient in and that allows for antigen preservation and detection in 60-300 µm-thick sections cut with a cryostat. Sections of this thickness provide enhanced visualization of tissue microarchitecture in a three-dimensional environment. In addition, the protocol preserves mesenchymal cells in a manner that enhances image quality when compared to standard cryostat or paraffin sections, thereby increasing the efficacy and reliability of immunostaining. We believe that this protocol will benefit all laboratories that visualize skin, and possibly other tissues and organs.
Keywords: Developmental Biology, Issue 126, Tissue cross-section, three-dimensional, skin, hair follicle, arrector pili muscle, cryosectioning, immunofluorescence, confocal microscopy
Introduction
The revolution of microscopic imaging equipment provides for sophisticated, high-resolution imaging instruments. However, when acquiring a microscopic image of a complete three-dimensional (3D) tissue cross-section, specimen preparation presents considerable challenges and can be the limiting factor in defining image quality. Each separate step deserves careful consideration in order to preserve tissue morphology and the antigenicity of target proteins, to minimize processing-induced artefacts, and to maximize the final image quality. For example, the traditional analysis of skin requires an image with a view of the epidermis and dermis, with hair follicles that are properly oriented, allowing for the anatomical analysis of stem cell compartment contributions to skin homeostasis1,2. This requires thorough concentration on how the skin is embedded and sectioned. Importantly, hair follicles can be thicker than 100 µm, which greatly surpasses the standard paraffin or frozen section thickness, resulting in a lower standard of analysis compared to whole mounts or thick cross-sections 3,4,5.
Taken together, each step of specimen preparation for microscopic analysis is a critical determinant that will affect image analysis. Here, a novel processing protocol for thick, 3D tissue cross-section analysis, which we call "horizontal whole mount," is presented. The protocol highly preserves antigenicity and enables the full exploitation of thick sections of skin by using standard confocal imaging equipment. This is a complete guide to using skin for thick tissue cross-section processing and imaging, including tissue harvest and paraformaldehyde (PFA)-assisted cryopreservation (step 1), the generation of 100 µm-thick tissue cross-sections with a cryostat (step 2), and immunofluorescent labeling and mounting (steps 3 and 4). The representative results compare confocal images of two distinct histological preparation techniques—classical cryosectioning and thick, 3D tissue cross-sectioning—highlighting the advantages of "horizontal whole mounts" for the potential user of this protocol.
Protocol
All animal experiments were subject to local ethical approval and performed under the terms of a UK government Home Office license.
1. Skin Harvest and Cryopreservation
- Preparations.
- Prepare one 100-mm culture dish with 25 mL of 4% PFA and two 100-mm culture dishes with 25 mL of 1x phosphate-buffered saline (PBS).
- Fill rectangular peel-away cryomolds by two-thirds with optimal cutting temperature compound (O.C.T.).
- Place a metal plate, on which the cryoblocks can be placed in a later step, into the -80 °C freezer.
- Skin harvesting, fixation, and cryopreservation.
- Clip the dorsal region of the animal cadaver with a dry electric shaver. NOTE: In this example, postnatal day 21 wildtype mice were used.
- Harvest the areas of interest on the skin. NOTE: The dorsomedial region of mouse skin (Figure 1a) contains the highest percentage of hair follicles that are evenly spaced and aligned, which allows for optimal orientation for sectioning. The removal of underlying non-dermal tissue is not necessary but can be performed if required.
- Trim the harvested skin into rectangular pieces of appropriate size to fit into the bottom of the cryomold, taking the directional growth of the hair follicle grain into account. NOTE: Smaller skin slices might be easier to handle for novices, since they are less likely to tangle during the incubation and mounting process. The example shown here is a ~1 cm2 area of dorsal skin, which fits into a 22 x 30 x 20 mm cryomold (Figure 2a).
- Fix the skin at room temperature in 25 mL of 4% PFA for 10-30 min, depending upon the thickness of the skin samples (Figure 1b).
- Wash the skin samples two times in 25 mL of PBS for at least 5 min each (Figure 1b).
- Dab the skin samples on a paper towel to carefully drain the tissue of excess PBS, which can result in crystallization during the freezing process and can affect cryosectioning results. NOTE: A standard sucrose gradient is not required. However, this may also be incorporated into the protocol at the user's discretion.
- Be aware of the orientation of the hair follicles for each skin sample. Use a dissecting microscope for visual assistance (especially anyone performing the protocol for the first time). Insert the skin sample into the O.C.T.-filled cryomold and equilibrate all areas of the skin with O.C.T. by removing any air bubbles attached to the surface of the clipped hair using forceps ( Figures 2a and 2b).
- Push the skin to the bottom of the O.C.T. filled block so that it lies flush with the bottom. NOTE: The skin can be oriented in any direction, as long as the grain of the hair follicle is noted for proper cutting procedures. The cryomolds will be re-oriented when the blocks are attached to the cryostat for cryosectioning. Mark the hair follicle orientation on the cryomold, since this step determines the subsequent orientation of the cryostat cut.
- Transfer the cryoblocks onto the metal plate in the -80 °C freezer to avoid floating and dislocation of the tissue.
- Monitor the freezing process to maintain the orientation of the skin at the bottom of the cryomold, since unseen air bubbles can cause the skin to rise to the surface of the cryomold. NOTE: Cryomolds with frozen tissue can be stored for more than a year at -80 °C and can be reused for additional sections.
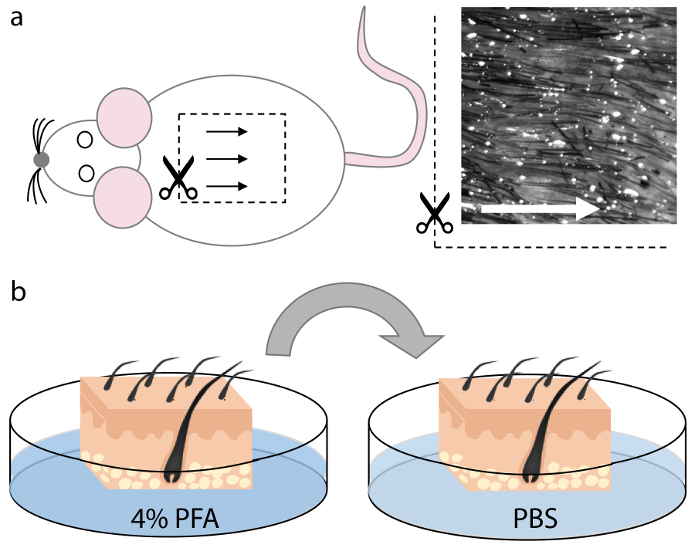

Figure 1. Harvest and fixation of mouse skin. (a) Skin tissue was harvested from the dorsomedial region of the animal cadaver. Hair follicles in this region are evenly spaced and aligned and therefore allow for optimal orientation during sectioning, as indicated by the arrows. (b) After cutting squares of an appropriate size that fits into the cryomold, the skin tissue was fixed in 4% PFA for 15 min and washed two times in PBS for 5 min each. Please click here to view a larger version of this figure.
2. Thick Tissue Cross-sectioning
- Preparation and tissue orientation to mount on the cryostat.
- Prepare a 100-mm culture dish with 15 mL of PBS. Place it on an easily-accessible area on the cryostat. In addition, prepare a 12-well plate with 2.5 mL of PBS per well; label according to the samples for the long-term storage of the sections at 4 °C. Use forceps to handle the sections.
- Adjust the temperature of the cryostat to -20 °C. NOTE: The temperature can affect sectioning, but a good guide is to start at -20 °C.
- To obtain sections approximately two hair follicles thick, adjust the cryostat to cut sections 150 µm thick. NOTE: The thickness of the section can be varied, depending upon the needs of the user and the limitations of the microscope that will be used for analysis. NOTE: The orientation of the sample is critical to obtaining skin sections with hair follicles in the appropriate orientation. This is achieved by mounting properly on the cryostat block. Make sure that the section plane is parallel to the hair follicle orientation (Figure 2c). As mentioned previously, the correct orientation of the section plane in the skin sample is a crucial step to determining the quality of the images that will be acquired in a later step.
Figure 2. Embedding, cryopreservation, and sectioning. (a)Marking the hair follicle (HF) direction on the cryomold, indicated by the black arrows, is important for proper orientation during cryosectioning. (b) The section plane needs to be aligned with the hair follicle orientation to generate sections in which the complete length of the hair follicles stays intact.(c) Sections were cut per the hair follicle orientation that was indicated by the black arrows on the cryomold. (d) The thick tissue cross-sections were collected with metal forceps and (e) transferred into a 100 mm culture dish containing 1x PBS. (f) At room temperature, the PBS dissolves away the O.C.T. compound that surrounds the thick tissue cross-sections, as indicated by the white arrows. The sections then float freely in the PBS. Please click here to view a larger version of this figure.
- Cryosectioning.
- Cut a section using the cryostat. Use forceps to collect the O.C.T. that contains the embedded piece of skin (Figure 2d). NOTE: Use a cryostat that enables independent movement and adjustment along the X, Y, and Z axes for optimal specimen positioning. This allows for the generation of ideally aligned tissue cross-sections.
- Transfer the section out of the cryostat into the 100 mm culture dish filled with PBS and continue with the next slice. Do not collect the samples on a slide (Figure 2e). NOTE: At room temperature, the PBS will dissolve away the O.C.T., leaving skin slices that are easy to handle with forceps (Figure 2f). NOTE: Fresh PBS may be needed in the 100 mm culture dish after dissolving many tissue sections 100 µm thick and can be changed accordingly.
- Use forceps to transfer the floating skin sections to the correctly labeled well in a 12 well plate filled with 2.5 mL of PBS (Figure 3a, left). NOTE: At 4 °C, the samples can be stored for at least two days. For the long-term storage of skin-containing O.C.T. blocks after sectioning, seal the cutting surface with a droplet of fresh O.C.T. After freezing of the O.C.T. droplet, wrap the used O.C.T. block in parafilm and place it back in the -80 °C freezer.
3. Immunofluorescent labeling.
Prepare the PB buffer (PBS supplemented with 0.5% skim milk powder, 0.25% fish skin gelatin, and 0.5% Triton X-100) at least 2 h in advance, as described previously5. NOTE: Sodium azide can be added to PB buffer for antibody preservation for the repeated use of the staining buffer.
- Label 1.5 mL microcentrifuge tubes and add 500 µL of PB buffer per tube. Carefully use forceps to transfer the skin slices from the PBS into separate tubes containing the PB buffer for blocking (Figure 3a, right). Make sure that all skin slices are fully submerged. Place the microcentrifuge tubes on a see-saw rocker at speeds no higher than 10 oscillations per minute, which should not disrupt the tissue integrity, for 1 h at room temperature. NOTE: It is critical that the speed does not exceed 10 oscillations per minute on the see-saw rocker, since it will induce tangling.
- While it is possible to add more than one slice per microcentrifuge tube, to save antibodies, only place one slice per tube. To decrease antibody usage, use a volume of 250 µL. NOTE: If needed, substitute microcentrifuge tubes with, for example, 96-well plates. However, the placement of the thick tissue cross-sections into 1.5-mL microcentrifuge tubes allows for the most effective antibody penetration into the tissue due to the enhanced liquid perturbation.
Label separate 1.5 mL microcentrifuge tubes for each skin slice and add 500 µL of PB buffer and the appropriate amount of primary antibody. After 1 h of blocking, transfer the skin slices into the freshly prepared tubes containing the antibodies. Incubate the slices at 4 °C overnight. NOTE: In this example, the following primary antibodies were used: FITC rat anti-human CD49f at a concentration of 1:50 and goat anti-mouse/rat integrin alpha 8 at a concentration of 1:100.
The next day, prepare two separate 1.5 mL microcentrifuge tubes containing 500 µL of PBS per sample. Wash the skin slices two times for 1 h at room temperature.
Prepare separate 1.5 mL microcentrifuge tubes containing 500 µL of PB buffer with the appropriate concentration of applicable secondary antibodies and 4',6-diamidino-2-phenylindole (DAPI). NOTE: The secondary antibodies used in this example are Alexa Fluor 488 donkey anti-rat IgG and Alexa Fluor 555 donkey anti-goat IgG at a concentration of 1:500. DAPI was used at a concentration of 1:100.
Carefully transfer the skin samples into the PB buffer, which contains the secondary antibody and DAPI, and incubate the skin slices at room temperature for 1 h at a low speed on a rotator or shaker.
Store the slices at 4 °C in the PB buffer containing the secondary antibody and DAPI for up to four days, and possibly longer if sodium azide is added to the PBS.
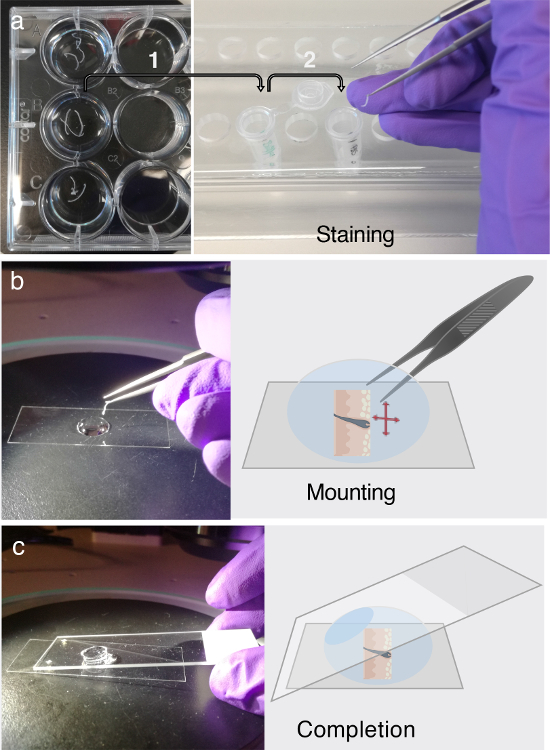
Figure 3. Immunofluorescent labeling and mounting. (a) The floating tissue cross-sections can be stored in 12-well plates for at least two days at 4 °C. Before immunofluorescent (IF) labeling, transfer the tissue cross-sections of interest into 1.5 mL microcentrifuge tubes containing PB buffer for blocking, as indicated by arrow 1. For IF labeling, adhere to the multi-step procedure elaborated upon in step 3. Each part of step 3 requires the careful transfer of the tissue cross-sections into freshly prepared microcentrifuge tubes containing primary antibody solution, secondary antibody solution, or washing buffer, which is indicated by arrow 2. (b) After IF labeling, the tissue cross-sections are unraveled and flattened in a droplet of glycerol, using the assistance of a dissecting microscope. (c) Once the tissue cross-section is entirely flattened onto the bottom of a cover slip, a regular microscope slide is used to mount the section. Please click here to view a larger version of this figure.
4. Mounting for Microscopic Visualization
Prior to imaging, transfer the skin slices to separate microcentrifuge tubes containing 500 µL of PBS to wash away the secondary antibodies and DAPI.
Use a 1,000 µL pipet, but cut off the first 0.5 cm of the pipette tip to enable the proper pipetting of the highly viscous glycerol. Place a 22 x 50 mm coverslip onto a dark background under a dissecting microscope (Figure 3b).
Add one droplet of 100% glycerol onto the cover slip (Figure 3b). Transfer the skin slice from the microcentrifuge tube onto the glycerol droplet. Use the dissecting microscope and pointed forceps to carefully unwind the skin slices that are curled up. NOTE: As the slice floats in the glycerol droplet, it can be untangled by coaxing the natural propensity of the tissue to return to its normal shape. Do not force the unnatural straightening of the slice, since this could cause damage to the tissue.
Mount the tissue once the entire length of the skin section is properly oriented and flattened on the cover slip; use a regular microscope slide. This step will further straighten the skin slice. NOTE: Avoid air entrapment (Figure 3c).
Image the in glycerol-mounted skin sections within the next two days. NOTE: Prolonged storage will negatively influence the tissue and imaging quality. In this example, all images were acquired with an upright confocal microscope using a 20x objective.
Representative Results
To emphasize the advantages of our technique, we compared our thick, 3D tissue cross-sectioning technique, "horizontal whole mount," to classical frozen sections. Classical frozen sections were cut as previously described5. To provide a visual structure for the epidermis in te microscopic images, we immunostained for integrin alpha-6 (Itga6), which is a component that anchors epidermal cells to the underlying basement membrane6. We also labeled the arrector pili muscle (APM), which is responsible for piloerection (also known as "goosebumps"), with integrin alpha-87. In the classical frozen sections, most hair follicles visualized with Itga6 were not sectioned along the entire length, generating predominantly incomplete hair follicles per section, as compared to horizontal whole mounts (Figure 4a-4d). Thick tissue cross-sections make it possible to acquire more Z-stack layers compared to conventional 10-µm sections, allowing for a more complete 3D image. This becomes even more apparent when studying the integrity of APMs, which are associated with the hair follicles and the overlying basement membrane. In classical cryosections, the vast portion of APMs were fractioned (Figure 4a-4d). Additionally, the tissue integrity of the hypodermal compartment is preserved in horizontal whole mounts, as compared to the destruction of adipocytes when cryosections are attached to warm glass slides, which is a well-known freeze-thawing artifact (Figure 4a-b, compare hypodermal regions)8.
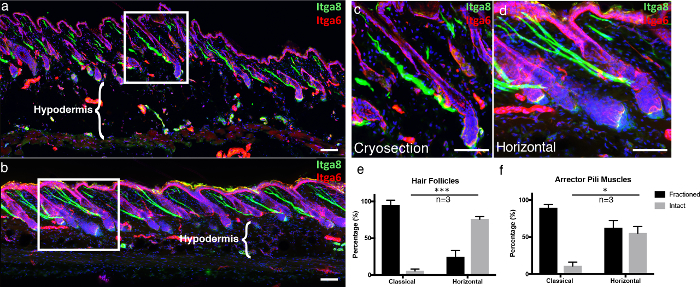
Figure 4. Horizontal whole mount compared to a classical cryosection. (a) Classically obtained skin cryosections 10 µm thick and (b) 100 µm-thick 3D tissue cross-sections were labeled with integrin alpha-6 (Itga6) and integrin alpha-8 (Itga8) to visualize the epidermal compartment and arrector pili muscles, respectively. The images of the thick tissue cross-sections are represented as maximum projections of a large Z stack. The white frames indicate the areas that are enlarged, displayed in (c) the classical and (d) horizontal whole mount sections. (e) Intact hair follicles and (f) intact arrector pili muscles were quantified in both the classical and the horizontal whole-mount sections. Scale bars indicate 100 µm. The data is represented as Mean ± Standard Error of the Mean (SEM). One section per biological replicate (n=3) was quantified. Unpaired t-test *P<0.05, ***P<0.0005. Please click here to view a larger version of this figure.
Discussion
Here, we present the "horizontal whole mount" technique, which has several advantages over classical frozen sections. One advantage of our technique is that it can easily be performed with standard histology equipment and a confocal microscope. Second, the tissue integrity is preserved in a superior manner when compared to standard cryosections. For example, adipocytes within the hypodermal layer (i.e., dermal white adipocytes (DWAT))9 are severely disrupted in standard cryosections, which makes studying tissues that are adipose-laden impossible. Our technique allows for the full analysis of the adipocytes in this layer and may be adapted to other tissues with high adipocyte contents. Furthermore, this allows for the improved detection of mesenchymal cells in lineage-tracing studies 2,10.
Standard cryosections, by their very nature, can never reveal the true 3D aspects of a tissue through confocal microscopy. This means that thicker sections are advantageous in the analysis of skin, since hair follicles, as well as their substructures, can traverse a depth that exceeds the standard thickness of 10 µm used in classical crysosections. We are intrigued that other tissues, such as the intestine and brain, for example, possess similar challenges with regard to viewing the tissue as a 3D structure2,7,11,12,13. Intriguingly, our technique has already been shown to be beneficial for use in certain applications in the intestines14. We believe that our horizontal whole-mount protocol has the potential be applied to any other tissue requiring 3D analysis.
Disclosures
The authors would like to disclose that the publication of this manuscript was funded by Thermo-Fisher Scientific Inc.
Acknowledgments
The authors acknowledge sponsorship from Thermo-Fisher Scientific and thank the Nikon Imaging Centre at Kings College London for support during confocal image acquisition.
References
- Plikus MV, et al. Cyclic dermal BMP signalling regulates stem cell activation during hair regeneration. Nature. 2008;451(7176):340–344. doi: 10.1038/nature06457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YV, Cheong J, Ciapurin N, McDermitt DJ, Tumbar T. Distinct self-renewal and differentiation phases in the niche of infrequently dividing hair follicle stem cells. Cell Stem Cell. 2009;5(3):267–278. doi: 10.1016/j.stem.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen UB, Lowell S, Watt FM. The spatial relationship between stem cells and their progeny in the basal layer of human epidermis: a new view based on whole-mount labelling and lineage analysis. Development. 1999;126(11):2409–2418. doi: 10.1242/dev.126.11.2409. [DOI] [PubMed] [Google Scholar]
- Braun KM, et al. Manipulation of stem cell proliferation and lineage commitment: visualisation of label-retaining cells in wholemounts of mouse epidermis. Development. 2003;130(21):5241–5255. doi: 10.1242/dev.00703. [DOI] [PubMed] [Google Scholar]
- Driskell RR, Giangreco A, Jensen KB, Mulder KW, Watt FM. Sox2-positive dermal papilla cells specify hair follicle type in mammalian epidermis. Development. 2009;136(16):2815–2823. doi: 10.1242/dev.038620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niculescu C, et al. Conditional ablation of integrin alpha-6 in mouse epidermis leads to skin fragility and inflammation. Eur J Cell Biol. 2011;90(2-3):270–277. doi: 10.1016/j.ejcb.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, et al. The basement membrane of hair follicle stem cells is a muscle cell niche. Cell. 2011;144(4):577–589. doi: 10.1016/j.cell.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desciak EB, Maloney ME. Artifacts in frozen section preparation. Dermatol Surg. 2000;26(5):500–504. doi: 10.1046/j.1524-4725.2000.99246.x. [DOI] [PubMed] [Google Scholar]
- Driskell R, Jahoda CA, Chuong CM, Watt F, Horsley V. Defining dermal adipose tissue. Exp Dermatol. 2014;23(9):629–631. doi: 10.1111/exd.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell RR, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. 2013;504(7479):277–281. doi: 10.1038/nature12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, Schepers AG, Delconte G, Siersema PD, Clevers H. Slide preparation for single-cell-resolution imaging of fluorescent proteins in their three-dimensional near-native environment. Nat Protoc. 2011;6(8):1221–1228. doi: 10.1038/nprot.2011.365. [DOI] [PubMed] [Google Scholar]
- Sada A, et al. Defining the cellular lineage hierarchy in the interfollicular epidermis of adult skin. Nat Cell Biol. 2016;18(6):619–631. doi: 10.1038/ncb3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung K, Deisseroth K. CLARITY for mapping the nervous system. Nat Methods. 2013;10(6):508–513. doi: 10.1038/nmeth.2481. [DOI] [PubMed] [Google Scholar]
- Tetteh PW, et al. Replacement of Lost Lgr5-Positive Stem Cells through Plasticity of Their Enterocyte-Lineage Daughters. Cell Stem Cell. 2016;18(2):203–213. doi: 10.1016/j.stem.2016.01.001. [DOI] [PubMed] [Google Scholar]