

Abstract
While it is recognized that aquaporin-4 (AQP4)-specific T cells and antibodies participate in the pathogenesis of neuromyelitis optica (NMO), a human central nervous system (CNS) autoimmune demyelinating disease, creation of an AQP4-targeted model with both clinical and histologic manifestations of CNS autoimmunity has proven challenging. Immunization of wild-type (WT) mice with AQP4 peptides elicited T cell proliferation, although those T cells could not transfer disease to naïve recipient mice. Recently, two novel AQP4 T cell epitopes, peptide (p) 135-153 and p201-220, were identified when studying immune responses to AQP4 in AQP4-deficient (AQP4-/-) mice, suggesting T cell reactivity to these epitopes is normally controlled by thymic negative selection. AQP4-/- Th17 polarized T cells primed to either p135-153 or p201-220 induced paralysis in recipient WT mice, that was associated with predominantly leptomeningeal inflammation of the spinal cord and optic nerves. Inflammation surrounding optic nerves and involvement of the inner retinal layers (IRL) were manifested by changes in serial optical coherence tomography (OCT). Here, we illustrate the approaches used to create this new in vivo model of AQP4-targeted CNS autoimmunity (ATCA), which can now be employed to study mechanisms that permit development of pathogenic AQP4-specific T cells and how they may cooperate with B cells in NMO pathogenesis.
Keywords: Immunology, Issue 126, neuromyelitis optica, aquaporin-4, flow cytometry, T cell adoptive transfer, CNS autoimmunity, optical coherence tomography
Introduction
Neuromyelitis optica (NMO) is a central nervous system (CNS) autoimmune inflammatory demyelinating disease that causes recurrent episodes of paralysis and visual loss leading to permanent neurologic disability1. NMO is currently considered primarily to be a humoral autoimmune disease2 as it is associated with antibodies (Igs) targeting aquaporin-4 (AQP4), a water channel expressed abundantly on astrocytes3,4. However, CNS inflammation is a prerequisite for CNS entry of AQP4 Ig5,6. Thus, it has not been possible to establish a model of NMO by transfer of anti-AQP4 Igs alone. The findings that (1) pathogenic AQP4-specific Igs in NMO patients are IgG11,2, a T cell-dependent Ig subclass7 (2) T cells are identified in NMO lesions8,9 (3) NMO is associated with certain MHC II genes (e.g. HLA-DR17 (DRB1*0301))10, and (4) proinflammatory AQP4-reactive DR-restricted Th17 cells are expanded in NMO patients11,12 all indicate that AQP4-specific T cells have a key role in NMO pathogenesis. Thus, it is important to develop animal models to determine how AQP4-specific T cells may contribute to NMO pathogenesis.
Several years ago, multiple AQP4 T cell epitopes were identified in wild-type (WT) mice13,14 and rats15. While it was observed that AQP4-reactive T cells could induce opticospinal inflammation in naïve recipient rats15,16, significant clinical signs of CNS disease were not observed. Similarly, direct immunization of WT mice with peptides containing AQP4 T cell epitopes14,17, or transfer of proinflammatory T cells targeting those determinants17, did not cause clinical signs or histologic evidence of CNS autoimmunity.
Recently, it was observed that immunization of C57BL/6 AQP4-deficient (AQP4-/-) mice with AQP4 peptide (p) 135-153 or p201-220, two determinants predicted to bind MHC II (I-Ab) with high affinity18, elicited strong CD4+ T cell responses17. In contrast, these two peptides elicited only modest proliferative responses in WT mice. Further, the T cell receptor (TCR) repertoire used for recognition of these determinants by T cells from AQP4-/- mice was unique. Collectively, these findings indicate that T cell recognition of AQP4 is regulated by thymic negative selection. AQP4 p135-153- or p201-220-specific Th17 cells from AQP4-/- donor mice induced paralysis in nearly 100% of naïve recipient WT mice; this was associated with opticospinal infiltrates of T cells, B cells and monocytes. Serial opticospinal coherence tomography (OCT) demonstrated dynamic visual system involvement. Mice with T cell-mediated AQP4-targeted CNS autoimmunity (ATCA) recovered from paralysis and visual system injury. In contrast to EAE induced by myelin oligodendrocyte glycoprotein (MOG) p35-55-specific T cells, which led to persistent clinical disease, ATCA induced by T cells alone was not associated with axonal loss or reduction in retinal ganglion cells (RGCs). Our results have clearly demonstrated that there are multiple pathogenic AQP4 T cell determinants. This new model of ATCA is useful for studying mechanisms controlling development of pathogenic AQP4-specific T cells, learning how those cells induce CNS inflammation and how they may cooperate with AQP4-specific B cells and antibodies to promote NMO pathogenesis.
In the present report, we describe the protocols used to induce and evaluate T cell-induced ATCA. We begin with the techniques used for immunization, T cell culture and Th17 polarization to generate pathogenic AQP4-specific T cells, flow cytometry analysis to confirm polarization and adoptive transfer of those T cells. We then describe methods used to evaluate clinical and histologic disease and the use of serial OCT to monitor visual system injury in recipient mice.
Protocol
All animal procedures were performed in compliance with experimental guidelines approved by the University of California, San Francisco Institutional Animal Care and Use Committee. C57BL/6 (H-2b) female mice, 8 weeks of age, were purchased and C57BL/6 AQP4-/- mice were provided by A. Verkman.
1. Immunization of Mice with AQP4 Peptides
- Prepare complete Freund's adjuvant (CFA) working stock.
- Finely grind a desiccated preparation of M. tuberculosis (H37Ra) using a mortar and pestle.
- Add ground H37Ra to incomplete Freund's adjuvant to a final concentration of 4 mg/mL. CFA can be stored at 4 °C for up to 6 months.
Dissolve lyophilized antigen (Ag) AQP4 peptide in phosphate buffered saline (PBS) to a final concentration of 1 mg/mL. Maintain at 4 °C or on ice.
- Prepare the assembly of 2 luer-lock syringes, joined with a stopcock, containing the CFA/peptide emulsion.
- Attach a glass luer-lock syringe without plunger to a 3-way nylon stopcock with 2 male luer-lock connections. Close the stopcock by switching the lever towards the syringe. Position the open end of the syringe up, allowing the syringe to act as a test tube. Place this in a tube rack for added stability.
- Vortex the CFA working stock and the Ag solution. Add a 1:1 volume of peptide / CFA to the open syringe. 200 µL of CFA/Ag (4 x 50 µL) is required per mouse. NOTE: Typically, about 1 mL of preparation is lost in the stopcock, which will reduce the amount available for immunization. Therefore, it is important to incorporate this into the calculation of total volume required. Example: For immunization of 5 mice: (200 µL/animal x 5 animals) + 1 mL extra volume = 2 mL total CFA/Ag required.
- Insert the glass plunger into the syringe and while holding it in place, invert the syringe so that the stopcock is oriented upward. Switch the stopcock lever to the unused female connection. Carefully apply pressure to the glass plunger in order to remove excess air from the syringe. The liquid fills the second male luer-lock connection of the stopcock.
- Attach a second glass syringe (plunger fully inserted) to the second male luer-lock connection of the stopcock. This should be a closed system of 2 glass syringes connected with a stopcock containing as little excess air as possible.
- To create an emulsion, mix by pushing the plungers to pass the liquid alternately between the 2 syringes for about 2 min. Chill the syringes on ice for about 10 min. Repeat the chilling and mixing until there is a clear change in the viscosity of the preparation, resulting in a very stiff, white emulsion. Refrigerate the syringes containing the emulsion at 4 °C or maintain on ice.
Transfer all of the emulsion to one glass luer-lock syringe, remove the empty glass syringe and replace it with a 1 mL luer-lock syringe for injection. Fill the new syringe with emulsion, remove it from the stopcock and attach a needle (25G x 5/8).
- "Water-test" the emulsion for consistency. Expel a drop of the emulsion into a small dish of water. The emulsion should not disperse, indicating that it is suitable for injection.
- If the emulsion disperses, it is not ready for injection; transfer the liquid back into the glass syringes and continue to mix and then chill again until the emulsion is stiff.
- Test the emulsion again until the proper consistency has been achieved.
Inject donor AQP4-/- mice subcutaneously (s.c.) with emulsion containing the AQP4 peptide. Each mouse receives 4 x 50 µL s.c. injections (200 µL total (100 µg peptide)). The 4 sites include both sides of the lower abdomen, which drain into the inguinal lymph nodes (LN), and both sides of the chest wall medial to each armpit, which drain into axillary LN. The adoptive transfer will require 1-2 immunized donor mice per recipient mouse.
2. T Cell Culture and Proinflammatory T cell Polarization
- 10-12 days after immunization, collect LN from the mice.
- In an ice tray, prepare 60 mm Petri dishes fitted with a cell strainer (mesh size 70 µm) and containing chilled RPMI media with 10% heat-inactivated fetal bovine serum (FBS) and 100 U/mL penicillin-100 µg/mL streptomycin (pen-strep) (RFPS).
- Euthanize mice by exposure to CO2, followed by cervical dislocation. Immerse mice in 70% ethanol and then pin each footpad to a dissection board.
- Cut a midline skin incision from the groin to the neck and down each leg. Pull the skin away from the peritoneum and pin it tautly to the board. Dissect inguinal and axillary LN and place into the Petri dish containing RFPS, on ice.19
- For adoptive transfer experiments, combine LN from all animals within the same immunization group. For flow cytometric analyses of Vβ usage, separate LN from individual animals.
Place the cell strainer containing the LN onto a 50-mL centrifuge tube containing RFPS. Use the flat end of a sterile 5 mL syringe plunger to press the LN cells through the strainer, washing with 20-30 mL of RFPS to facilitate recovery of the cells. Maintain LN cells on ice throughout the processing until time for culture.
Wash twice, centrifuging at 393 x g for 5 min. After the second wash, resuspend the LN cells in T cell media (TCM). TCM contains RPMI with 10% heat-inactivated FBS, 292 µg/mL L-glutamine, 110 µg/mL sodium pyruvate, and 55 µM 2-mercaptoethanol and pen-strep. Typically, a volume of 5 mL TCM per donor mouse is used.
Count the LN cells. Expected yield is approximately 3-6 x 107 LN cells per immunized mouse donor. Set aside 3-4 x 106 for a proliferation assay (see Section 3 below).
- Set up polarizing cultures for adoptive transfer (Section 6).
- Prepare a Th17 polarizing culture of 5 x 106 LN cells per mL with 10 µg/mL Ag, 20 ng/mL recombinant mouse interleukin (IL)-23 and 10 ng/mL recombinant mouse IL-6 in TCM.
- Alternatively, for Th1 polarization, prepare a culture of 5 x 106 LN cells per mL with 10 µg/mL Ag and 10 ng/mL recombinant mouse IL-12 in TCM.
- Add 2 mL (1 x 107 cells) per well of the polarizing culture mixture into a 12-well plate. The LN cells from 2-4 donor mice will fill one 12-well plate. Maintain cultures in a humidified incubator at 37 °C with 5% CO2 for 72 h.
- Visually check the LN cells in culture for activation at 48 h and 72 h. Activated cells will form extensive clusters and the T cells will increase in size, becoming more pleomorphic. The increase in metabolism will cause a lowering of the pH in the media, changing from peach to orange. If the pH is low enough to turn the media yellow, add 1 mL of fresh TCM to prevent toxicity to the cells in the remaining 24 h.
- Proceed to Section 6 for adoptive transfer. Polarization efficiency may be verified by intracellular cytokine staining (ICS), as described in Section 5.
- Set up cultures using LN from individual mice for flow cytometric analysis of surface Vβ expression (Section 4).
- Prepare 5 x 106 LN cells per mL with 10 µg/mL Ag in TCM.
- Add 2 mL (1 x 107 cells) of the culture per well into a 12-well plate. Cultures should be maintained in a humidified incubator at 37 °C with 5% CO2 for 10 days. Proceed to Section 4.
3. Proliferation Assay
NOTE: This continues from Section 2.4.
Prepare 3-4 mL of LN cells in a tube, at 2 x 106 cells per mL in TCM.
Gently mix the cells by inverting the tube several times, and then transfer to a sterile trough.
Use a multichannel pipettor to plate 100 µL of cells per well into a 96-well round bottom tissue culture plate. Typically, plate 15 wells for triplicate testing of 4 Ag concentrations and a no Ag reference.
Dilute the Ag in TCM at various concentrations, typically 80, 20, 5, 1 and 0 µg/mL (for 2x stocks). Add 100 µL of each concentration in triplicate for a final concentration of 40, 10, 2.5. 0.5 and 0 µg/mL. Incubate for approximately 72 h at 37 °C. NOTE:Steps 3.5 through 3.7 involve radioactive materials. Proper usage, monitoring and disposal of radioactive materials should be conducted, according to institutional and governmental regulations.
Prepare a working stock solution of 3H-thymidine (40 µCi/mL) by adding 1 mCi 3H-thymidine to 25 mL RPMI, and store this at 4 °C. Pipet 0.5 mL of the working solution into a sterile trough.
Add 25 µL of 3H-thymidine (1 µCi) per well using a multichannel pipet. Incubate for an additional 18 h at 37 °C.
Harvest cells onto a glass filter mat using a 96-well cell harvester and rinse with 70% ethanol. After drying, seal the filter mat into a plastic sample bag with 5 mL scintillation fluid. Measure radioactivity in each well using a scintillation counter.
4. Flow Cytometry Analysis for Cell Surface TCR Vβ Expression
NOTE: This continues from Section 2.6. Antibodies for mouse TCR Vβ 2, 3, 4, 5.1 and 5.2, 6, 7, 8.1 and 8.2, 8.3, 9, 10b, 11, 12, 13, 14, and 17a are available commercially for testing the TCR Vβ expression.
For detection of TCR Vβ, dislodge T cells from the culture plate by pipetting several times and transfer cells to a tube.
Wash with FACS buffer (FB) containing 2% FBS, 0.1% sodium azide and 2 mM EDTA in PBS.
Transfer cells to a V-bottom 96-well plate at a density of 1 x 105 to 3 x 106 cells per well. If the entire Vβ panel is being tested, the cells should be distributed into multiple wells (one well per Vβ being tested).
Exclude dead cells from flow cytometric analysis by staining with a LIVE/DEAD viability dye, which reacts with free amines exposed by necrotic cells. Follow the manufacturer's instructions for centrifugation and resuspension in viability dye. Incubate on ice for 10-20 min. Throughout the cell staining procedures, protect cells from light and keep on ice.
After incubation, wash the plate once in FB. To detect TCR Vβ, suspend the cells in 100 µL of FB containing a 1:100 dilution of antibodies for CD4 (clone RM4-5) and TCR Vβ. Incubate for 15-60 min on ice.
Wash the plate once in FB and fix the cells by adding 200 µL of 4% paraformaldehyde diluted in PBS. Incubate for 20 min on ice. Wash the plate in FB and analyze by flow cytometry.
5. Flow Cytometric Analysis for Intracellular Cytokine Production
For intracellular cytokine staining (ICS), treat T cell cultures with a 1:1,000 dilution of protein transport inhibitor reagent (e.g., GolgiPlug) in TCM, 4 h prior to ICS to prevent cytokine secretion. At that time, activate T cells with 50 ng/mL phorbol 12-myristate 13-acetate (PMA) and 500 ng/mL ionomycin in order to induce protein production.
After 4 h of culture at 37 °C, dislodge cells from the culture plate by pipetting up and down, and transfer to a centrifuge tube (15 or 50 mL).
Wash with FB. Resuspend cells in FB and transfer to a V-bottom 96-well plate at a density of 5 x 105 to 3 x 106 cells per well.
Stain cells with viability dye as described above (section 4.2). After the incubation, wash the plate once in FB.
- Add antibodies for detection of surface markers as follows:
- For detection of surface markers, suspend the cells in 100 µL of FB containing a 1:100 dilution of antibody for CD4 (clone RM4-5).
- Optionally, include antibodies for B220 (clone 30-F11) and CD11b (clone M1/70) at a 1:100 dilution to improve gating of B cells and monocytes/macrophages, respectively. Generally, PE-Cy7 anti-CD4, FITC anti-B220 and PerCP-Cy5.5 anti-CD11b work well. Choice of antibody/fluorochrome conjugates should be guided by configuration of flow cytometer to be used for analysis, and optimal concentrations of antibodies should be determined empirically.
- Incubate for 15-60 min on ice.
Wash the plate with FB and resuspend the cells in 200 µL of Fixation/Permeabilization solution for 20 min on ice. This will fix the cells and permeabilize the membranes.
In order to maintain the permeabilization of the cell membranes during the intracellular staining, use Perm/Wash buffer (PW) (diluted 1:10 from the 10X stock) instead of FB. Wash the wells in 200 µL of PW.
For ICS, prepare a 1:100 dilution of the antibodies for interferon (IFN)-γ and IL-17A using 1x PW. One option is APC anti-IFN-γ (clone XMG1.2) and PE anti-IL-17A (clone eBio17B7) which works well. Resuspend cells in 100 µL of the intracellular antibody cocktail and then incubate for at least 15 min. It is also possible to continue the staining overnight at 4 °C.
Wash the wells in 200 µL of PW and resuspension in FB for flow cytometry.
In parallel, prepare an unstained sample (cells in FB without antibodies) to optimize detector voltages, and single-stained samples for each individual fluorochrome (i.e., cells or compensation beads stained with only one antibody/fluorochrome) to determine compensation spillover values.
Using a flow cytometer, acquire ≥10,000 cells (events), gating on viable (LIVE/DEAD-negative) CD4+ cells. To ensure accurate gating of T cells, exclude the B220+ and CD11b+ cells (B cells and monocytes/macrophages, respectively). Within the CD4+ T cell subpopulation, determine the percentage of cells expressing IFN-γ (Th1 lineage) or IL-17A (Th17 lineage).
6. Adoptive Transfer of Pathogenic AQP4-specific T cells
NOTE: This step follows from Section 2.5: T cell culture and proinflammatory T cell polarization.
Recover polarized cells from 12-well plates by pipetting up and down to dislodge, and transferring them to a 50 mL tube. Maintain on ice until injections.
Count the cells, wash twice in cold PBS, and prepare a suspension of 1 x 108 cells/mL in PBS. Generally, inject cells within the hour.
Gently mix the cells by swirling and transfer to a 1 mL syringe at the time of injection. Administer 200 µL of the cells (2 x 107) intravenously (i.v.) to each mouse, through the tail vein.
Inject each recipient mouse i.p. with 200 ng of B. pertussis toxin diluted in 200 µL of PBS that day and 2 days later. Monitor mice daily for signs of clinical disease.
7. Clinical Assessment of ATCA
Examine recipient mice daily for clinical signs of CNS autoimmunity. In general, mice will show signs of CNS autoimmunity 5-8 days after adoptive transfer of AQP4-specific T cells. The mice will show clinical symptoms of neurological dysfunction. It is helpful to perform the evaluations on a naive mouse to define the normal ability of a mouse.
Evaluate mice for loss of tail tone. Hold the mice gently at the base of the tail and lift upright. A tail that droops continuously is described as loss of tail tone (score of 1). Loss of tail tone is often the first sign of clinical disease.
Test for righting reflex by placing mouse on its back on a flat surface. Slow righting is a sign of truncal incoordination (score of 2). A naïve mouse will completely resist any attempt to place it on its back, so any slowness in the ability to right is scored as a dysfunction. It is typical to test the mouse 3-4 times for this reflex.
Evaluate posture and ambulation. Mice begin to show a change in posture resulting in a lowering or dragging of the hips during ambulation (score of 2.5). Further disease results in monoplegia, complete paralysis of one hind limb (score of 3.0) and paraplegia, full paralysis of both hind limbs (score of 3.5). Moderate quadraparesis, weakness of all four extremities, results in very slow forward movement (score of 4). Severe quadraparesis allows almost no forward movement (score of 4.5). Finally, with severe disease they may become moribund or die (score of 5).
Administer to mice that lose the ability to acquire fluid or solid food 1 mL of 5% glucose in saline i.p. Alternatively, use supplementation with fluid gel cups, high caloric gel and bacon softies to counteract dehydration and food deprivation. Euthanize moribund mice.
8. Tissue Preparation and Histology
Anesthetize mice with 100 mg/kg ketamine and 10 mg/kg xylazine, and then pin each footpad to a dissection board.
Open up the chest, exposing the heart. Incise the liver to permit blood outflow. Attach a butterfly needle to the tubing of a miniflow variable speed pump using a separate reservoir for PBS and 10% formalin. Insert the needle into the left ventricle of the heart and perfuse with 5 mL of PBS, followed by 25 mL of 10% formalin.
- Remove the head and then the eyes. Process eyes and retinas as described previously20. Cut across the eye sockets, insert scissors at the nose and cut the skull anterior to posterior on each side.
- Remove the skull, expose the optic nerves, cut the optic chiasm and place in a cassette between 2 foam pads.
- Immerse in 10% formalin (24 h), and then 50% ethanol (24 h) and then transfer to 70% ethanol.
Remove brain and spinal column and place in 10% formalin. Serially section brains in the coronal plane; section spinal cords in sagittal (approximately half) and serial coronal planes (approximately 20 cross-sections per mouse).
Process CNS samples routinely for paraffin embedding. Stain 8 µm thick sections with hematoxylin and eosin (optic nerves) or Luxol fast blue-hematoxylin and eosin (brain and spinal cord).
Evaluate optic nerves for the presence of inflammation within the nerve and surrounding meninges (optic neuritis) or predominantly in the surrounding meninges (optic perineuritis).
Count meningeal and parenchymal inflammatory foci (>10 clustered mononuclear cells) in the brain and spinal cord samples for each mouse.
9. In Vivo Retinal Imaging by Optical Coherence Tomography (OCT)
NOTE: Spectral domain OCT retinal imaging of mice is performed using commercial equipment (e.g., Spectralis with TruTrack eye tracker) to achieve consistent ocular orientation and reduce motion artifacts.
Five min prior to imaging, dilate pupils with 1% tropicamide (one drop per eye) and place mouse to be imaged in the anesthesia induction chamber, providing a steady flow of 2 liters per min (1.5% isoflurane). Turn on heat mat and prepare the post-anesthesia recovery cage.
Place the mouse on the imaging cylinder and redirect the anesthesia flow accordingly. Protect eyes with 0.3% hydroxypropyl methylcellulose to keep the eye moist and to ensure refraction continuity. Place a custom contact lens on the eye to be examined.
Guided by the infra-red fundus image, direct the laser to the eye, ensuring the beam is centered on the optic nerve head. Vertical and horizontal OCT scans should confirm that the retina lays perpendicular to the laser.
Perform 25 B-scans in high-resolution mode and rasterize from 30 averaged A-Scans.
After imaging both eyes, remove contact lens and apply ophthalmic gel. Leave mouse in the warm recovery cage.
10. Processing and Analysis of OCT
- Use the modular imaging software for automated segmentation.
- Manually correct segments corresponding to the inner limiting membrane (ILM) and inner plexiform layer (IPL), representing the limits of the inner retinal layers (IRL)21.
- Verify that the retinal nerve fiber layer (RNFL) and the ganglion cell layer (GCL) reside within the limits of the IRL and do not overlap.
Calculate IRL thicknesses using the Early Treatment Diabetic Retinopathy Study (ETDRS)2 grid with diameters of 1, 2, and 3 mm centered on the optic disc, and export into a spreadsheet file. The software calculates thickness of each retinal layer by averaging each grid sector. Therefore, the central segment, corresponding to the optic nerve head, is excluded.
Analyze statistical differences between groups at each time point. Analyze both eyes for each mouse, using generalized estimating equations with an exchangeable correlation matrix and adjustments for intra-subject inter-eye correlations21.
Representative Results
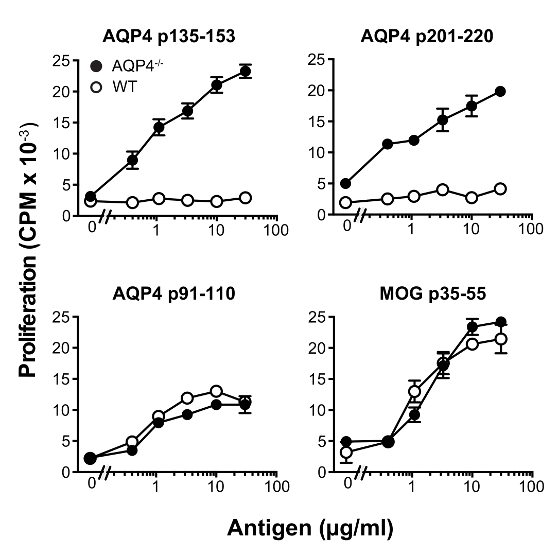
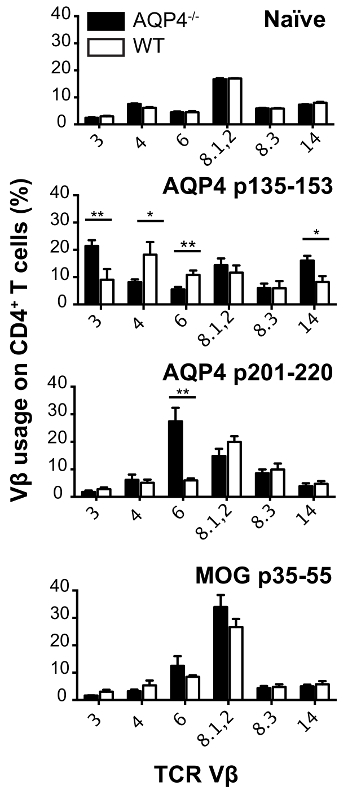
In this protocol, we used donor T cells from C57BL/6 AQP4-/- mice. Subcutaneous immunization of these mice with AQP4 p135-153 or p201-220, which contain pathogenic T cell epitopes, elicited strong proliferative T cell responses in draining lymph nodes (Figure 1), whereas these two peptides induced much weaker T cell proliferation in WT mice. In comparison, immunization with AQP4 p91-110, containing a non-pathogenic AQP4 T cell determinant13, or MOG p35-55, a myelin peptide that activates T cells that cause experimental autoimmune encephalomyelitis (EAE)22,23,24, induced similar magnitude of T cell proliferation in AQP4-/- and WT mice. Analysis of TCR utilization by flow cytometry staining for individual Vβ's or Vβ families, demonstrated that p135-153- and p201-220-specific T cells from AQP4-/- mice utilized unique TCR repertoires. Selective hyper-proliferation of AQP4 p135-153 and p201-220 in AQP4-/- mice, as well as the unique TCR utilization (Figure 2), indicated that pathogenic T cell responses to these determinants is normally regulated by thymic negative selection, underscoring the importance for using AQP4-/- donor T cells in this protocol.
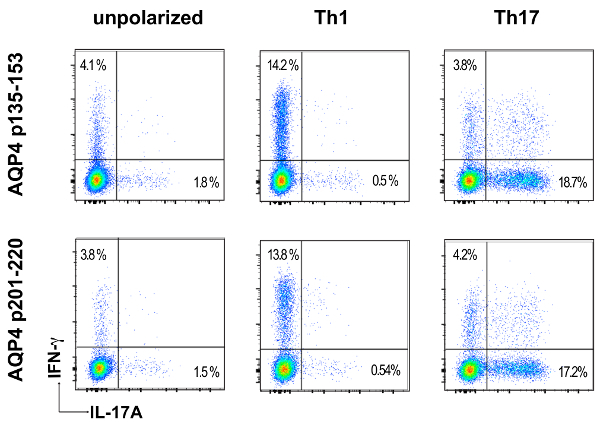
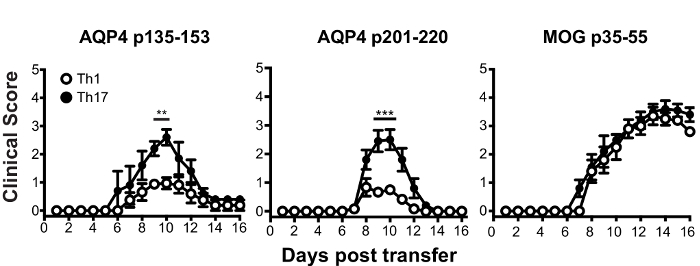
Prior to adoptive transfer for induction of ATCA, lymph node cells from AQP4 peptide-primed mice were cultured in vitro in Th17- or Th1-polarizing conditions for three days. The extent of polarization of donor CD4+ T cells was confirmed by intracellular cytokine staining (ICS) and measured by flow cytometry (Figure 3). Naïve recipient mice were injected intravenously with 2 x 107 donor AQP4 peptide-specific T cells. After approximately six days, nearly 100% of recipient mice developed clinical signs of CNS autoimmune disease, including limp tail and hind limb paralysis (Figure 4). Th17-polarized AQP4-specific T cells induced more severe clinical disease than Th1-polarized AQP4-specific T cells. A representative mouse that received Th17 AQP4-peptide-specific and developed complete hind limb paralysis (paraplegia) is shown in Video 1. In contrast with mice that developed EAE after administration of MOG-specific Th17 cells, recipient mice recovered from clinical disease induced by AQP4-specific Th17 cells. As for EAE induced by MOG p35-55-specific Th17 cells, clinical disease induced by AQP4 p135-153-specific or p201-220-specific Th17 cells was associated with infiltration of mononuclear cells in the CNS parenchyma and meninges (Figure 5). Lesions were more abundant in the meninges than in the parenchyma for CNS autoimmunity induced by AQP4-specific Th17 cells.
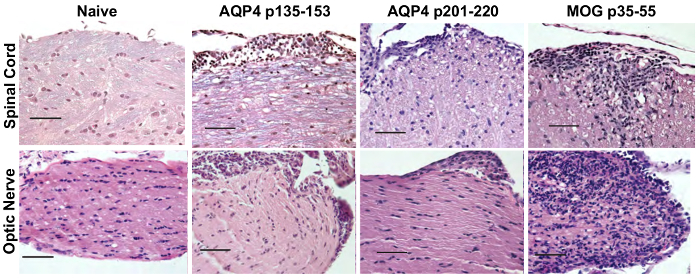
Optic nerve involvement was demonstrated by histological evaluation and by serial OCT. AQP4-specific and MOG-specific Th17 cells both induced optic nerve inflammation, which was characterized by presence of mononuclear cells. Whereas AQP4-specific Th17 cells caused optic perineuritis, MOG-specific Th17 cells induced severe optic neuritis (Figure 5). Using serial OCT, optic nerve inflammation was evident by swelling and increased inner retinal layer (IRL) thickness for clinical disease induced by AQP4-specific or MOG-specific Th17 cells (Figure 6). For AQP4 Th17-induced CNS autoimmunity, IRL thickness returned to baseline as mice recovered from clinical disease. In contrast, persistence of EAE induced by MOG-specific Th17 corresponded with IRL thinning and, as we demonstrated previously17, was associated with loss of retinal ganglion cells.
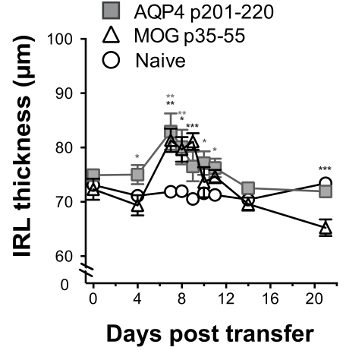
Figure 1: AQP4 p135-153 and p201-220 elicit robust T cell proliferation in AQP4-/- mice, but not WT mice. Mice were immunized s.c. with the indicated peptides in CFA. Eleven days later, lymph nodes were removed, and then cultured with either no antigen, or with the peptide used for immunization. Proliferation was measured by 3H-thymidine incorporation (mean ± SEM, representative of 5 experiments). Please click here to view a larger version of this figure.
Figure 2: AQP4-specific T cells from AQP4-/- mice utilize unique TCR repertoires. AQP4-/- and WT mice were immunized with the indicated peptides. Eleven days later, lymph nodes were removed and cultured with peptide used for immunization. Cells were harvested. TCR Vβ utilization was analyzed by flow cytometry (mean ± SEM, n = 5). Please click here to view a larger version of this figure.
Figure 3: Proinflammatory polarization of donor AQP4-specific T cells. Eleven days after immunization with AQP4 p135-153 or p201-220, lymph node cells were harvested and cultured with the peptide used for immunization in non-polarizing conditions, Th1- or Th17-polarizing conditions. Th17 or Th1 polarization was examined by ICS and flow cytometry for IL-17 or IFN-γ, respectively. Please click here to view a larger version of this figure.
Figure 4: Th17-polarized AQP4-specific T cells induce paralysis in WT recipient mice. WT recipient mice received 2 x 107 donor Th17-polarized AQP4 p135-153 or p201-220-specific T cells from AQP4-/- mice. Th17-polarized MOG-specific T cells served as a positive control. Results are representative of 8 experiments (n = 5/group). * p <0.05, ** p <0.01, *** p <0.001. Please click here to view a larger version of this figure.
Figure 5: AQP4-specific Th17 cells induce opticospinal inflammation in WT mice. Recipient mice received 2 x 107 donor Th17 AQP4 p135-153- or p201-220-primed Th17 cells from AQP4-/- mice or Th17 MOG p35-55-specific cells and were sacrificed 10 days later. Spinal cord and optic nerve tissues were prepared and stained with H&E/LFB to evaluate for evidence of inflammation and demyelination, respectively. Results are representative of 5 mice/group. Scale bar = 50 µm. Please click here to view a larger version of this figure.
Figure 6: Optic nerve inflammation induced by AQP4-specific Th17 cells can be monitored by longitudinal retinal OCT. WT recipient mice received 2 x 107 donor Th17-polarized AQP4 p201-220-specific or MOG p35-55-specific T cells on day 0. They were examined by OCT on day 0 (before administration of T cells) and then on days 4, 7, 8, 9 10, 11 14 and 21. IRL thickness was measured (mean ± SEM). Statistics indicate a comparison with naïve control. Results are representative of 3 experiments (5 mice/group). * p <0.05, ** p <0.01, *** p <0.001. Please click here to view a larger version of this figure.
Discussion
AQP4 was identified as the primary target in NMO IgG in 20053. Then, it was recognized that it would be important to establish an AQP4-targeted animal model of CNS autoimmunity. Such a model could be useful to investigate how AQP4-specific T and B cells participate in development of CNS autoimmunity and to test candidate therapeutics for NMO. Although identification of AQP4-specific T cell epitopes in wild-type mice was first reported in 201013, T cells responding to those epitopes did not cause clinical or histologic disease14,17. The inability to generate a model of CNS autoimmunity based upon immune reactivity to AQP4 remained an enigma until 2015 when Jones, et al.25 discovered that donor AQP4 p135-153-primed T cells from AQP4-/- mice were capable of causing clinical and histologic signs of CNS autoimmunity in WT mice. Of interest, AQP4 p135-153 is predicted to bind MHC II (I-Ab) with high affinity17. Only one other AQP4 amino acid sequence, 201-220, is predicted to bind I-Ab with similar high affinity. Indeed, we observed that AQP4 p135-153 and p201-220 both elicit robust proliferation in AQP4-/-, but not WT, mice. Here, we have shown how one can isolate and expand encephalitogenic Th17 AQP4 p135-153- and p201-220-reactive T cells from AQP4-/- mice. When transferred into WT recipient mice, donor AQP4-reactive Th17 cells induced paralysis, which was accompanied by mononuclear cell infiltrates in spinal cord and optic nerve. Afferent visual system injury is well known in patients with AQP4-seropositive NMOSD26. Here, we observed that optic nerve inflammation induced by AQP4-reactive and MOG-reactive Th17 cells was distinct. Whereas AQP4-specific Th17 cells induced optic perineuritis, MOG-specific Th17 cells induced severe optic neuritis. We have also described the techniques used to monitor optic nerve inflammation induced by AQP4-specific and MOG-specific T cells by serial OCT. Other investigators should now be able to apply the protocols described here to advance their own studies focused on pathogenic mechanisms of ATCA.
One can easily avoid three potential pitfalls in our protocol. First, adoptive transfer of ATCA requires use of AQP4-specific T cells from AQP4-/- donor mice. Specifically, donor WT AQP4 p135-153-specific T cells did not cause ATCA in recipient WT mice. Secondly, it is important to perform a "water-test" with a drop of the peptide/CFA emulsion before immunization of AQP4-/- mice (Protocol Step 1.5). The emulsion should not disperse in water when it is suitable for s.c. injection. If the emulsion disperses, one should mix the emulsion once more, chill again and repeat the water-test. Lastly, activated donor CNS antigen-specific T cells induce CNS autoimmunity more efficiently than resting T cells. One should visually inspect those cultures under the light microscope prior to harvesting the donor T cells for adoptive transfer. Rapidly dividing cells may form clusters, which are easily identified. Also, when cultures contain many activated T cells, the media may transition from pink to orange or even to yellow, due to reduction in pH. One can also assess activation of donor AQP4-primed lymph node T cells for proliferation by 3H-thymidine incorporation, as described in Protocol Step 3.
Our discovery that the two pathogenic AQP4 T cell epitopes are (1) predicted to bind MHC II with high affinity and (2) elicit potent proliferative responses in AQP4-/-, but not WT, mice suggest that T cells targeting those determinants are normally controlled by thymic negative selection17. The TCR repertoires utilized for recognition of AQP4 p135-153 and p201-220 in AQP4-/- mice are unique (Figure 2), which is also consistent with clonal deletion mediated by thymic medullary epithelial cells. Other tolerogenic mechanisms may normally restrain immune responses to AQP4. Subsequent to our initial report17, another group also demonstrated that AQP4 p201-220 contains an encephalitogenic T cell determinant27. When α/β (TCRα-/-) T cell-deficient mice were reconstituted with AQP4-/- CD4+ T cells, it was possible to elicit an encephalitogenic AQP4-specific T cell response, but not an AQP4-specific humoral response, implying that in WT mice AQP4-specific B cell responses, similar to AQP4-specific T cell responses, are subject to negative selection. Indeed, loss of spinal cord axons and RGCs, which was not observed in WT mice with ATCA induced by AQP4-specific T cells alone, may require participation of pathogenic AQP4-specific antibodies. It is clear that mouse models of AQP4-targeted CNS autoimmunity will continue to evolve as we learn more regarding the tolerogenic mechanisms normally controlling AQP4-specific T cell and B cell immunity.
Other models of AQP4-targeted CNS autoimmunity are also being developed6,16,28,29,30. Each one may offer advantages for studying particular aspects that are relevant to NMO pathogenesis. AQP4-specific T cells have been identified in WT rats6,16,29. Those AQP4-specific T cells caused histologic changes of CNS autoimmunity but, similar to observations in mice, WT rat AQP4-specific T cells do not cause significant signs of clinical disease. Therefore, the mechanisms of tolerance restricting T cell and B cell AQP4-specific immune responses in WT mice are also operational in rats. Regardless, one should not underestimate the power in using mouse models for studying mechanisms involved in pathogenesis of disease. The wealth of knock-out, transgenic and reporter mice can be advantageous. It should also be recognized that several fundamental discoveries in autoimmunity were made using mouse EAE models. For example, demonstration that T cell clones specific for a self-antigen can mediate autoimmune disease31,32, identification of the role of T cell costimulation in autoimmunity33 and the discovery of the developmental pathway for Th17 differentiation34 were first described using mouse EAE models. Using the mouse model of ATCA that we have developed, one now has the means to study development and regulation of pathogenic AQP4-specific immune responses in vivo, which should provide important insights related to NMO pathogenesis.
Disclosures
S.A. Sagan, A. Cruz-Herranz, C.M. Spencer, P.P. Ho, L. Steinman, A.J. Green, and R.A. Sobel report no disclosures. S.S. Zamvil is Deputy Editor of Neurology, Neuroimmunology and Neuroinflammation and is a member of the advisory board for the International Society of Neuroimmunology. He has served on the Editorial Board of the Journal of Clinical Investigation, The Journal of Immunology and The Journal of Neurological Sciences, and has been a charter member of the grant review committee for the National Institutes of Health (NIH) Clinical Neuroimmunology and Brain Tumors (CNBT) study section and the National Multiple Sclerosis Society (NMSS). He has served as a consultant and received honoraria from Biogen-Idec, EMD-Serono, Genzyme, Novartis, Roche/Genentech, and Teva Pharmaceuticals, Inc., and has served or serves on Data Safety Monitoring Boards for Lilly, BioMS, Teva and Opexa Therapeutics. Currently, Dr. Zamvil receives research grant support from the NIH, the NMSS, The Maisin Foundation, Biogen and Celgene.
Acknowledgments
Support was provided to S.S.Z. by the National Institute of Health (RO1 AI073737 and RO1 NS092835-01), National Multiple Sclerosis Society (RG 4768, RG 5179 and RG 5180), Maisin Foundation and Guthy Jackson Charitable Foundation.
References
- Hardy TA, et al. Atypical inflammatory demyelinating syndromes of the CNS. Lancet Neurol. 2016;15(9):967–981. doi: 10.1016/S1474-4422(16)30043-6. [DOI] [PubMed] [Google Scholar]
- Lennon VA, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364(9451):2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202(4):473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder. Neurol Neuroimmunol Neuroinflamm. 2015;2(1):62. doi: 10.1212/NXI.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett JL, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66(5):617–629. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradl M, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66(5):630–643. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- Nurieva RI, Chung Y. Understanding the development and function of T follicular helper cells. Cell Mol Immunol. 2010;7(3):190–197. doi: 10.1038/cmi.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti CF, et al. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol. 2014;24(1):83–97. doi: 10.1111/bpa.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm. 2015;2(4):110. doi: 10.1212/NXI.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum DG, et al. HLA-DRB association in neuromyelitis optica is different from that observed in multiple sclerosis. Mult Scler. 2010;16(1):21–29. doi: 10.1177/1352458509350741. [DOI] [PubMed] [Google Scholar]
- Varrin-Doyer M, et al. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol. 2012;72(1):53–64. doi: 10.1002/ana.23651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaknin-Dembinsky A, et al. T-cell responses to distinct AQP4 peptides in patients with neuromyelitis optica (NMO) Mult Scler Relat Disord. 2016;6:28–36. doi: 10.1016/j.msard.2015.12.004. [DOI] [PubMed] [Google Scholar]
- Nelson PA, et al. Immunodominant T cell determinants of aquaporin-4, the autoantigen associated with neuromyelitis optica. PLoS One. 2010;5(11):15050. doi: 10.1371/journal.pone.0015050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri SR, et al. Functional characterization of aquaporin-4 specific T cells: towards a model for neuromyelitis optica. PLoS One. 2011;6(1):16083. doi: 10.1371/journal.pone.0016083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl M, et al. Pathogenic T cell responses against aquaporin 4. Acta Neuropathol. 2011;122(1):21–34. doi: 10.1007/s00401-011-0824-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeka B, et al. Highly encephalitogenic aquaporin 4-specific T cells and NMO-IgG jointly orchestrate lesion location and tissue damage in the CNS. Acta Neuropathol. 2015;130(6):783–798. doi: 10.1007/s00401-015-1501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagan SA, et al. Tolerance checkpoint bypass permits emergence of pathogenic T cells to neuromyelitis optica autoantigen aquaporin-4. Proc Natl Acad Sci U S A. 2016;113(51):14781–14786. doi: 10.1073/pnas.1617859114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita R, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015;43:405–412. doi: 10.1093/nar/gku938. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell MI, Iritani BM, Ruddell A. Lymph node mapping in the mouse. J Immunol Methods. 2008;332(1-2):170–174. doi: 10.1016/j.jim.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova E, Toychiev AH, Yee CW, Sagdullaev BT. Optimized protocol for retinal wholemount preparation for imaging and immunohistochemistry. J Vis Exp. 2013. p. e51018. [DOI] [PMC free article] [PubMed]
- Cruz-Herranz A, et al. The APOSTEL recommendations for reporting quantitative optical coherence tomography studies. Neurology. 2016;86(24):2303–2309. doi: 10.1212/WNL.0000000000002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. 1995;25(7):1951–1959. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- Shetty A, et al. Immunodominant T-cell epitopes of MOG reside in its transmembrane and cytoplasmic domains in EAE. Neurol Neuroimmunol Neuroinflamm. 2014;1(2):22. doi: 10.1212/NXI.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnarfi N, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. 2013;210(13):2921–2937. doi: 10.1084/jem.20130699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Huang H, Calabresi PA, Levy M. Pathogenic aquaporin-4 reactive T cells are sufficient to induce mouse model of neuromyelitis optica. Acta Neuropathol Commun. 2015;3:28. doi: 10.1186/s40478-015-0207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel FC, et al. Microstructural visual system changes in AQP4-antiboby-seropositive NMOSD. Neurol. Neuroimmunol. Neuroinflamm. 2017;4:72. doi: 10.1212/NXI.0000000000000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel AL, et al. Deletional tolerance prevents AQP4 directed autoimmunity in mice. Eur J Immunol. 2017. [DOI] [PMC free article] [PubMed]
- Zhang H, Verkman AS. Longitudinally extensive NMO spinal cord pathology produced by passive transfer of NMO-IgG in mice lacking complement inhibitor CD59. J Autoimmun. 2014;53:67–77. doi: 10.1016/j.jaut.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeka B, et al. Aquaporin 4-specific T cells and NMO-IgG cause primary retinal damage in experimental NMO/SD. Acta Neuropathol Commun. 2016;4(1):82. doi: 10.1186/s40478-016-0355-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix CM, Levin MH, Verkman AS. Complement-independent retinal pathology produced by intravitreal injection of neuromyelitis optica immunoglobulin G. J Neuroinflammation. 2016;13(1):275. doi: 10.1186/s12974-016-0746-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamvil S, et al. T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature. 1985;317(6035):355–358. doi: 10.1038/317355a0. [DOI] [PubMed] [Google Scholar]
- Zamvil SS, et al. Encephalitogenic T cell clones specific for myelin basic protein. An unusual bias in antigen recognition. J Exp Med. 1985;162(6):2107–2124. doi: 10.1084/jem.162.6.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchroo VK, et al. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80(5):707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]