

Abstract
Within the innate immune system, effector lymphocytes known as natural killer (NK) cells play an essential role in host defense against aberrant cells, specifically eliminating tumoral and virally infected cells. Approximately 30 known monogenic defects, together with a host of other pathological conditions, cause either functional or classic NK cell deficiency, manifesting in reduced or absent cytotoxic activity. Historically, cytotoxicity has been investigated with radioactive methods, which are cumbersome, expensive and potentially hazardous. This article describes a streamlined, clinically applicable flow cytometry-based method to quantify NK cell cytotoxic activity. In this assay, peripheral blood mononuclear cells (PBMCs) or purified NK cell preparations are co-incubated at different ratios with a target tumor cell line known to be sensitive to NK cell-mediated cytotoxicity (NKCC). The target cells are pre-labeled with a fluorescent dye to allow their discrimination from the effector cells (NK cells). After the incubation period, killed target cells are identified by a nucleic acid stain, which specifically permeates dead cells. This method is amenable to both diagnostic and research applications and, thanks to the multi-parameter capabilities of flow cytometry, has the added advantage of potentially enabling a deeper analysis of NK cell phenotype and function.
Keywords: Immunology and Infection, Issue 126, natural killer, NK cell, cytotoxicity, cell lysis, cell killing, K562, flow cytometry, chromium release, NK cell cytotoxicity, CFSE
Introduction
Natural killer (NK) cells are a sophisticated subset of human innate lymphocytes critically involved in the elimination of virally infected cells, transformed cells, and other pathogenic threats 1,2. NK cell lytic granules house cytotoxic proteins, such as perforin and granzymes. Upon activation, NK cells form a complex interaction with their targets known as immunological synapse, whereby these cytolytic molecules are locally released, resulting in direct target cell lysis and apoptosis, together with cytokine and chemokine release and ultimately in the induction of an inflammatory state 1,3,4.
NK cell activation involves a complex string of activating and inhibitory interactions between NK cell receptors and ligands expressed on the surface of target cells, forming a tightly regulated system. One of the most studied mechanisms of NK cell activation is the "missing self". Indeed, lack of detection of class I major histocompatibility complex (MHC), or human leukocyte antigen (HLA) molecules, on infected or transformed cells triggers NK cell cytotoxicity. Tumor and virus-infected cells generally downregulate these antigens to escape T cell-mediated immunity, thus becoming primary NK cell targets 1,3,4.
Assessment of NK cell function is primarily categorized into degranulation or cytotoxicity assays. However, degranulation assays, such as flow cytometric detection of the degranulation-associated marker CD107a, are only indicative of NK cell activation and not of their ultimate function, the direct killing of target cells 5,6,7,8. Hence, this limitation has drawn investigators to cytotoxicity assays as a more telling and more direct alternative.
The long-time "gold standard" for assessing cell-mediated cytotoxic activity of both T and NK cells is the chromium release assay (CRA). CRA involves radioactively labeling of target cells with 51Cr and co-incubating them with effector cells. This assay is steeped in the principle that cell lysis results in the release of protein-bound 51Cr into the supernatant, which can be measured by gamma counting. This assay, while effective, is problematic for a variety of reasons: high material costs, handling and disposal of radioactive 51Cr, spontaneous release of 51Cr, and difficult standardization - making it altogether impractical 9,10.
A number of non-radioactive assays, involving fluorescent labeling, enzyme release, and even bioluminescence, have since been developed as alternatives to CRA 11,12,13,14. We describe here a flow cytometry-based method for measurement of NK cell cytotoxic activity on K562 target cells that is simple, sensitive, and reproducible. K562 cells are a human erythroleukemic cell line with reduced expression of HLA class I and heightened expression of ligands for activatory NK receptors, which makes them particularly susceptible to NK cell-mediated cytotoxicity 15. In this assay, K562 cells are pre-labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) and co-cultured at various ratios with either peripheral blood mononuclear cells (PBMCs) or purified NK cells 1. CFSE is a stable, protein-binding fluorescent dye that allows discrimination of target cells from effector NK cells 16,17. After the co-incubation, a nucleic acid stain, specifically permeating the membrane of dead cells, is used to identify killed target cells (see table of materials). The samples are then acquired on a flow cytometer to determine the percentage of dead (i.e., stain+) CFSE+ target cells.
This assay can be used as a routine diagnostic screening for monogenic defects affecting the NK cell compartment, of which there are approximately 30 known defects causing either functional or classic NK cell deficiency, and for primary or secondary hemophagocytic lymphohistiocytosis. It is also useful to investigate NK cell activity in patients with recurrent, severe herpes viral infections, to evaluate immune reconstitution following hematopoietic cell transplantation or post immunomodulatory therapy 18,19,20, and for a host of basic research applications.
Protocol
Samples were collected according to the ethical guidelines established by the UCLA Human Research Protection Program and IRB approved.
1. Preparation of reagents
NOTE: Unless otherwise stated, all reagents should be allowed to equilibrate at room temperature prior to use. All reagents must remain sterile.
- Prepare a 2x working solution of Tween-20 (i.e., 0.2%) by adding 10 µL of Tween-20 solution to 5 mL of phosphate-buffered saline (PBS) without calcium and magnesium with a p20 pipette.
- Given the high viscosity of Tween-20, take the following steps to ensure accuracy: collect slowly to avoid bubble formation, do not submerge the entire tip to avoid carry-over, and pipette up and down several times in PBS to wash out all Tween-20 from the tip.
Add 2 µL of IL-2 stock solution (2.1E6 IU/mL) to 198 µL of complete media (i.e., 1:100 dilution), which is RPMI with 1% penicillin-streptomycin and 10% fetal bovine serum (FBS), then proceed to an additional 1:100 dilution by adding 2 µL of IL-2 intermediate solution to 198 µL of complete media to prepare a 7x working solution (210 IU/mL).
Add 2 µL of CFSE stock solution (5 mM) to 500 µL of plain media (RPMI). Vortex, spin down to remove drops from the cap, then proceed with an additional 1:20 dilution by adding 50 µL of the CFSE intermediate solution to 950 µL of plain RPMI to obtain a 2x working solution of 1 µM. Vortex well and keep protected from light.
2. Isolation of PBMCs as effector cells
NOTE: This assay has been validated for effective use with total PBMCs from healthy controls. However, it is recommended that NK cell content be verified with each PBMC preparation (Figure 1). Also, the volume of whole blood for collection is based on the frequency of NK cells in peripheral whole blood and this may vary from person to person, particularly between healthy donors and patients.
Collect a minimum of 4 mL of human whole blood in sodium or lithium heparinized blood collection tubes. Store blood samples at room temperature upon collection and use within 30 h of collection.
Dilute whole blood with 4 mL of PBS (i.e. equal volume of PBS to whole blood).
Add 4 mL of density gradient solution (see table of materials) to a 15-mL conical tube. Carefully overlay diluted whole blood over the density gradient solution by tilting the tube.
Centrifuge at 650 x g for 24 min with the brake off.
Carefully collect the layer of mononuclear cells, which is the thin white layer below the top layer of plasma and platelets, into a new 15-mL conical tube. Bring the volume to 15 mL with PBS.
Centrifuge at 450 x g for 10 min with the brake on. Aspirate the supernatant and resuspend cells in 0.5 mL of complete media.
Use an automated cell counter or hemocytometer to count the cells, and adjust the PBMC concentration to 5x106 cells/mL with complete media.
Check the NK cell content by flow cytometry (see step 4).
Start the assay within 30 min. Keep cells at room temperature for up to 10 min, or place in a humidified 5% CO2 incubator at 37 °C until ready for use.
3. Isolation of NK cells as effector cells
NOTE: This portion of the protocol is an alternative to using total PBMCs as effector cells. The typical yield from 4 mL of whole blood from a healthy individual is approximately 4x105 NK cells, or approximately 5-15% of PBMCs, though this frequency varies between donors 21. It is recommended that a purity check be performed after the isolation.
Collect a minimum of 4 mL of human whole blood in sodium or lithium heparinized blood collection tubes. Take 50 µL of whole blood to stain as a pre-enrichment sample.
Add the enrichment cocktail to blood samples at 50 µL/mL (see table of materials). Mix well and incubate for 10 min at room temperature.
Add an equal volume of PBS + 2% FBS to dilute the sample and gently mix well.
Add 4 mL of density gradient solution to a 15-mL conical tube. Carefully overlay the diluted sample on top of the density gradient medium.
Centrifuge at 1,200 x g for 10 min with the brake off.
Carefully collect the layer of enriched cells into a new 15-mL conical tube. Wash the cells by bringing the volume up to 50 mL with PBS + 2% FBS.
Centrifuge at 300 x g for 10 min. Discard the supernatant and repeat the wash.
Aspirate the supernatant and resuspend cells in 0.5 mL of complete media.
Using an automated cell counter or hemocytometer, count the cells and adjust the concentration to 5x105 cells/mL. Take 50 µL of purified cell suspension to stain as a post-enrichment sample.
Keep cells at room temperature for up to 10 min, or place in a humidified 5% CO2 incubator at 37 °C until ready for use.
4. Immunophenotyping for assessment of NK cell content
Using 50 µL total isolated PBMCs, pre-enrichment whole blood, or purified NK cells, stain with anti-CD56 fluorochrome-conjugated antibody as an NK cell marker. Markers for other cell types and lineages (i.e. CD3+ T cells, CD19+ B cells) may also be included to assess the origin of contamination, if any.
Incubate for 20 min at 4 °C, and wash by adding 2 mL of PBS + 2% FBS.
Centrifuge at 450 x g for 5 min.
For PBMCs or NK cells, aspirate supernatant and resuspend cells in 100 µL of PBS + 2% FBS.
- For whole blood samples, perform red blood cell (RBC) lysis:
- After centrifugation, aspirate the supernatant and resuspend in 2 mL red blood cell lysis buffer (31.75 g of ammonium chloride + 4.16 g of Tris base + 6.65 g of EDTA, dissolved in 5000 mL of deionized water).
- Incubate for 10 min at room temperature.
- Repeat wash twice, aspirate the supernatant, and resuspend lysed sample in 100 µL of PBS + 2% FBS. NOTE: RBC lysis may also be performed prior to staining.
- Proceed with flow cytometric analysis.
5. Thawing, culturing, and harvesting of K562 target cells
Aliquot 10 mL of complete media into a 15-mL conical tube and allow to warm in a 37 °C water bath. For the thawing and culturing of K562 cells, use only media pre-warmed to 37 °C for all steps.
Place one vial of frozen K562 cells in the 37 °C water bath until most of it is thawed.
Transfer the semi-thawed K562 cells to the complete media, pre-warmed to 37 °C, in the 15-mL conical tube. Wash the inside of the cryogenic vial with some pre-warmed media, and add the wash to the 15-mL tube.
Centrifuge at 140 x g for 7 min with the brake on. Remove the supernatant and resuspend the pellet in 1 mL of complete media.
Aliquot an appropriate amount of cell suspension to count cells using an automated cell counter or hemocytometer.
Adjust the concentration to 1x105-2x105 cells/mL and culture in a humidified 5% CO2 incubator at 37 °C for at least 2 days before use. Maintain the culture at this density range for no longer than 3 months, then thaw a new aliquot. Never exceed a cell concentration of 1x106 cells/mL in culture.
- For use in the assay, mix the K562 cell culture well with a 10-mL serological pipette and remove an aliquot to count and assess cell viability using an automated cell counter or hemocytometer and Trypan Blue staining.
- If the viability is less than 85%, proceed with the following dead cell removal steps.
- Add 15 mL of density gradient solution to 50-mL conical tubes, as needed. Collect all cells from culture and slowly overlay the cell suspension over the density gradient solution by tilting the tube.
- Centrifuge at 650 x gfor 24 min with brake off. Carefully collect the layer of live cells into a new 15-mL conical tube. Bring the volume to 50 mL with PBS.
- Centrifuge at 450 x g for 10 min with the brake on. Remove the supernatant and resuspend the pellet in 1 mL of complete media. Count the cells and check cell viability (as in 5.7).
- Culture in a humidified 5% CO2 incubator at 37 °C for at least 2 days before use.
Collect 5x105 K562 target cells and centrifuge at 140 x g for 7 min. Aspirate the supernatant and resuspend the cell pellet in 500 µL in media with 1% FBS.
6. Labeling of K562 target cells
NOTE: Make sure the K562 cells are well resuspended before adding the CFSE working solution.
Pipette 500 µL of the CFSE working solution to the 500 µL of K562 cells for a final concentration of 0.5 µM. Using the same 1000-1250 µL tip, mix immediately by gently pipetting up and down 3-5 times.
Incubate in the 37 °C water bath or in a humidified 5% CO2 incubator at 37 °C for 10 min in the dark. NOTE: Since the temperature equilibration lag time is longer in the incubator compared to the water bath, incubation time might need to be adjusted especially for volumes > 1 mL.
Quench the labeling reaction with 10 mL of complete media for 10 min in the dark at room temperature.
Centrifuge the labeled K562 cells at 140 x g for 7 min. Remove the supernatant and resuspend in 1 mL of complete media.
Count the cells using an automated cell counter or hemocytometer and bring the concentration to 1x105 cells/mL in complete media.
Place cells in a humidified 5% CO2 incubator at 37 °C until ready for use. Cells can be kept at room temperature for up to 10 min, or in in a humidified CO2 incubator at 37 °C until ready for use. Do not refrigerate.
7. Plating
NOTE: If NK cells are limiting, the concentration of both K562 and NK cells can be halved, so that half of the cells are plated in the assay. The assay yields comparable results with half the amount of cells (as long as the ratios are kept consistent).
If using total PBMCs, label a 96-well, U-bottom, tissue culture-treated plate for the following conditions per well: (1) Effector (E) : Target (T) = 50 : 1 + IL-2 - positive control for NK cell cytotoxicity; (2) E : T = 50 : 1; (3) E : T = 25 : 1; (4) E : T = 12.5 : 1; (5) E : T = 6.25 : 1; (6) T - target cells only - negative control for K562 death; (7) E - effector cells only; (8) T + Tween - Positive control for K562 death
If using purified NK cells, label a 96-well, U-bottom, tissue culture treated plate for the following conditions per well: (1) Effector (E) : Target (T) = 5: 1 + IL-2 - positive control for NK cell cytotoxicity; (2) E : T = 5 : 1; (3) E : T = 2.5 : 1; (4) E : T = 1.25 : 1; (5) E : T = 0.625: 1; (6) T - target cells only - negative control for K562 death; (7) E - effector cells only; (8) T + Tween - positive control for K562 death
Add 100 µL of complete media to conditions 3, 4, 5, 6, 7. For this and all subsequent steps, (recommended) use a multichannel pipette.
- Add 100 µL of effector cells (PBMCs at 5x106 cells/mL or purified NK cells at 5x105 cells/mL) to conditions 1, 2, 3. Add effector cells for conditions 4-7 by serial dilution. Do not change tips from condition to condition.
- Mix condition 3, take 100 µL, and transfer to condition 4.
- Mix condition 4, take 100 µL, and transfer to condition 5.
- Mix condition 5, take 100 µL, and transfer to condition 7; Condition 6 does not have effector cells.
Add 100 µL of prepared 2x 0.2% Tween working solution to condition 8 (final concentration: 0.1% Tween-20); condition 8 does not have effector cells.
Add 30 µL of IL-2 working solution to condition 1. IL-2 should be added to the effector cell suspension prior to the addition of target cell suspension (final concentration: 27 IU/mL).
Add 100 µL of target cells (labeled K562 cells at 1x105 cells/mL) to conditions 1, 2, 3, 4, 5, 6, 8. Condition 7 does not have target cells.
Mix all wells with a multichannel pipette and centrifuge the plate at 120 x g for 2 min.
Incubate in a humidified CO2 incubator at 37 °C for 4 h. At the end of the incubation, place the plate on ice and proceed to viability staining within 30 min.
8. Staining for viability and acquisition
Prepare dead cell stain (see table of materials) by adding 3 µL of the 5 µM stock solution to 600 µL of PBS.
Add 50 µL of diluted dead cell stain to each well for a final volume of 250 µL and a final concentration of 0.005 µM. Mix well with a multichannel pipette.
Proceed with flow cytometric analysis. Using a flow cytometer, collect at least 1,000 events in the target cell gate (Figure 2) for statistically meaningful results.
Representative Results
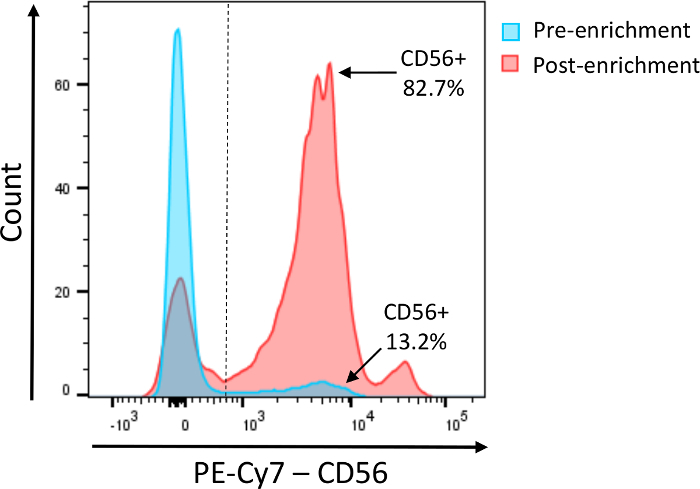
Before setting up the assay, it is highly recommended that NK cell content be assessed in the effector population of choice. Figure 1 shows a typical CD56 staining before (light blue) and after (red) NK cell enrichment. NK cells comprise up to 15% of PBMCs and should be at least 80% pure after enrichment.
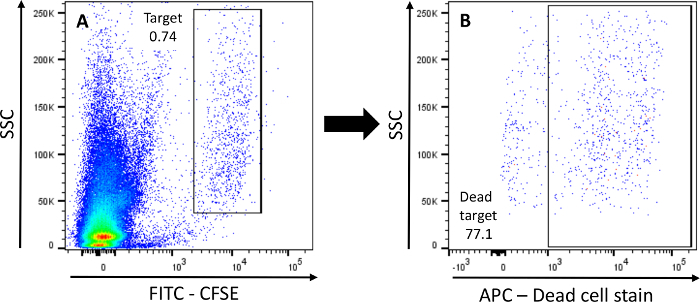
Flow cytometric analysis in this assay involves detection of two parameters: CFSE, detectable in the same channel as FITC; and a dead cell stain, detectable in the same channel as APC (Figure 2A,B). After data acquisition, the gating strategy in Figure 2 is used to analyze data. Dead (i.e., dead cell stain+) K562 target cells are gated out of the CFSE+ population, providing the % of killed cells within the target population.
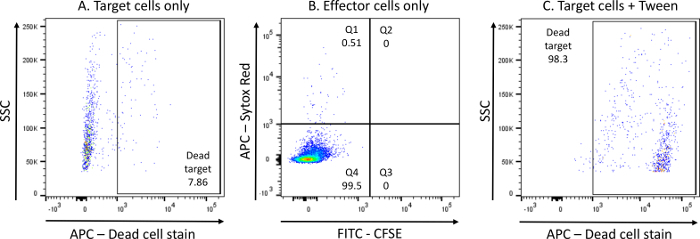
Control conditions are crucial in ensuring the effectiveness of the assay itself and its separate components. In order for the assay to be considered valid, the following should be expected in the control conditions (6-8): Target cells only (condition 6) should result in cell death < 15% (Figure 3A). No CFSE signal should be detected for effector cells only (condition 7), and cell death should be < 5% (Figure 3B). For Tween-mediated killing of target cells (condition 8), cell death should be > 85% (Figure 3C).
A patient with defect in NK cell function is expected to have reduced killing activity at most or all ratios tested as compared with a healthy individual. The parallel of healthy donor and patient cells in Figure 4 shows differential, significant reduction in target cell death with decreasing effector-target cell ratio.
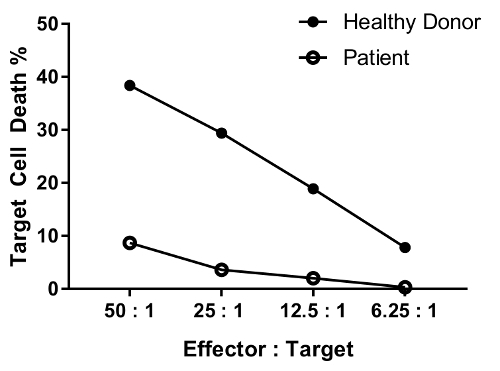
Figure 1: Assessment of NK cell content within the effector population. Representative histogram after staining total PBMCs (light blue) or purified NK cells (red) for CD56. Please click here to view a larger version of this figure.
Figure 2: Gating strategy for detection of target cells.A) The initial gate is set in a FITC-A/SSC-A plot. K562 cells are gated as CFSE+ events. B) Dead K562 cells (i.e., positive for dead cell stain) are gated in the subsequent APC-A/SSC-A plot within the CFSE+ target cell population. Please click here to view a larger version of this figure.
Figure 3: Representative data for control conditions.A) Percentage of dead cell stain+ K562 cells within the CFSE+ gate (gating strategy as in Figure 2) in condition 6 (target cells only) - negative control for K562 cell death. B) CFSE and dead cell stain detection in condition 7 (effector cells only - total PBMCs). C) Percentage of dead cell stain+ K562 cells within the CFSE+ gate (gating strategy as in Figure 2) in condition 8 (target cells + Tween) - positive control for K562 cell death. Please click here to view a larger version of this figure.
Figure 4: Representative data for the dilution effect of various effector-target cell ratio. The effector:target cell ratios tested are as follows: 50:1, 25:1; 12.5:1; 6.25:1. Data are background (condition #6)-removed. Please click here to view a larger version of this figure.
| Sample | Condition | Effector (E) : Target (T) |
| PBMCs | 1 - positive control for NK cell cytotoxicity | 50 : 1 + IL-2 |
| 2 | 50 : 1 | |
| 3 | 25 : 1 | |
| 4 | 12.5 : 1 | |
| 5 | 6.25 : 1 | |
| 6 | T only | |
| 7 - negative control for K562 death | E only | |
| 8 - positive control for K562 death | T + Tween | |
| NK cells (purified) | 1 - positive control for NK cell cytotoxicity | 5 : 1 + IL-2 |
| 2 | 5 : 1 | |
| 3 | 2.5 : 1 | |
| 4 | 1.25 : 1 | |
| 5 | 0.625 : 1 | |
| 6 | T only | |
| 7 - negative control for K562 death | E only | |
| 8 - positive control for K562 death | T + Tween |
Table 1
Discussion
The method described here provides a straightforward and cost-effective alternative to the traditional 51Cr release assay to assess NK cell cytotoxic activity. This method is sensitive, reproducible, and less time-consuming than previous standard methods, like CRA, and can be used for both clinical and research applications.
While the assay works with both total PBMCs and enriched NK cells, the option to use PBMCs without the need to purify cell populations is a great benefit when dealing with small volumes of collected blood or few or poor quality cells from patients' samples. This assay utilizes only CFSE and dead cell exclusion viability stain. Looking at these two parameters alone provides succinct and sufficient information for diagnostic purposes, with the added benefit of not requiring further compensation in flow cytometric analysis.
This method can also be used to study basic NK cell biology and test novel NK cell-targeted therapies. In this regard, the ever-expanding capabilities of flow cytometers introduce an invaluable element of versatility to this assay, allowing for multiparameter analysis of NK cells and their activity not previously possible with assays like CRA. Alternative cell tracking and viability dyes, fluorescing in other channels, are available to meet the investigators' needs.
This protocol is optimized for use with the prototypical NK cell target K562 cell line. However, it can be adapted to alternative suspension target cell lines with varying degrees of sensitivity to NK cell cytotoxicity, such as HL-60, Daudi, U937 and Raji. In this regard, it is important to note that the concentration of fluorescent dye and of serum used during target cell labeling might have to be optimized for each target cell line, as different cell lines might take up the fluorescent label differently, and for each flow cytometer. While it is typically recommended to avoid using serum during labeling, we opted for slight quenching with 0.5% of FBS for two reasons. First, it helps reducing target cell stress and thus background stain; second, it brings CFSE signal within the appropriate range of detection of our flow cytometer without requiring daily settings adjustments, thus aiding in the reproducibility of the assay. Additional protocol variations might include testing different E:T ratios and incubation times to adapt the assay to different activation conditions. The duration of the co-culture of effector and target cells to test NK cytotoxicity has historically ranged from 4-16 h, though longer periods tend to result in increased spontaneous release 9,16. In our method, longer incubation might also result in loss of fluorescent labeling by the target cells due to killing or proliferation, as these fluorescent dyes are diluted upon cell division. Thus, incubations in the lower end of the time window are generally preferred and they should not exceed overnight incubation 6,16,22.
It is important to take note of which variables can potentially affect the outcome of this assay. In our experience, the K562 target cells are especially sensitive to temperature changes. Therefore, the K562 cells should not be refrigerated but rather kept at room temperature or placed in a humidified CO2 incubator at 37 °C before their use. For the same reason, all reagents should be brought to the appropriate temperatures as indicated in the protocol. Another parameter affecting these cells is the centrifugation speed, which should be reduced to minimize cellular stress. Moreover, limiting the lag time between effector/target preparation and the start of the co-incubation to less than 30 min is essential to ensure maximal killing activity and assay reproducibility. Likewise, as CFSE signal is generally robust, accurate pipetting, proper mixing of cells and precise incubation time when labeling and quenching are crucial to ensure consistency in the staining. Finally, NK cell cytotoxic ability is highly variable even in healthy individuals and is influenced by a number of factors, including developmental stage, sex, age, and weight 23,24,25. In addition, up to 30% of healthy controls display a significant reduction or complete loss of cytotoxic ability if tested more than 24 hours after blood draw. Therefore, it is recommended to use freshly purified PBMCs or NK cells whenever possible.
Disclosures
The authors declare no conflicts of financial interest.
Acknowledgments
We would like to thank Jill Narciso, UCLA Immunogenetics Center, for her assistance with manuscript preparation.
References
- Iannello A, Debbeche O, Samarani S, Ahmad A. Antiviral NK cell responses in HIV infection: I. NK cell receptor genes as determinants of HIV resistance and progression to AIDS. J Leukoc Biol. 2008;84(1):1–26. doi: 10.1189/jlb.0907650. [DOI] [PubMed] [Google Scholar]
- Caligiuri MA. Human natural killer cells. Blood. 2008;112(3):461–469. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology. 2009;128(1):7–15. doi: 10.1111/j.1365-2567.2009.03123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren HS, Smyth MJ. NK cells and apoptosis. Immunol Cell Biol. 1999;77(1):64–75. doi: 10.1046/j.1440-1711.1999.00790.x. [DOI] [PubMed] [Google Scholar]
- Tognarelli S, Jacobs B, Staiger N, Ullrich E. Flow Cytometry-based Assay for the Monitoring of NK Cell Functions. J Vis Exp. 2016. [DOI] [PMC free article] [PubMed]
- Somanchi SS, McCulley KJ, Somanchi A, Chan LL, Lee DA. A Novel Method for Assessment of Natural Killer Cell Cytotoxicity Using Image Cytometry. PLoS One. 2015;10(10):e0141074. doi: 10.1371/journal.pone.0141074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294(1-2):15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Atkinson EA, Gerrard JM, Hildes GE, Greenberg AH. Studies of the mechanism of natural killer (NK) degranulation and cytotoxicity. J Leukoc Biol. 1990;47(1):39–48. doi: 10.1002/jlb.47.1.39. [DOI] [PubMed] [Google Scholar]
- Kim GG, Donnenberg VS, Donnenberg AD, Gooding W, Whiteside TL. A novel multiparametric flow cytometry-based cytotoxicity assay simultaneously immunophenotypes effector cells: comparisons to a 4 h 51Cr-release assay. J Immunol Methods. 2007;325(1-2):51–66. doi: 10.1016/j.jim.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane KL, Ashton FA, Schmitz JL, Folds JD. Determination of natural killer cell function by flow cytometry. Clin Diagn Lab Immunol. 1996;3(3):295–300. doi: 10.1128/cdli.3.3.295-300.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YY, et al. An improved flow cytometry-based natural killer cytotoxicity assay involving calcein AM staining of effector cells. Ann Clin Lab Sci. 2012;42(1):42–49. [PubMed] [Google Scholar]
- Korzeniewski C, Callewaert DM. An enzyme-release assay for natural cytotoxicity. J Immunol Methods. 1983;64(3):313–320. doi: 10.1016/0022-1759(83)90438-6. [DOI] [PubMed] [Google Scholar]
- Karimi MA, et al. Measuring cytotoxicity by bioluminescence imaging outperforms the standard chromium-51 release assay. PLoS One. 2014;9(2):e89357. doi: 10.1371/journal.pone.0089357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim DE, et al. Glyco-engineered anti-EGFR mAb elicits ADCC by NK cells from colorectal cancer patients irrespective of chemotherapy. Br J Cancer. 2014;110(5):1221–1227. doi: 10.1038/bjc.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West WH, Cannon GB, Kay HD, Bonnard GD, Herberman RB. Natural cytotoxic reactivity of human lymphocytes against a myeloid cell line: characterization of effector cells. J Immunol. 1977;118(1):355–361. [PubMed] [Google Scholar]
- Jedema I, van der Werff NM, Barge RM, Willemze R, Falkenburg JH. New CFSE-based assay to determine susceptibility to lysis by cytotoxic T cells of leukemic precursor cells within a heterogeneous target cell population. Blood. 2004;103(7):2677–2682. doi: 10.1182/blood-2003-06-2070. [DOI] [PubMed] [Google Scholar]
- Lecoeur H, Fevrier M, Garcia S, Riviere Y, Gougeon ML. A novel flow cytometric assay for quantitation and multiparametric characterization of cell-mediated cytotoxicity. J Immunol Methods. 2001;253(1-2):177–187. doi: 10.1016/s0022-1759(01)00359-3. [DOI] [PubMed] [Google Scholar]
- Carotta S. Targeting NK Cells for Anticancer Immunotherapy: Clinical and Preclinical Approaches. Front Immunol. 2016;7:152. doi: 10.3389/fimmu.2016.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A, Viswanathan C. Natural killer cells: In health and disease. Hematol Oncol Stem Cell Ther. 2015;8(2):47–55. doi: 10.1016/j.hemonc.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Rezvani K, Rouce RH. The Application of Natural Killer Cell Immunotherapy for the Treatment of Cancer. Front Immunol. 2015;6:578. doi: 10.3389/fimmu.2015.00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo LS, et al. Practical NK cell phenotyping and variability in healthy adults. Immunol Res. 2015;62(3):341–356. doi: 10.1007/s12026-015-8664-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zons P, et al. Comparison of europium and chromium release assays: cytotoxicity in healthy individuals and patients with cervical carcinoma. Clin Diagn Lab Immunol. 1997;4(2):202–207. doi: 10.1128/cdli.4.2.202-207.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yovel G, Shakhar K, Ben-Eliyahu S. The effects of sex, menstrual cycle, and oral contraceptives on the number and activity of natural killer cells. Gynecol Oncol. 2001;81(2):254–262. doi: 10.1006/gyno.2001.6153. [DOI] [PubMed] [Google Scholar]
- Laue T, et al. Altered NK cell function in obese healthy humans. BMC Obes. 2015;2:1. doi: 10.1186/s40608-014-0033-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazeldine J, Lord JM. The impact of ageing on natural killer cell function and potential consequences for health in older adults. Ageing Res Rev. 2013;12(4):1069–1078. doi: 10.1016/j.arr.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]