

Abstract
Background
The aim of this study was to investigate the role of tilianin in modulating mitochondrial functions and mitochondria-mediated apoptosis during cardio-protection.
Material/Methods
Myocardial ischemia/reperfusion (I/R) injury was induced by 30 minutes coronary occlusion followed by two hours reperfusion in Sprague-Dawley rats. To investigate the cardio-protective effects of tilianin, apoptosis was evaluated by TUNEL. Mitochondrial ultrastructure and function were assessed by transmission electron microscopy, dynamics of mitochondrial permeability transition pore (mPTP) opening and ATP production in the myocardium; Ca2+ content and reactive oxygen species (ROS) were measured to evaluated the level of damage factors in the mitochondria. The related apoptotic proteins were analyzed through immunoblot.
Results
Pretreatment with tilianin significantly reduced apoptosis after I/R injury in rats (p<0.05). In addition, tilianin could alleviate mitochondrial damage, markedly inhibited mPTP opening and improved ATP production (p<0.05). There was also a significant reduction for content of Ca2+ and ROS in the mitochondria (p<0.01). Apoptosis protein analysis found that treatment with tilianin led to the downregulation of apoptosis-inducing factor (AIF) (p<0.01), and suppressed the leakage of cytochrome c and activation of caspase-3 (p<0.05).
Conclusions
This study showed that tilianin can alleviate apoptosis of cardiomyocytes and protect myocardium, possibly via the protection of mitochondria and repression of mitochondrial apoptotic pathways. Mechanistically, inhibition of Ca2+ overload, mPTP opening, and ROS production in mitochondria may explain the observed tilianin-mediated treatment effects.
MeSH Keywords: Apoptosis; Ischemic Preconditioning, Myocardial; Mitochondria, Heart
Background
Myocardial ischemia/reperfusion (I/R) injury occurs in the process of severe impairment of coronary blood supply and subsequent reperfusion, which simultaneously causes further damage to ischemic myocardium [1]. Increasing evidence suggests that apoptosis is one of the major mechanisms that leads to myocardial I/R injury [2]. Hence, illuminating underlying apoptotic mechanisms and identifying new strategies is crucial to the treatment of I/R injury.
Apoptosis is a highly programmed process of cell death and closely related to mitochondria. The reason is that mitochondria are not only at the center of energy metabolism, but are also involved in apoptotic signal transduction, thus contributing to apoptotic modulation [3]. It has previously been reported that both the generation of reactive oxygen species (ROS) and overload of Ca2+ are the major contributors of apoptosis during myocardial I/R injury [4,5]. On the one hand, a massive accumulation of Ca2+ and sudden bust of ROS in mitochondria can cause the dysfunction of mitochondria, which leads to enhanced permeability of mitochondrial permeability transition pore (mPTP), which is a high-conductance pore of inner membranes in mitochondria [6,7]. The opening of mPTP allows for the release of pro-apoptotic factors, such as apoptosis-inducing factor (AIF) and cytochrome c, which transfer from the mitochondrial matrix into the cytosol [8]. There is a difference between AIF and cytochrome c. Cytochrome c is the promoter of caspase activation, in which caspase-3 is processed into cleaved caspase-3 and ultimately results in cell death [9]. However, AIF in the cytoplasm tanslocates into the nucleus and leads to chromatin condensation and molecular weight DNA fragmentation, which doesn’t rely on the participation of caspase [10]. On the other hand, these stimulations of damage factors also result in depolarization of mitochondrial membrane potential and uncoupling of the respiratory chain, which decreases the synthesis of ATP [11]. This in turn affects the functions of cardiomyocytes, results in energy depletion, and causes necrotic cell death. Therefore, the preservation of mitochondria is a potential therapeutic strategy to reduce apoptosis and limit myocardial I/R injury.
Tilianin is a flavonoid that was isolated from Moldavican dragonhead [12]. A previous study showed that tilianin was capable of correcting energy metabolic dysfunction, and inhibiting cell apoptosis and anti-oxidation in a myocardial I/R injury model [13]. However, these studies did not explore the potential effect of tilianin on mitochondrial function and its apoptotic signaling pathway. Therefore, the anti-apoptotic mechanism of tilianin and influence on mitochondria still remain unclarified in I/R injury.
In this study, we hypothesized that tilianin induces cardio-protection by ameliorating mitochondrial function and inhibiting the mitochondrial-mediated apoptotic signaling pathway. To verify this hypothesis, we examined the effects of tilianin on mitochondria and the expression of apoptosis related proteins in rats with I/R injury.
Material and Methods
Animals
Male adult Sprague-Dawley rats (six months old, 230–280 g) were purchased from the Experimental Animal Centre of Xin Jiang Medical University (Certificate No: syxk2003-0001). All rats were raised (five per cage) at 23°C, and had free access to feed and water. All procedures were approved by the Committee for the Ethical Use of Experimental Animals at Xin Jiang Medical University.
Reagents
Tilianin (purity >98%) was provided by Xin Jiang Institute of Medicine (Xin Jiang, China). Propranolol was obtained from Li sheng Pharma (Tianjin, China). ATP assay kits were purchased from Nanjing Jian Cheng Bioengineering Institute (Nanjing, China). DCFH-DA and cyclosporine A (Cs-A) were all purchased from Sigma Chemical (St. Louis, MO, USA). TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) apoptosis assay kits were obtained from Roche (IN, USA). Rabbit polyclonal anti-active + pro caspase-3 (ab13847) and Anti-AIF-[E-20] (ab32516) antibodies were from Abcam (London, Britain). Rabbit polyclonal anti-cytochrome c (10993-1-AP) was from Proteintech (Wuhan, China). β-actin was obtained from ZSGB (Beijing, China). Cell mitochondria isolation kit and BCA (bicinchoninic acid assay) protein assay were purchased from Solarbio (Shanghai, China)
Induction of myocardial I/R injury
The myocardial I/R injury model were modified from a previous study [14]. Briefly, rats were anesthetized by an intraperitoneal (ip) injection of 25% urethane (5.0 mL/kg) and fixed on a constant temperature operating table. The electrocardiogram was recorded by BL-420E (Taimeng, Chengdu, China). A respirator was provided to maintain normal breathing rate at 60 breathes per minute. In these conditions, a heart left thoracotomy was performed to expose the third or fourth intercostal space. The pericardium was opened, and a 4-0 Prolene suture was ligated around the proximal left anterior descending (LAD) coronary artery. The ligature was released to allow reperfusion for 120 minutes after 30 minutes of LAD ischemia as previously described [15]. Ischemia was confirmed based on a significant ST-segment elevation in lead II of electrocardiogram following ligation of the LAD and a ≥50% ST-segment elevation drop; recovery of T wave indicated successful reperfusion.
Experimental design
Adult Sprague-Dawley rats (SPF grade, male, 230–280 g) were used in this study. All animals were administered adaptive feed for one week and were randomly divided into the following seven groups (n=84; 12 rats/group): (1). Sham group: rats were gavaged with saline for seven days; (2). Model control group: 30 minutes of ischemia was followed by reperfusion for 120 minutes; (3–5). Tilianin groups: rats were gavaged for one week with low, medium, and high tilianin solution in water (T-L, 1.25 mg/kg/day; T-M, 2.5 mg/kg/day; T-H, 5.0 mg/kg/day), according to our previous study [13]; (6) Cyclosporine A group: rats were treated with infusion of Cs-A (10 mg/kg) ip 10 minutes before reperfusion [16]; (7) Propranolol group: rats were gavaged with propranolol (Prop, 25 mg/kg/day) for seven days before reperfusion. Propranolol was chosen as the positive control to assess the curative effect of tilianin preliminarily. At the end of the reperfusion period, the hearts were subsequently excised and processed for morphological and molecular studies. Six rats in each group were chosen to determine mitochondrial functions. The other six rats in each group were randomly divided for apoptotic assessment and immunoblot analysis.
Assessment of cardiomyocyte apoptosis
Initially, samples of tissue from AAR (area-at-risk) were cut and fixed in 4% formaldehyde for 48 hours, processed through a xylene and an ethanol gradient, embedded in paraffin wax and cut into 3-μm thick transverse sections. Then, we performed TUNEL analysis with an apoptosis detection kit from Roche, as per the manufacturer’s instructions. Finally, we observed stained sections under light microscopy (Olympus, Shimadzu, Japan). Ten stained fields were chosen randomly from each group and about 100 cells were scored at 400×. The index of apoptosis was determined (number of apoptotic myocytes/total number of myocytes counted ×100%) from a total of 70 fields/heart, and the assays were performed in a blinded manner.
Measurement of ATP contents in rat myocardia
After the operation, fresh myocardial tissues from AAR were harvested and weighed. ATP (adenosine triphosphate) concentrations were measured according manufacturer’s instructions of ATP assay kits and absorbance was measured at 636 nm.
Isolation of mitochondria and cytosol
Mitochondrial fractions were isolated according to a previously described procedure [17]. Experiments were conducted at 4°C. Briefly, the left ventricle tissue (150 mg) was finely minced with scissors and a homogenate machine and then suspended in lysis buffer. After centrifugation at 1,000 g for 10 minutes, the supernatant was centrifuged again at 12,000 g for 10 minutes to obtain the cytosolic fraction. The extraction of cytoplasm was immediately frozen at −80°C for immunoblot analysis. The precipitation was collected again and centrifuged at 12,000 g for 10 minutes, and eventually the high purity of mitochondria in the bottom of the centrifuge tube was stored with 100 uL storage buffer. The protein concentration of mitochondria and cytoplasm were measured using the BCA protein assay.
Identification of mitochondrial with transmission electron microscopy (TEM)
At the end of reperfusion, we chose the left ventricle to isolate and slice into thick slices, and fixed slices in 2% glutaraldehyde for 24 hours. After rinsing with phosphate buffer, specimens were fixed in osmium tetroxide for 2 hours at room temperature. Then the tissue specimens were dehydrated in a graded series of acetone solutions. Finally the specimens were embedded in Epon Araldite and sliced into ultrathin sections. We observed stained areas using an EOL-JEM-1230 TEM (JEOL, Tokyo, Japan). The mitochondria were imaged at 20,000×.
Concentration of Ca2+ in mitochondria of myocardia
The method was adjusted slightly based on previous studies by Elizabeth et al. [18]. The collected mitochondria were resuspended in 1% HC1. The supernatant was obtained by centrifugation at 30,000 g for 60 minutes, and colored by phenolphthalein complexing ketone.
ROS production in mitochondria of myocardia
We respectively mixed 1.995 mL PBS and the mitochondrial suspension (0.5 mg/mL−1) with 50 uL DCFH-DA solution and incubated at 37°C for 30 minutes. Fluorescence intensity was measured using an emission wavelength of 527 nm, while the excitation wavelength was 488 nm [19].
mPTP opening in mitochondria
Briefly, isolated mitochondria were resuspended with 2 mL of buffer. The initial absorbance of mitochondria at 520 nm was recorded, and then the sample was incubated for one minute in 96-well plates at 37°C. CaCl2 pulses (200 umol/L) were applied to these to induce the opening of mPTP [20]. A520 was measured for six minutes until the value did not change any more. The absorbance of A520 and initial A520 were calculated. A reduction between A520 and initial A520 was indicative of the degree of mPTP opening.
Immunoblot analysis
The cytosolic protein concentration was measured with the BCA assay kit. The protein were separated on a 15% SDS-PAGE (100 ug/lane) in process of electrophoresis and electrophoretically transferred to PVDF membranes (Millipore, USA). Membranes were blocked with TBST (TBS and 0.1% Tween 20) buffer containing 5% non-fat dried milk at room temperature for one hour, and then were incubated overnight at 4 °C with the following primary antibodies against AIF at (1: 7,000), pro-Caspase-3 and cleaved-caspase-3 (both at 1: 330), cytochrome c at (1: 3,000), β-actin at (1: 3,000). Protein bands were washed three times with TBST and then incubated with HRP-conjugated secondary antibody at room temperature for another one hour. For quantitative analysis, the optical densities of bands were analyzed by the ImageJ software (National Institutes of Health, MD, USA). β-actin was chosen as a loading control to normalize the densitometric value of target proteins.
Statistical analysis
Statistical analysis was performed with OriginPro8 software. For each experimental series, data were presented as means ±SD. Results were analyzed with one-way ANOVA followed by Tukey post-test for multiple comparisons. Statistical significance were considered when p<0.05.
Results
Effect of tilianin post-conditioning on apoptosis after I/R injury
As shown in Figure 1, TUNEL-positive cardiomyocytes had brown-staining nuclei indicated by the red arrows. Compared with the sham group, the model group had dramatically improved TUNNEL-positive myocardial cells (p<0.01), which showed that there were increased cardiomyocytes undergoing apoptosis after 120 minutes of reperfusion. In comparison to the model group, treatment with T-M, T-H, Prop, and Cs-A resulted in a significant decrease in the apoptotic index of cells (28.17±3.1%, 20.68±2.8%, 27.46±2.5%, 26.19±2.5%, respectively, p<0.05). These data clearly showed that tilianin inhibited cell apoptosis induced by I/R. Cs-A, which is a known inhibitor of mPTP in mitochondrial membranes, also showed a reduction in cardiomyocyte apoptosis with a dose of 10 mg/kg−1 (p<0.05) (Figure 2). So, tilianin displayed an effective protection to myocardial tissue that suffered from I/R injury, which was similar to propranolol and CS-A in therapeutic results.
Figure 1.

Effect of tilianin on myocardial mitochondrial structure in myocardial I/R injury rats. (A) Sham group; (B) Model group; (C) Tilianin low dose group; (D) Tilianin medium dose group; (E) Tilianin high dose group; (F) Cyclosporine A (Cs-A) group; (G) Propranolol group.
Figure 2.

Effect of tilianin on apoptosis in myocardial I/R injury in rat hearts by TUNEL staining. (A) Sham group; (B) Model group; (C) Tilianin low dose group; (D) Tilianin medium dose group; (E) Tilianin high dose group; (F) Cyclosporine A (Cs-A) group; (G) Propranolol group. All data are expressed as mean ±SD, n=6. Compared with the Sham group, @@ p<0.01; Compared with the Model group, * p<0.05, ** p<0.01; Compared with Cs-A group, # p<0.05.
The improvement of mitochondrial structure after tilianin post-conditioning
As revealed by TEM analysis, the outer membrane of mitochondria was smooth and not swollen in the sham group. There was definite damage of mitochondrial ultrastructure after I/R injury in the model group compared to the sham group. Specifically, some mitochondria suffered high-amplitude swelling, disorders and fracture of ridges; the others were characterized by vacuolar degeneration and seemed to dissolve into abnormal structure as indicated by the red arrows in Figure 1. Furthermore, the mitochondrial structure was generally improved in a dose-dependent manner with tilianin treatment, and there was a complete matrix and significant decreased swelling in the T-H group. In addition, the T-H group exhibited similar therapeutic effects as the Prop and Cs-A group, with slight swelling and complete ridges.
The reduction of energy depletion in myocardium after ischemia/reperfusion
In general, a decrease of ATP levels reflects damage or dysfunction of mitochondria in cardiomyocytes. In comparison with the sham group, mitochondria from the model group showed a significant decrease in ATP content (p<0.01), which implied that the mitochondrial dysfunction led to metabolism disturbance in myocytes after I/R injury. ATP production generally rose with dosage increase of tilianin. Compared with the model group, the T-H group and Cs-A group had reduced dissipation of energy in the mitochondria after I/R (p<0.05) (Figure 3). Those data suggested that tilianin has the function of improving mitochondrial energy metabolism status. Moreover, propranolol can also reduce energy consumption significantly (p<0.01).
Figure 3.
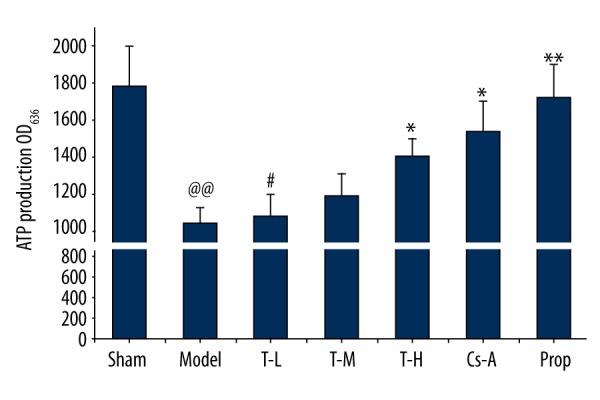
Effect of tilianin on the concentration of ATP. All data are expressed as mean ±SD, n=6. Compared with the Sham group, @@ p<0.01; Compared with the Model group, * p<0.05, ** p<0.01; Compared with Cs-A group, # p<0.05.
Tilianin treatment decreases ROS in mitochondria during reperfusion
The ROS content of the model group significantly increased in myocardial mitochondria compared with the sham group (p<0.01). Compared with the model group, each drug treatment group reduced the content of ROS following I/R in varying degrees (Figure 4). The T-L group showed a remarkable decrease of ROS production (p<0.05). Specifically, the T-H group, Cs-A group, and Prop group exhibited remarkable effect on inhibiting ROS production (p<0.01). These results suggest that tilianin can inhibit accumulation of ROS in mitochondria and thus produce anti-oxidative stress damage effect during I/R.
Figure 4.
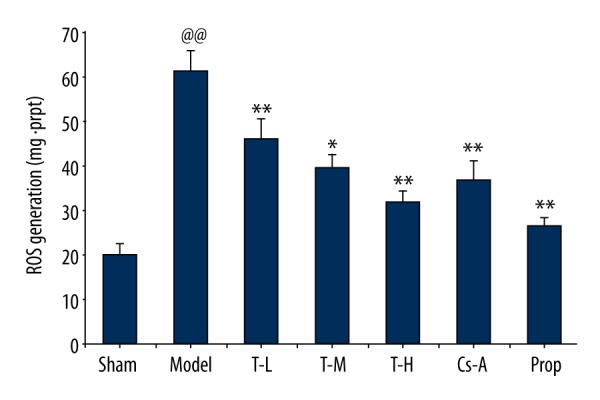
Effect of tilianin on the production of ROS in mitochondria. All data are expressed as mean ±SD, n=6. Compared with the Sham group, @@ p<0.01; Compared with the Model group, * p<0.05, ** p<0.01; Compared with Cs-A group, # p<0.05.
Tilianin treatment inhibits mPTP opening during reperfusion
Mitochondrial swelling is an important indicator of opening of mPTP [21], which was measured by absorbance change in mitochondrial after induction of 200 μmol/L−1 CaCl2 in our study. In comparison to the sham group, the model group displayed a significant increase in OD520 (p<0.05), implying an evident opening of mPTP after I/R injury. The T-H group and Cs-A group showed a significant reduction of OD520 within six minutes in comparison with the model group (p<0.05) (Figure 5), which adequately revealed the effects of tilianin on mPTP, implying that tilianin can inhibit the opening of mPTP.
Figure 5.
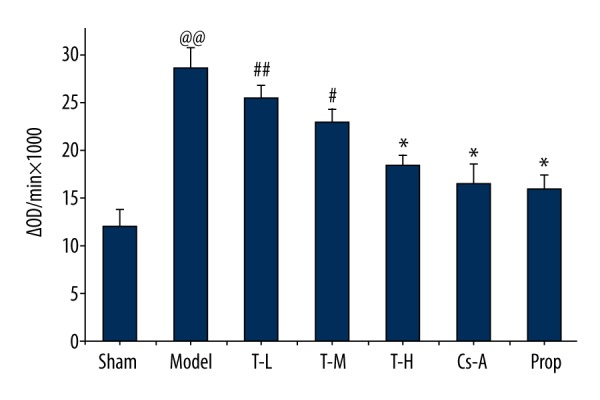
Effect of tilianin on the mPTP opening. All data are expressed as mean ±SD, n=6. Compared with the Sham group, @@ p<0.01; Compared with the Model group, * p<0.05, ** p<0.01; Compared with Cs-A group, # p<0.05.
Tilianin treatment inhibits increase of Ca2+ in mitochondria during reperfusion
In comparison with the sham group, the concentration of Ca2+ in mitochondria markedly increased in model group (p<0.01). However, the rats treated with tilianin and Cs-A were found to have an effective inhibition of Ca2+ in mitochondria as compared with the model group (p<0.05) (Figure 6), especially in the T-H group (p<0.01). These results fully revealed the specific mechanism of cardio-protective effects of tilianin lies in relief of Ca2+ overload in mitochondria compared with a previous study.
Figure 6.
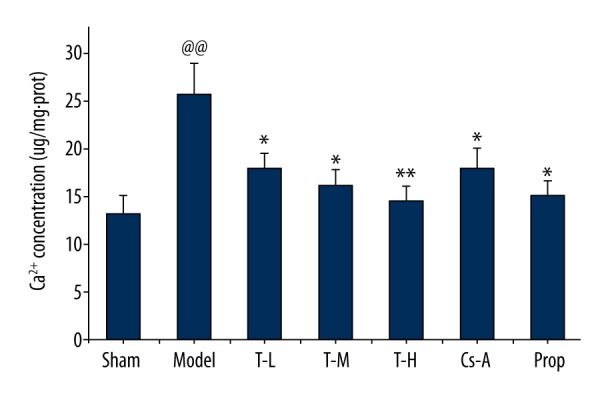
Effect of tilianin on the Ca2+ production in mitochondria. All data are expressed as mean ±SD, n=6. Compared with the Sham group, @@ p<0.01; Compared with the Model group, * p<0.05, ** p<0.01; Compared with Cs-A group, # p<0.05.
Effects of tilianin on caspase-3 and translocation of AIF and cytochrome c in the cytosol
We investigated the effects of tilianin on caspase-3 activation. As depicted in immunoblot analyses (Figure 7), caspase-3 was similar among groups, whereas the expression of cleaved caspase-3 significantly increased in the model group as compared with the sham group (p<0.01). The T-H group, Cs-A group, and Prop group effectively decreased the levels of cleaved caspase-3 compared to the model group (p<0.05). In addition, we observed the expression of pro-apoptotic protein cytochrome c and AIF in the cytoplasm. With the exception of the T-L group, there were no significant differences to be found between the T-H group and the Prop group compared to the Cs-A group in expression of pro-apoptotic protein cytochrome c and AIF (p>0.05) (Figures 8, 9). To sum up, our results suggested that tilianin-mediated inhibition of apoptosis might be via the mitochondrial signaling pathway.
Figure 7.
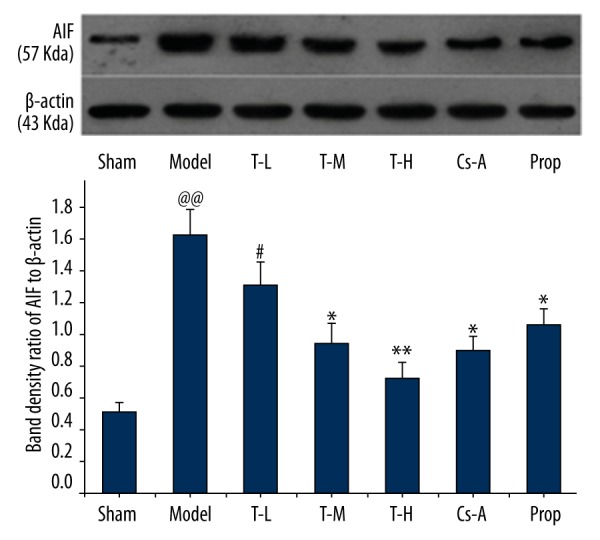
Relative expression of AIF in the cytoplasm. All data are expressed as mean ±SD, n=6. Compared with the Sham group, @@ p<0.01; Compared with the Model group, * p<0.05, ** p<0.01; Compared with Cs-A group, # p<0.05.
Figure 8.
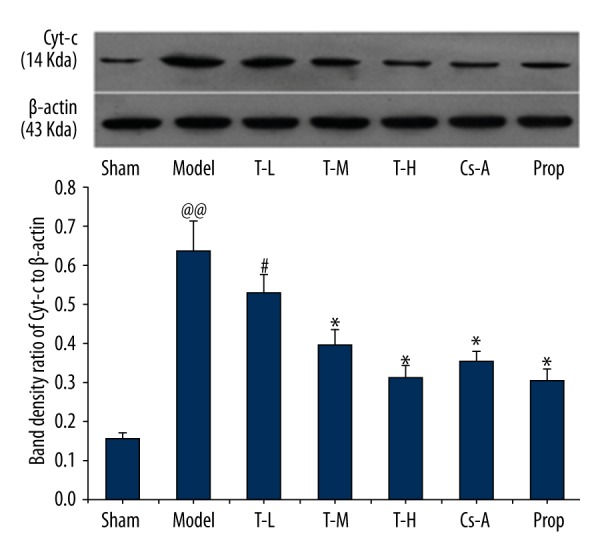
Effect of tilianin on the cytochrome c in the cytoplasm. All data are expressed as mean ±SD, n=6. Compared with the Sham group, @@ p<0.01; Compared with the Model group, * p<0.05, ** p<0.01; Compared with Cs-A group, # p<0.05.
Figure 9.

Relative expression of caspase-3 and cleaved caspase-3 in the myocardium. All data are expressed as mean ±SD, n=6. Compared with the Sham group, @@ p<0.01; Compared with the Model group, * p<0.05, ** p<0.01; Compared with Cs-A group, # p<0.05.
Discussion
In this study, the relationship between tilianin and mitochondrial function was explored for the purpose of elucidating the underlying mechanism for the cardio-protective effect of tilianin. We demonstrated two findings, first, tilianin post-conditioning significantly decreased cardiomyocyte apoptosis after I/R injury in rats in vivo. The protection of tilianin was preliminarily verified, which was similar to propranolol in reduction of cardiomyocyte apoptosis. Second, we confirmed the alleviation of mitochondrial damage and the reduction of related destructive stimulus to mitochondria, which might partly explain the downregulation of apoptotic proteins. These findings suggested that the protective effects of tilianin on myocardial tissue closely relates to mitochondrial protection and mitigation of apoptosis during I/R injury.
It is generally known that I/R injury is associated with mitochondrial dysfunction and cell death via both necrosis and apoptosis [22]. Mitochondria are vulnerable to attack from ROS and Ca2+, which are considered to be two key activators of mPTP during I/R. In early reperfusion, the burst of ROS is generated from electronic respiratory chain due to leakage of electrons during the period of ischemia, and it, taken together with the overload of Ca2+ accumulated in ischemia, promotes the opening of mPTP in reperfusion, which generally leads to swelling and rupture of mitochondria [23]. It in turn affects the functions of cardiomyocytes; resulting in the depletion of ATP and causing necrotic cell death that contributes to myocardial infract [24]. However, mitochondria not only are major sites of energy metabolism, but also regulate cell apoptosis in the high energy-consuming organ [25]. There is growing evidence that indicates that mitochondria dysfunction is closely related to cardiomyocyte apoptosis induced by I/R injury [26]. With stimulation to mitochondria of ROS and Ca2+, transient mPTP opening may lead to apoptosis. Mitochondria-related apoptotic pathways can be activated after the release of pro-apoptotic proteins. In the present study, we showed that myocardial cell apoptosis was reduced in rats with pretreatment of tilianin. Meanwhile, we confirmed the improvement of mitochondrial status in term of mitochondrial structure, ATP production, and the opening of mPTP. These results supported the hypothesis that the effect of cardio-protection of tilianin is associated with mitochondrial protection.
Previous research has shown the effects of tilianin on anti-I/R injury includes relief of calcium overload and correction of energy metabolism, which are closely related to the regulation of mitochondrial function after I/R injury [27]. Therefore, we focused on mitochondrial to further explain the specific mechanism of myocardial protection effect in this experiment. The improvement of Ca2+-ATPase and Na+-K+-ATPase activity has been approved in previous research, which indirectly reflected the energy metabolism status of cardiomyocytes and Ca2+ transport capacity. However, our results directly confirmed the increase of ATP production and decrease of Ca2+ content in mitochondria with tilianin post-conditioning after I/R injury, these results are consistent with a previous study. Mitochondria not only play a major role in generation of ROS, but also are vulnerable to oxidative stress injury from ROS. In particular, under conditions of excess ROS and overload of Ca2+ during reperfusion, the structure and function of mitochondria are susceptible to severe damage, which can lead to energy metabolism disorder and trigger apoptosis. In this study, we first demonstrated reduction of the accumulation of ROS and Ca2+ in mitochondria after post-conditioning with tilianin, which might explain the improvement of mitochondrial structure, energy metabolism status, and the opening of mPTP.
mPTP is a highly dynamic, non-selective pore which plays a crucial role in triggering the apoptosis of cardiomyocytes induced by myocardial I/R injury [28]. Irreversible opening of mPTP leads to the leakage of pro-apoptotic proteins from the mitochondrial intermembrane into the cytosol, which has been accepted as a key procedure to initiate the mitochondrial apoptotic pathway [29,30]. These pro-apoptotic proteins, including cytochrome c, AIF, Endo G, and Smac/DIABLO display a different point at whether caspase is involved in the regulation of apoptotic signaling pathway. The release of cytochrome c promotes caspase-3 activation, which is then activated into cleaved caspase-3 and leads ultimately to DNA damage [9]. In contrast, AIF translocates from cytosol into the nucleus where it mediates large-scale DNA fragmentation and cell death in a caspase-independent manner [31]. In our study, we found that tilianin post-conditioning involved the inhibition of mitochondrial apoptotic pathway by use of the inhibitor of mPTP (Cs-A), which could reduce the release of pro-apoptotic proteins. The mPTP may be an important target of tilianin. Evidence also indicated the downregulation of cleaved caspase-3 and reduction of cytochrome c and AIF in cytosol, which is consistent with findings that pretreatment with tilianin inhibits the opening of mPTP and apoptosis. To summarize, the decreased expression of cleaved caspase-3, cytochrome c, and AIF in cytosol suggested that the inhibition mechanisms of tilianin on apoptosis were to inhibit the opening of mPTP and thus block mitochondria-mediated apoptotic pathways.
Conclusions
The present study demonstrated that the protective effect of tilianin during I/R injury was associated with mitochondrial protection, as evidenced by the improvement of mitochondrial structural integrity, a reduction in energy exhaustion, and inhibition of mPTP opening. Furthermore, the reduced expression of cytochrome c and AIF in the cytosol and downregulation of cleaved caspase-3 in the myocardium showed that pretreatment with tilianin can inhibit cardiomyocyte apoptosis during I/R injury, possibly in virtue of the repression of mitochondria-mediated apoptosis pathways. However, the concrete mechanism of mitochondrial protection and anti-apoptotic could be partly involved in inhibition of Ca2+ overload, ROS production, and mPTP opening in mitochondria.
Footnotes
Conflict of interest
None.
Source of support: This work was supported by Grants from the National Natural Science Foundation of China (Nos. 81360671, 81460638, and 81660704)
References
- 1.Buja LM. Myocardial ischemia and reperfusion injury. Cardiovasc Pathol. 2005;14(4):170–75. doi: 10.1016/j.carpath.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Anversa P, Kajstura J, Leri A, Bolli R. Life and death of cardiac stem cells: A paradigm shift in cardiac biology. Circulation. 2006;113:1451–63. doi: 10.1161/CIRCULATIONAHA.105.595181. [DOI] [PubMed] [Google Scholar]
- 3.Li X, Liu J, Lin L, et al. Traditional chinese medicine shuang shen ning xin attenuates myocardial ischemia/reperfusion injury by preserving of mitochondrial function. Evid Based Complement Alternat Med. 2014;2014:180965. doi: 10.1155/2014/180965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li D, Liu M, Tao TQ, et al. Panax quinquefolium saponin attenuates cardiomyocyte apoptosis and opening of the mitochondrial permeability transition pore in a rat model of ischemia/reperfusion. Cell Physiol Biochem. 2014;34:1413–26. doi: 10.1159/000366347. [DOI] [PubMed] [Google Scholar]
- 5.Webster KA. Mitochondrial membrane permeabilization and cell death during myocardial infarction: Roles of calcium and reactive oxygen species. Future Cardiol. 2012;8(6):863–84. doi: 10.2217/fca.12.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia-Dorado D, Ruiz-Meana M, Inserte J. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res. 2012;94(2):168–80. doi: 10.1093/cvr/cvs116. [DOI] [PubMed] [Google Scholar]
- 7.Zhao ZQ. Oxidative stress-elicited myocardial apoptosis during reperfusion. Curr Opin Pharmaco. 2004;4:159–65. doi: 10.1016/j.coph.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Broughton BRS, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40(5):e331–39. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- 9.Narula J, Pandey P, Arbustini E, et al. Apoptosis in heart failure: Release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc Natl Acad Sci USA. 1999;96(14):8144–49. doi: 10.1073/pnas.96.14.8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Norberg E, Orrenius S, Zhivotovsky B. Mitochondrial regulation of cell death: Processing of apoptosis-inducing factor (AIF) Biochem Biophys Res Commun. 2010;396(1):95–100. doi: 10.1016/j.bbrc.2010.02.163. [DOI] [PubMed] [Google Scholar]
- 11.Camara AKS, Bienengraeber M, Stowe DF. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front Physiol. 2011;2:13. doi: 10.3389/fphys.2011.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan Y, Xing JG, Zhang YJ. [HPLC method for determining the content of moldavica dragonhead extract the thistle glycosides]. Chin J Exp Tradit Med Formul. 2010;12(13):68. [in Chinese] [Google Scholar]
- 13.Guo XH, Cao WJ. Mechanism and protective effects of tilianin on myocardial ischemia reperfusion injury in rats. Chin J Exp Tradit Med Formul. 2013;05:168–72. [in Chinese] [Google Scholar]
- 14.Hu XR, Xu CW, Zhou XY, et al. Sodium butyrate protects against myocardial ischemia and reperfusion injury by inhibiting high mobility group box 1 protein in rats. Biomed Pharmacother. 2010;9(5):1–4. doi: 10.1016/j.biopha.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Li Q, Wang F, Zhang YM, et al. Activation of CB2 Receptor by JWH133 protects heart against Ischemia/Reperfusion-induced apoptosis. Cell Physiol Biochem. 2013;31:693–702. doi: 10.1159/000350088. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Iorga A, Sharma S, et al. Intralipid, a clinically safe compound, protects the heart against ischemia-reperfusion injury more efficiently than cyclosporine-A. J Am Soc Anesthesiol. 2012;117(4):836–46. doi: 10.1097/ALN.0b013e3182655e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Argaud L, Gateau-Roesch O, Ovize M, et al. Preconditioning delays Ca+2-induced mitochondrial permeability transition. Cardiovasc Res. 2004;61:115–22. doi: 10.1016/j.cardiores.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Elizabeth AF, James DM, Takttro T. Myocardial mitochondrial calcium accumulation modulates nuclearcalcium accumulation and DNA Fragmentation. Ann Thorac Sur. 1995;60(2):338–44. doi: 10.1016/0003-4975(95)00446-r. [DOI] [PubMed] [Google Scholar]
- 19.Wang SX, Wang J, Shao JB, et al. Plumbagin mediates cardioprotection against myocardial ischemia/reperfusion injury through Nrf-2 signaling. Med Sci Monit. 2016;22:1250–57. doi: 10.12659/MSM.897618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Javadov SA, Clarke S, Das M, et al. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol. 2003;549(2):513–24. doi: 10.1113/jphysiol.2003.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernardi R, Colonna P, Costantini, et al. The mitochondrial permeability transition. Biofactors. 1998;8(3–4):273–81. doi: 10.1002/biof.5520080315. [DOI] [PubMed] [Google Scholar]
- 22.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88(2):581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orogo AM, Gustafsson ÅB. Cell death in the myocardium: My heart won’t go on. IUBMB Life. 2013;65(8):651–56. doi: 10.1002/iub.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halestrap AP. A pore way to die: The role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans. 2010;38(4):841–60. doi: 10.1042/BST0380841. [DOI] [PubMed] [Google Scholar]
- 25.Li R, Liu Y, Chen N, et al. Valproate attenuates nitroglycerin-induced trigeminovascular activation by preserving mitochondrial function in a rat model of migraine. Med Sci Monit. 2016;22:3229–37. doi: 10.12659/MSM.900185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ong SB, Hall AR, Hausenloy DJ. Mitochondrial dynamics in cardiovascular health and disease. Antioxid Redox Sign. 2013;19(4):400–14. doi: 10.1089/ars.2012.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo X, Cao W, Yao J, et al. Cardioprotective effects of tilianin in rat myocardial ischemia-reperfusion injury. Mol Med Rep. 2015;11(3):2227–33. doi: 10.3892/mmr.2014.2954. [DOI] [PubMed] [Google Scholar]
- 28.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion – a target for cardioprotection. Cardiovasc Res. 2004;61(3):372–85. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 29.Kinnally KW, Peixoto PM, Ryu SY, et al. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta. 2011;1813(4):616–22. doi: 10.1016/j.bbamcr.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 31.Broughton BRS, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40(5):e331–39. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]