

Abstract
The analysis of circulating tumor DNA (ctDNA) using next-generation sequencing (NGS) has become a valuable tool for the development of clinical oncology. However, the application of this method is challenging due to its low sensitivity in analyzing the trace amount of ctDNA in the blood. Furthermore, the method may generate false positive and negative results from this sequencing and subsequent analysis. To improve the feasibility and reliability of ctDNA detection in the clinic, here we present a technique which enriches rare mutations for sequencing, Enrich Rare Mutation Sequencing (ER-Seq). ER-Seq can distinguish a single mutation out of 1 x 107 wild-type nucleotides, which makes it a promising tool to detect extremely low frequency genetic alterations and thus will be very useful in studying disease heterogenicity. By virtue of the unique sequencing adapter's ligation, this method enables an efficient recovery of ctDNA molecules, while at the same time correcting for errors bidirectionally (sense and antisense). Our selection of 1021 kb probes enriches the measurement of target regions that cover over 95% of the tumor-related driver mutations in 12 tumors. This cost-effective and universal method enables a uniquely successful accumulation of genetic data. After efficiently filtering out background error, ER-seq can precisely detect rare mutations. Using a case study, we present a detailed protocol demonstrating probe design, library construction, and target DNA capture methodologies, while also including the data analysis workflow. The process to carry out this method typically takes 1-2 days.
Keywords: Cancer Biology, Issue 126, Next Generation Sequencing, cfDNA (Circulating cell free DNA), Rare Mutations, ER-Seq (enrich rare mutation sequencing), baseline database
Introduction
Next-generation sequencing (NGS), a powerful tool to investigate the mysteries of the genome, can provide a large quantity of information, which may reveal genetic alterations. The application of NGS analysis in the clinic has become more common, especially for personalized medicine. One of the greatest limitations of NGS, however, is a high error rate. Although it is deemed suitable for studying inherited mutations, the analysis of rare mutations is greatly limited1,2, especially when analyzing DNA obtained from a "liquid biopsy".
Circulating tumor DNA (ctDNA) is cell-free DNA (cfDNA) in the blood that is shed from tumor cells. In most cases, the quantity of ctDNA is extremely low, which make its detection and analysis very challenging. However, ctDNA has many attractive features: its isolation is minimally invasive, it can be detected in the early stages of tumor growth, the ctDNA level reflects therapeutic efficiency, and ctDNA contains DNA mutations found in both primary and metastatic lesions3,4,5. Therefore, given the rapid development of the NGS technique and analysis, the application of ctDNA detection has become more attractive.
Different massively parallel sequencing approaches have been utilized for ctDNA detection but none of these approaches have been accepted for routine use in clinics due to their limitations: low sensitivity, lack of versatility, and a relatively high cost6,7,8. For example, duplex sequencing, based on a unique identifier tag (UID), repeatedly corrects errors in the consensus bidirectionally, rectifying most sequencing errors. However, the feasibility of this method is lost due to its high cost and low data utilization9,10. Similarly, CAPP-Seq and its improved iteration, CAPP-IDES11,12, have greater practicality in cfDNA detection, though the accuracy and universality of these methods need improvement.
To meet the current need for accurate ctDNA detection and analysis, we developed a new strategy, Enrich Rare Mutation Sequencing (ER-Seq). This approach combines the following: unique sequencing adapters to efficiently recover ctDNA molecules, with bidirectional error correction and the ability to distinguish a single mutation out of > 1 × 107 wild-type nucleotides; 1021 kb probes which enrich measurement of target regions that cover over 95% of the tumor-related mutations from 12 tumors, including lung cancer, colorectal cancer, gastric cancer, breast cancer, kidney cancer, pancreatic cancer, liver cancer, thyroid cancer, cervical cancer, esophageal cancer, and endometrial carcinoma (Table 1); and baseline database screening making it efficient and easy to precisely detect rare mutations in ctDNA.
To build a baseline database, find all the gene mutations by ER-Seq from a number of the same type of samples (~1000 at the beginning). These real mutations must be verified by several other reliable detection methods and analysis. Next, summarize the pattern of false mutations and cluster all the false mutations to build the initial baseline database. Continue adding false mutations found from subsequent sequencing experiments to this database. Therefore, this baseline database becomes a dynamic expanded database, which significantly improves sequencing accuracy.
To promote progress in tumor diagnosis and monitoring, we present ER-Seq, a low cost and feasible method for the acquisition of universal data. We present a case study which underwent ER-Seq analysis, demonstrating its accuracy for detecting rare mutations and feasibility for use in the clinic.
Protocol
Tumor specimens and blood samples were obtained according to a protocol approved by the Ethics Committee of Peking University People's Hospital. Written informed consent was obtained from the patients to use their samples. Participants were screened according to the following criteria: female, advanced Non-Small Cell Lung Cancer, EGFR p.L858R mutation indicated by previous Sanger Sequencing, disease progression following two session of EGFR targeted therapy with Erlotinib, and ER-Seq which was applied to ctDNA to analyze the cause of resistance and find new target drugs.
1. DNA Extraction from Peripheral Blood for cfDNA and genomic DNA (gDNA)
Collect 10 mL of peripheral blood in a collection tube, invert gently up and down 6-8 times to mix. Avoid cell shearing. Blood samples can be stored in the collection tube at 6-37 °C for up to 72 hours.
Centrifuge the collection tube at room temperature for 10 min at 1600 (±150) x g.
Transfer the supernatant (plasma) from the collection tube to four clean 2 mL appropriately labelled centrifuge tubes using a disposable pipet without disturbing the pellet layer (white blood cells).
Transfer1 mL of the cells using a clean disposable pipet to a 2 mL appropriately labelled centrifuge tube. The cells can be stored at -20 °C or colder before step 1.8.
Centrifuge the plasma (from step 1.3) for 10 min at 4 °C, and 16000 (±150) x g.
Transfer the plasma to clean centrifuge tubes properly labelled as "plasma", leave ~0.1 mL of residual volume at the bottom to avoid contamination. Store the plasma at -80 °C until step 1.7.
Use 3 mL of plasma from step 1.6 to isolate cfDNA (contains ctDNA) using a commercially available kit, following the manufacturer's instructions.
Use 200 µL of white blood cells from step 1.3 to isolate gDNA using a commercially available kit, following the manufacturer's instructions. NOTE: Both cfDNA and gDNA can be stored at -20 °C before use.
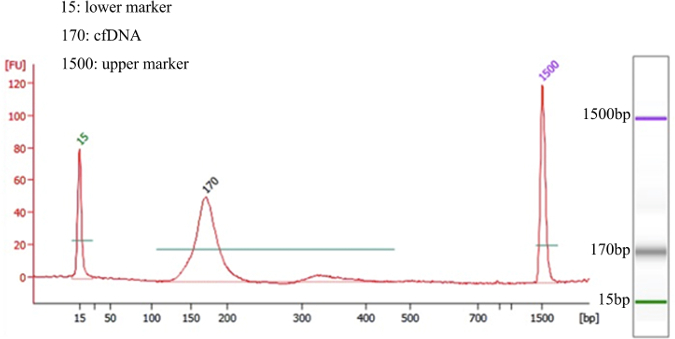
Apply different quality controls for cfDNA and gDNA (Table 2). Perform quality control by a standardized method for the bioanalyzer to determine levels of sample degradation and/or gDNA contamination. Peak analysis of the quality of cfDNA extraction should appear similar to Figure 1. NOTE: The peak size of cfDNA is around 170 bp. Note that increasing the amount of DNA will result in a higher quality library for the effective detection of rare mutations in cfDNA. We recommend using at least 30 ng cfDNA (30,000 copies).
2. Library Preparation
NOTE: Regarding base library construction on fragmented DNA, cfDNA exists in fragments with a peak size ~170 bp and thus does not need to be fragmented.
- Fragment the control sample (gDNA) by sonication to obtain 200-250 bp fragments.
- Prepare 1 µg of gDNA sample in 100 µL of Tris-EDTA buffer in a clean sonication tube.
- Set the sonication program as 30 s on and 30 s off for 12 cycles for a total of 12 min.
- After sonication, confirm the product (5 µL) contains 200-250 bp fragments by running an analysis with 2% agarose gel.
- Transfer all fragmentation products to a new 1.5 mL tube and incubate with 150 µL of magnetic beads for 5 min to select the correct fragments.
- Remove the supernatant by putting the tube on a magnetic rack for 30 s.
- Wash the beads with 200 µL of 80% ethanol (freshly prepared) with the tube staying on the magnetic rack. Incubate for 30 s before removing the supernatant. Repeat once.
- Air dry the beads with the tube lid open while staying on the magnetic rack. Avoid overdried beads to make sure that the DNA target recovers well. Elute DNA fragments from the beads by adding 32 µL of 10 mM Tris-HCl (pH 8) to the beads to resolve the DNA fragments followed by quantification by UV/Vis spectroscopy. NOTE: At least 200 ng is needed for the following experiments.
- End Prep Reaction NOTE: Follow manufacturer's instruction and modify as needed. Use 30 ng of cfDNA; and 1 µg of gDNA as the control.
- Add 7 µL of reaction buffer, 3 µL of the enzyme mix, 30 ng of cfDNA in a sterile nuclease-free tube, and add ddH2O to total volume of 60 µL. In a separate tube, add the above components but with 1 µg of gDNA instead of cfDNA.
- Mix and incubate the mixture at 20 °C for 30 min followed by 65 °C for 30 min in a thermocycler without the lid heated. Proceed immediately to the next step.
- Adapter Ligation
- Add 30 µL of the ligation master mix, 1 µL of the ligation enhancer, and 4 µL of the unique sequencing adapters to the mixture from step 2.2.
- Incubate at 20 °C for 15 min in a thermocycler without the lid heated.
- Vortex to resuspend the magnetic beads and leave the beads at room temperature for at least 30 min before the next step.
- Add 87 µL of the resuspended magnetic beads to the previously mentioned mixture; mix well by pipetting up and down and then incubate at room temperature for 5 min.
- Wash and air dry the beads as described in Step 2.1.
- Elute the DNA target from the beads by adding 20 µL of 10 mM Tris-HCl (pH 8).
- Enrichment of DNA fragments ligated with adapter
- Add 20 µL of adapter ligated DNA fragments, 5 µL of Index Primer/i7 (2.5 µL Primer-P7 (10 pM) and 2.5 µL of Primer-P5 (10 pM), for sequencing purposes), 25µL of library amplification master mix, and ddH2O to a total volume of 50 µL.
- PCR amplify the adapter ligated DNA and thermal cycle as follows: initial denaturation at 98 °C for 45 s, followed by 8 cycles (cfDNA) or 4 cycles (gDNA) of annealing °C for 15 s, 65 °C for 30 s, 72 °C for 30 s), and a final extension at 72 °C for 60 s, then holding the temperature at 4 °C.
- Add 45 µL of suspended magnetic beads to the PCR-enriched DNA to get acquired fragments.
- Elute DNA fragments with adapters in 30 µL of 10 mM Tris-HCl (pH 8).
- Library Quality Control
- Follow the manufacturer's protocol for the commercial kit to measure adapter ligated DNA concentration. Use the bioanalyzer to determine the DNA quality.
3. Targeted DNA capture
NOTE: Target enrichment was performed using a custom sequence capture-probe which is specifically designed for a 1021 kb target enrichment region covering known tumor-associated driver mutations from 12 different types of tumors. Modifications to the manufacturer's protocol are detailed in the following steps.
- Blocking
- Add 1.5 µg of pooling libraries (maximum number of libraries to pool for cfDNA is 6, gDNA is 20) from step 2, 8 µL of P5 Block(100P), 8 µL of P7 Block(100P), and 5 µL of Cot-1 DNA (1 µg/µL) in a sterile tube. Dry the contents of the tube using a vacuum concentrator set at 60 °C.
- Hybridize the DNA capture probes with the library.
- Add the following components to the tube from step 3.1: 8.5 µL of 2X Hybridization buffer, 2.7 µL of Hybridization enhancer, and 1.8 µL of nuclease-free ddH2O.
- Mix by pipetting up and down and incubate in a thermomixer at 95 °C for 10 min.
- Following incubation, immediately add 4 µL of the Custom Probe. Incubate samples in a thermal cycler at 65 °C with lid heated to 75 °C for 4 h.
- DNA fragments capture
- Incubate the hybridized target DNA with streptavidin beads followed by washing the beads to remove unbound DNA. Use the commercial kit according to manufacturer's instructions to get a final 20 µL of resuspended beads with captured DNA fragments.
- Amplification of the captured DNA fragments
- Perform PCR using the commercial kit with 2 µM backbone oligonucleotides, according to manufacturer's instructions.
- When the PCR reaction is completed, add 45 µL of suspended beads directly to the PCR product and enrich the amplified target DNA fragments bound to the beads by elution with 30 µL of 10 mM Tris-HCl (pH 8).
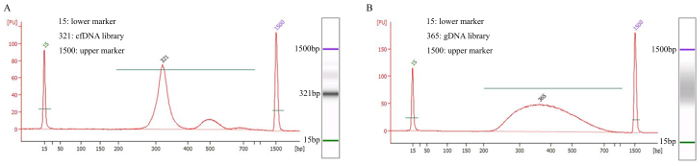
Quantify the DNA fragments with the library quantification kit and determine the average fragment length by the Bioanalyzer (Figure 2).
4. Sequencing
Sequence multiplexed libraries using 75 bp paired-end runs on a benchtop sequencer for 18 h. Total data produced is up to 60 Gigabytes, 15G is required for patient sample cfDNA and 1G for control sample gDNA.
5. Data analysis workflow
NOTE: Figure 3 displays the general work flow and data analysis process. Data analysis parameters and commands are shown below.
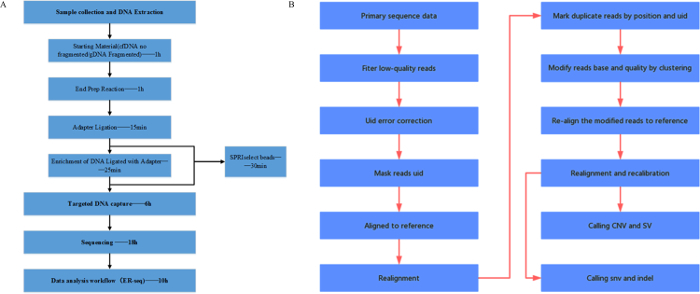
Filter low-quality reads and correct for errors and masking of UID using NCfilter. NCfilter(own) NCfilter filter -l 5 -q 0.5 -n 0.1 -T 3 -G -1 fastq1 -2 fastq2 realSeq(own) python bin/realRealSeq MaskUID -1 fq1 -2 fq2 -o output dir -p prefix -u 4 -s 7 -m 3 -i uidFile -f.
Referencing genome hg19 (human genome 19), locate the sequencing results using BWA (version 0.7.12-r1039) mem and realign with Genome Analysis Toolkit (GATK) (version 3.4-46-gbc02625). bwa mem -t 3 hg19 fastq1 fastq2 java -d64 -server -XX:+UseParallelGC -XX:ParallelGCThreads=8 -Xms2g -Xmx10g - Djava.io.tmpdir=/tmp/ -jar GenomeAnalysisTK.jar -T RealignerTargetCreator -R hg19 -L targetRegion -known dbsnp — allowPotentiallyMisencodedQuals -nt 10 -I cancerBam -I normalBam java -d64 -server -XX:+UseParallelGC -XX:ParallelGCThreads=8 -Xms2g -Xmx10g — Djava.io.tmpdir=/tmp/ -jar GenomeAnalysisTK.jar -T IndelRealigner -R hg19 -known dbsnp -allowPotentiallyMisencodedQuals — targetIntervals intervals -I cancerBam -I normalBam
Cluster the duplicates according to the UID and position of the template fragments, which will correct the error bases introduced by PCR/sequencing and modify the base quality. realSeq(own) python realSeq/bin/realSeq CLUSTER -b unmarkdup_sort_merge.bam -r chipRegion -o output_dir -p prefix -m 6
Re-align the clustered duplicates by BWA mem. bwa mem -t 3 hg19 fastq1 fastq2
Perform a local realignment followed by a recalibration of base quality score using GATK (version 3.4-46-gbc02625). Loacal realignment: java -d64 -server -XX:+UseParallelGC -XX:ParallelGCThreads=8 -Xms2g -Xmx10g -Djava.io.tmpdir=/tmp/ -jar GenomeAnalysisTK.jar -T RealignerTargetCreator -R hg19 -L targetRegion -known dbsnp -allowPotentiallyMisencodedQuals -nt 10 -I cancerBam -I normalBam java -d64 -server -XX:+UseParallelGC -XX:ParallelGCThreads=8 -Xms2g -Xmx10g -Djava.io.tmpdir=/tmp/ -jar GenomeAnalysisTK.jar -T IndelRealigner -R hg19 -known dbsnp -allowPotentiallyMisencodedQuals -targetIntervals intervals -I cancerBam -I normalBam base quality-score recalibration: java -d64 -server -XX:+UseParallelGC -XX:ParallelGCThreads=8 -Xms2g -Xmx10g -Djava.io.tmpdir=/tmp/ -jar GenomeAnalysisTK.jar -T BaseRecalibrator -R hg19 -L targetRegion -knownSites dbsnp -knownSites cosmic -allowPotentiallyMisencodedQuals -nct 10 -I bam -o grp java -d64 -server -XX:+UseParallelGC -XX:ParallelGCThreads=8 -Xms2g -Xmx10g -Djava.io.tmpdir=/tmp/ -jar GenomeAnalysisTK.jar -T PrintReads -I bam -BQSR grp -o outbam -R hg19 -nct 8 -allowPotentiallyMisencodedQuals
For SNV (single-nucleotide variant) and INDEL (small insertions or deletions) calling, the accuracy of the low frequency mutations is further confirmed by GSR (Good Support Reads) with both forward and reverse strand read pairs and filtration through control group (germline mutations) and the baseline database. realDcaller(own) python realDcaller -f hg19 -r chipRegion -C baselineDB -O -L 2 cancerBam normalBam -p 30
Use baseline samples as a control for CNV (copy number variation) calling. NCcnv(own) python NCcnv -o cnvdir -t tmpdir -r chipRegion cnv --normgt normalBam --tumgt cancerBam --repdb repeatMarsk
Realigned unmapped, clips and discordant pair reads to call SV (Structural Variations). NCsv(own) python NCsv -t tmpdir -r hg19 -o outputdir -x transcript -w bwapath -n 4 normalBam cancerBam
Representative Results
The 1021 kb probes enriched target regions used in ER-Seq are shown in Table 1, which covers over 95% of the gene mutations in 12 common tumors. The wide range of these probes makes this process applicable to a majority of cancer patients. Additionally, our unique sequencing adapters and baseline database screening make it possible to detect rare mutations precisely.
Due to the different properties of gDNA and cfDNA extracted from peripheral blood, there is a range of data quality, which is determined by the different instruments and quality controls shown in Table 2. Successfully extracted cfDNA should display a peak size of ~170 bp, as analyzed by the bioanalyzer QC and shown in Figure 1. Large fragments indicate contamination from genomic DNA, which should not appear in the final product. DNA extracted from this patient sample was of sufficient quality and quantity for ER-Seq (cfDNA - 42.6 ng; gDNA - 3.426 µg).
Target DNA capture using a custom probe was then amplified by PCR. The average fragment size was evaluated by the bioanalyzer and representative data for a patient's cfDNA in Figure 2A showed a peak around ~320 bp and a unique band on agarose gel. gDNA should appear as a smear on an agarose gel, as shown in Figure 2B. The qualities of both libraries were acceptable for the subsequent sequencing.
After sequencing, the data was analyzed in order according to the work flow in Figure 3B. To illustrate the advantages of our ER-Seq versus the traditional method, we performed both analyses using the same sample. Results from both analyses are shown in Table 3 and Table 4. We showed that ER-Seq improves coverage depth by 23% compared with the traditional method (Table 3, 3214.3 reads in ER-Seq vs 2475 reads in traditional analysis), which was due to its efficient recovery of cfDNA molecules, thus greatly enhancing the analysis of rare mutations. It is also clear that the unique sequencing adapters used in ER-Seq enable easy differentiation of natural and PCR-induced duplications (Table 3, 0 PCR induced duplicated read in ER-Seq). Additionally, we found in the calling results that analysis based on ER-Seq was 100% consistent for EGFR p.L858R detection and that the frequency of detection was a bit higher (2.7% vs 1.2%) when compared with traditional analysis (Table 4). Importantly, ER-Seq analysis enabled the detection of other relatively low-frequency mutations, including EGFR p.T790M (variation frequency 0.53%) (Table 4), and this was not recognized by traditional analysis due to high background noise.
Figure 1. Representative QC Result of Successfully Isolated cfDNA For the left graphs, the X axis shows fragment size (bp) and the Y axis indicates relative fluorescence units (FU). The primary size of cfDNA is ~170 bp and there are no large fragments of contamination. A simulated electrophoresis gel is shown on the right. Please click here to view a larger version of this figure.
Figure 2. Example of Libraries QC Analyzed. The X axis shows the fragment size (bp) and the Y axis indicates the relative fluorescence units (FU). A simulated electrophoresis gel was shown on the right of each graph. (A) Patient cfDNA sample showed a sharp peak after it was amplified with adapters. (B) Control gDNA sample showed evenly distributed fragments with no sharp peak and no bias toward one side. Both samples were acceptable for subsequent sequencing. Please click here to view a larger version of this figure.
Figure 3. ER-Seq Work Flow. (A) General work flow. (B) Data analysis work flow. Please click here to view a larger version of this figure.
| 2735 exons from all the exon region of 70 genes | |||||||||
| ABL1 | ABL2 | AKT1 | AKT2 | AKT3 | ALK | APC | AR | ARAF | ATM |
| ATR | AURKA | AURKB | AXL | BAP1 | BCL2 | BRAF | BRCA1 | BRCA2 | BRD2 |
| BRD3 | BRD4 | BTK | C11orf30 | C1QA | C1S | CBL | CCND1 | CCND2 | CCND3 |
| CCNE1 | CD274 | CDH1 | CDK13 | CDK4 | CDK6 | CDK8 | CDKN1A | CDKN1B | CDKN2A |
| CDKN2B | CHEK1 | CHEK2 | CRKL | CSF1R | CTNNB1 | DDR1 | DDR2 | DNMT3A | EGFR |
| EPHA2 | EPHA3 | EPHA5 | ERBB2 | ERBB3 | ERBB4 | ERCC1 | ERG | ESR1 | EZH2 |
| FAT1 | FBXW7 | FCGR2A | FCGR2B | FCGR3A | FGFR1 | FGFR2 | FGFR3 | FGFR4 | FLCN |
| FLT1 | FLT3 | FLT4 | FOXA1 | FOXL2 | GAB2 | GATA3 | GNA11 | GNAQ | GNAS |
| HDAC1 | HDAC4 | HGF | HRAS | IDH1 | IDH2 | IGF1R | IL7R | INPP4B | IRS2 |
| JAK1 | JAK2 | JAK3 | KDR | KIT | KRAS | MAP2K1 | MAP2K2 | MAPK1 | MAPK3 |
| MCL1 | MDM2 | MDM4 | MED12 | MET | MITF | MLH1 | MLH3 | MPL | MS4A1 |
| MSH2 | MSH3 | MSH6 | MTOR | MYC | MYD88 | NF1 | NF2 | NOTCH1 | NOTCH2 |
| NOTCH3 | NOTCH4 | NRAS | NTRK1 | NTRK3 | PALB2 | PDGFRA | PDGFRB | PDK1 | PIK3CA |
| PIK3CB | PIK3R1 | PIK3R2 | PMS1 | PMS2 | PRKAA1 | PSMB1 | PSMB5 | PTCH1 | PTCH2 |
| PTEN | PTPN11 | RAF1 | RARA | RB1 | RET | RHEB | RHOA | RICTOR | RNF43 |
| ROCK1 | ROS1 | RPS6KB1 | SMARCA4 | SMARCB1 | SMO | SRC | STAT1 | STAT3 | STK11 |
| SYK | TMPRSS2 | TOP1 | TP53 | TSC1 | TSC2 | VEGFA | VHL | XPO1 | XRCC1 |
| introns, promoters and breakpoints or fusion region from the following 24 genes | |||||||||
| ALK | FGFR1 | FGFR2 | FGFR3 | NTRK1 | NTRK3 | PDGFRA | PDGFRB | ROS1 | RET |
| MET | BRAF | ABL1 | BRD3 | BRD4 | EGFR | RAF1 | BCR | ERG | TMPRSS2 |
| RARA | KIF5B | BCL2L11 | TERT | ||||||
| other relative genes: 1122 exons from 847 genes |
Table 1. 1021 kb Probes Enriched Target Regions. These regions included: 2735 exons from 170 genes; introns, promoter regions and breakpoint regions from 24 genes; 1122 exons from 847 related genes. These cover over 95% of the tumor related driver mutations and target mutation sites from the 12 most common tumors (lung cancer, colorectal cancer, gastric cancer, breast cancer, kidney cancer, pancreatic cancer, liver cancer, thyroid cancer, cervical cancer, esophageal cancer, and endometrial carcinoma).
| DNA | Result | Indication | Ideal Range |
| cfDNA | Fragment Size | Identification of DNA Fragment distribution | 168bp±20(a) |
| Concentration | More accurate DNA quantification | Total cfDNA ≥30ng(b) | |
| gDNA | A260/A230 | Identification of chemical contaminants (e.g., ethanol) | 1.50 – 2.2 |
| A260/A280 | Identification of protein contaminants | 1.60 – 2.2 | |
| Concentration | DNA quantification | Total gDNA >3ug |
Table 2. Quality Analysis of cfDNA and gDNA Extracted from Peripheral Blood. The peak size of cfDNA is around 170Bp. Quality control should be performed by a standardized method for the 2100 Bioanalyzer to determine levels of sample degradation and/or gDNA contamination. Peak analysis of the quality of the cfDNA extraction should appear similar to Figure 1. Note that increased DNA amount will result in a library with a higher quality for the effective detection of rare mutations in cfDNA. We recommend using at least 30 ng cfDNA (30,000 copies).
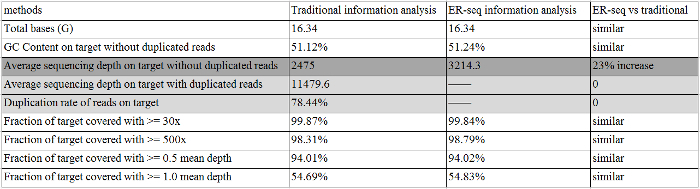 Table 3. The QC of ER-Seq Information Analysis and Traditional Information Analysis. The dark gray highlight indicates a coverage depth increase occurred in ER-Seq compared with traditional methods; a light gray highlight indicates no PCR induced duplicated read in ER-Seq. Please click here to view a larger version of this figure.
Table 3. The QC of ER-Seq Information Analysis and Traditional Information Analysis. The dark gray highlight indicates a coverage depth increase occurred in ER-Seq compared with traditional methods; a light gray highlight indicates no PCR induced duplicated read in ER-Seq. Please click here to view a larger version of this figure.
| Gene | Chr | Start | End | cHGVS | pHGVS | Function | caseAltIsHot | ER-Seq var_freq | Traditional var_freq |
| TP53 | 17 | 7577534 | 7577535 | c.746G>T | p.R249M | Missense | HighFreq | 0.0400 | 0.0378 |
| EGFR | 7 | 55259514 | 55259515 | c.2573T>G | p.L858R | Missense | Actionable | 0.0270 | 0.0120 |
| ATM | 11 | 108203618 | 108203619 | c.7919delC | p.T2640Ifs*6 | frameshift | ND | 0.0169 | 0.0180 |
| PTCH1 | 9 | 98221917 | 98221918 | c.2851G>T | p.D951Y | Missense | ND | 0.0154 | 0.0260 |
| EGFR | 7 | 55249070 | 55249071 | c.2369C>T | p.T790M | Missense | Actionable | 0.0053 | ——(0.0013) |
| RB1 | 13 | 49039151 | 49039152 | c.2230A>T | p.I744F | Missense | ND | 0. 0036 | ——(0.0014) |
Table 4. The Calling Result of ER-Seq Information Analysis and Traditional Information Analysis.
Discussion
The existence of circulating tumor DNA (ctDNA) was discovered more than 30 years ago, however the application of ctDNA analyses is still not routine in clinical practice. Interest in the practical application of ctDNA methods has increased with the development of technologies for ctDNA detection and analysis. Tumor monitoring with ctDNA offers a minimally-invasive approach for the assessment of microscopic residual disease, response to therapy, and tumor molecular profiles under the background of tumor evolution and intratumoral heterogeneity13,14. Improvements in the sensitivity and specificity of analysis will facilitate all these applications15,16.
In this manuscript, we introduced ER-Seq, a promising method aimed at improving the sensitivity of ctDNA detection, using a NSCLC case as an example. Compared with traditional analysis, the application of our unique sequencing adapters in ER-Seq significantly improved its sensitivity and specificity, indicated by a coverage depth increase and an ability to detect low-frequency mutations (such as EGFR p.T790M (0.53%)). The existence of T790M in this patient sample may be a contributing factor to Erlotinib resistance, and gave insight into how to better treat this patient, suggesting the use of a third-generation EGFR inhibitor specific for T790M, such as AZD929117-20. Another critical point in ER-Seq that contributes to its sensitivity and specificity is the baseline database screening, which facilitates the precise detection of low-frequency mutations in ctDNA by filtering out background noise.
Although ER-Seq has many advantages that make it superior to traditional methods, there are still some challenges that need to be overcome. One of the major challenges for ER-Seq is the extremely low level of ctDNA present in the blood. For those low-frequency mutations, the acquisition of sufficient template ctDNA is critical. Our unique adapters significantly increase the recycle rate of ctDNA, though they still need to be improved. Another challenge for ER-Seq, and for all the other sequencing techniques, is the limitation of coverage of target regions. Our selected 1021 kb probe enriched regions, which can cover about 95% of tumor-related mutations, are more comprehensive compared with other small-plane methods21,22,23. However, as tumor heterogeneity has been well recognized and more and more tumor biomarkers have been discovered, the relative coverage rate of our probes enriched regions decreases. To better serve as a tool for early diagnosis and disease monitoring of tumor patients, more comprehensive sequencing and analysis techniques are needed. Currently, ER-Seq still relies on a high volume of data for detecting low-frequency mutations. Further improvement of data utilization could be favorable for the application of ER-Seq.
As discussed above, many limitations associated with ER-Seq exist. However, our unique analysis technique still has many advantages compared with other top-ranking methods in this field. The pros and cons of this technique merit further research. The potential of ER-Seq for routine clinical use will be of great importance for clinical physicians, providing them with a powerful tool to diagnose tumors and monitor tumor dynamics and response to therapy. Novel mutations associated with resistance to conventional and targeted therapy found by our analysis might offer new avenues of treatment for cancer patients with advanced disease.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work is supported by Geneplus–Beijing Institute.
References
- Kennedy SR, et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc. 2014;9:2586–2606. doi: 10.1038/nprot.2014.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Bettegowda C, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014;6 doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratman SV, Newman AM, Alizadeh AA, Diehn M. Potential clinical utility of ultrasensitive circulating tumor DNA detection with CAPP-Seq. Expert Rev. Mol. Diagn. 2015;15:715–719. doi: 10.1586/14737159.2015.1019476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J. Clin. Oncol. 2014;32:579–586. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013;10:472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- Su Z, et al. A platform for rapid detection of multiple oncogenic mutations with relevance to targeted therapy in non-small-cell lung cancer. J. Mol. Diagn. 2011;13:74–84. doi: 10.1016/j.jmoldx.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukita Y, et al. High-fidelity target sequencing of individual molecules identified using barcode sequences: de novo detection and absolute quantitation of mutations in plasma cell-free DNA from cancer patients. DNA Res. 2015;22:269–277. doi: 10.1093/dnares/dsv010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt MW, et al. Sequencing small genomic targets with high efficiency and extreme accuracy. Nat. Methods. 2015;12:423–425. doi: 10.1038/nmeth.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt MW, et al. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA. 2012;109:14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014;20:548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34(5):547–555. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alix-Panabières C, Schwarzenbach H, Pantel K. Circulating tumor cells and circulating tumor DNA. Annu. Rev. Med. 2012;63:199–215. doi: 10.1146/annurev-med-062310-094219. [DOI] [PubMed] [Google Scholar]
- Dawson SJ, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013;368:1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short-read alignment with Burrows- Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn A, Simon R. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics. 2011;27:175–181. doi: 10.1093/bioinformatics/btq630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshew T, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Kuang Y, Rogers A, Yeap Y, Wang L, Makrigiorgos M, Vetrand K, Thiede S, Distel RJ, Jänne PA. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin. Cancer Res. 2009;15:2630–2636. doi: 10.1158/1078-0432.CCR-08-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Uchida J, Nishino K, Kumagai T, Okuyama T, Okami J, Higashiyama M, Kodama K, Imamura F, Kato K. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin. Cancer Res. 2011;17:7808–7815. doi: 10.1158/1078-0432.CCR-11-1712. [DOI] [PubMed] [Google Scholar]
- Leary RJ, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci. Transl. Med. 2010;2 doi: 10.1126/scitranslmed.3000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshew T, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- Bratman SV, Newman AM, Alizadeh AA, Diehn M. Potential clinical utility of ultrasensitive circulating tumor DNA detection with CAPP-Seq. Expert Rev. Mol. Diagn. 2015;15:715–719. doi: 10.1586/14737159.2015.1019476. [DOI] [PMC free article] [PubMed] [Google Scholar]