

Abstract
The goal of this protocol is to isolate herpes simplex virus type 1 (HSV-1) DNA from infected cells for the identification of associated viral and cellular proteins by mass spectrometry. Although proteins that interact with viral genomes play major roles in determining the outcome of infection, a comprehensive analysis of viral genome associated proteins was not previously feasible. Here we demonstrate a method that enables the direct purification of HSV-1 genomes from infected cells. Replicating viral DNA is selectively labeled with modified nucleotides that contain an alkyne functional group. Labeled DNA is then specifically and irreversibly tagged via the covalent attachment of biotin azide via a copper(I)-catalyzed azide-alkyne cycloaddition or click reaction. Biotin-tagged DNA is purified on streptavidin-coated beads and associated proteins are eluted and identified by mass spectrometry. This method enables the selective targeting and isolation of HSV-1 replication forks or whole genomes from complex biological environments. Furthermore, adaptation of this approach will allow for the investigation of various aspects of herpesviral infection, as well as the examination of the genomes of other DNA viruses.
Keywords: Immunology, Issue 126, Herpes simplex virus (HSV), isolation of proteins on nascent DNA (iPOND), accelerated native iPOND (aniPOND), microbiology, molecular biology, virology, DNA isolation, click chemistry, mass spectrometry, viral genome, DNA replication, isolation of nuclei
Introduction
Viruses have a limited capacity to carry out essential functions and therefore depend on host factors to facilitate critical aspects of infection including viral gene expression, replication, repair, recombination, and transport. The activities of these host factors are often augmented by virally encoded proteins. In addition, viruses must avoid detection and interference by cellular responses to viral infection. Therefore, virus host interactions dictate the outcome of infection. Of paramount importance is understanding how viruses alter the cellular environment to adapt the cellular machinery to facilitate viral processes. Of particular interest is identifying which factors and processes act on viral genomes throughout the infectious cycle.
Herpes simplex virus type 1 (HSV-1) is a double stranded DNA virus that infects a substantial proportion of the human population. Within the first hour of infection, the viral genome enters the nucleus, where an ordered cascade of viral gene expression ensues in coordination with viral DNA (vDNA) replication1. In the nucleus, genomes are subject to epigenetic regulation, undergo repair and recombination, and are packaged into capsids, such that the first progeny virions are produced within less than six hours. The comprehensive evaluation of viral genome associated proteins throughout the course of infection will lay the groundwork to investigate the molecular details of processes that act on viral genomes, and will provide insight into which viral and cellular factors are involved in various stages of infection.
Previous methods for the investigation of host factors involved in viral infection include affinity purification of viral proteins for the analysis of associated cellular proteins2,3,4,5,6,7,8,9. These assays have been instrumental for the identification of cellular factors involved in host antiviral responses, as well as viral chromatin modification, gene expression, and DNA repair. However, it is difficult to ascertain whether interactions depend on the association with vDNA, and proteomics only provide insight into interactions that occur as a function of a specific viral factor. Chromatin immunoprecipitation (ChIP) has been used to identify where specific viral and cellular proteins bind to viral genomes10,11,12,13,14,15 and fluorescent in situ hybridization (FISH) combined with immunocytochemistry has enabled the visualization of cellular factors that colocalize with vDNA16,17,18,19,20. These assays allow for spatial and temporal analysis. However, limitations include the need for highly specific antibodies, limited sensitivity, and the need for previous insight into virus host interactions. We therefore developed a method based on iPOND (isolation of proteins on nascent DNA)21 and aniPOND (accelerated native iPOND)22 to selectively label and purify vDNA from infected cells for the unbiased identification of viral genome associated proteins by mass spectrometry. iPOND has been instrumental for the investigation of cellular replication fork dynamics.
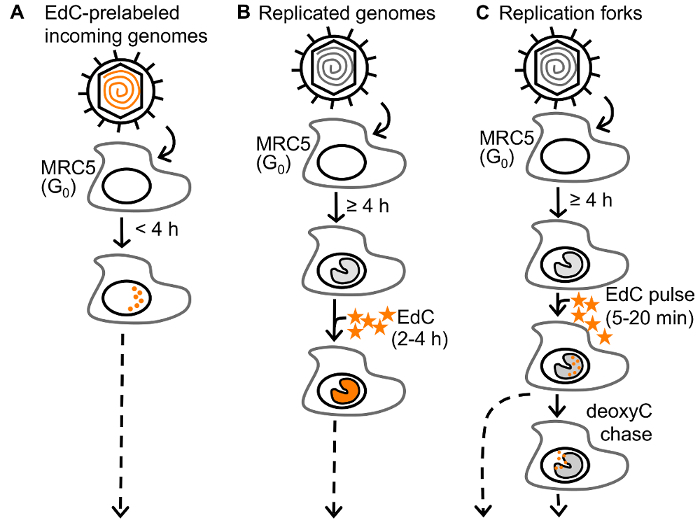
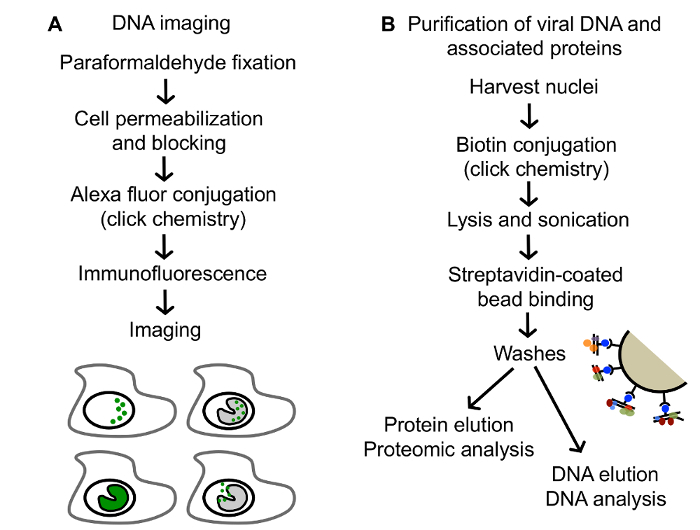
For the selective purification of viral genomes from infected cells, replicating vDNA is labeled with ethynyl modified nucleosides, 5-ethynyl-2´-deoxyuridine (EdU) or 5-ethynyl-2´-deoxycytidine (EdC) (Figure 1), followed by covalent conjugation to biotin azide via click chemistry to facilitate single step purification of viral genomes and associated proteins on streptavidin-coated beads (Figure 2B). Importantly, infections are carried out in stationary cells, which are not engaged in cellular DNA replication to enable specific labeling of vDNA. Furthermore, HSV-1 infection causes cell cycle arrest and inhibits cellular DNA replication23,24. Virus can be prelabeled before infection for the analysis of proteins associated with incoming viral genomes (Figure 1A) or labeled during DNA replication for the analysis of proteins associated with newly synthesized vDNA (Figure 1B)25. Furthermore, pulse chase analysis can be used to investigate the nature of proteins associated with viral replication forks (Figure 1C)26. In addition, ethynyl-modified vDNA can be covalently conjugated to a fluorophore for spatial investigation of protein dynamics (Figure 2A and Figure 3). Imaging allows for the direct visualization of vDNA and is a complimentary approach for the validation of vDNA-protein interactions, and can be adapted to track viral genomes throughout infection. We anticipate that these approaches could be further modified to study any aspect of herpesviral infection, including latency and reactivation, and to study other DNA viruses. Moreover, labeling with 5-ethynyl uridine (EU) could allow for the analysis of RNA viral genomes.
Protocol
1. Cell Culture, Viral Infection, and EdC Labeling (Figure 1)
The following protocol involves working with viruses. Please refer to your institution's bio-safety protocols regarding safe handling of viruses and other biological agents. This protocol was approved by the Institutional Review Board of the University of Pittsburgh.
Trypsinize a confluent 150 cm2 tissue culture flask of MRC-5 cells and transfer the cells to a 600 cm2 tissue culture plate containing 100 mL DMEM plus 10% FBS. Incubate at 37 °C in the presence of 5% CO2 for 3 - 4 days to reach confluency and stationary phase. A confluent 600 cm2 dish contains ~7 x 107 cells. Prepare 1 plate for each condition and 1 plate for each corresponding negative control. NOTE: Cells must reach the stationary phase to inhibit cellular DNA replication to ensure that only vDNA is labeled and purified in subsequent steps. NOTE: Each sample requires a complimentary unlabeled negative control prepared using the same infection conditions and time point without the addition of EdC. NOTE: A scaled down version of this experiment should be carried out in tandem to test for the labeling of cellular DNA by imaging using comparable cell growth, infection, EdC labeling conditions, and time points (see Step 2).
Dilute 7 x 108 PFU HSV-1 in 7 mL cold Tris-Buffered Saline (TBS). Remove growth medium and store at 37 °C. To infect, add the 7-mL diluted virus to MRC-5 cells and rock for 1 h at room temperature. Following adsorption, remove inoculum, rinse with 50 mL room temperature TBS, and replace the original growth medium. Incubate at 37 °C in the presence of 5% CO2. This step should be carried out for each experimental condition and negative control. NOTE: Increased EdC labeling can be achieved using HSV-1 mutants defective for the expression of the viral dUTPase (UL50 gene) and/or uracil glycosylase (UL2 gene). HSV-1 dUTPase has low substrate specificity and can decrease the activation of several nucleoside analogs25,27.
- Label viral genomes with EdC. Follow the directions to label incoming genomes (1.3.1.), replicating genomes (1.3.2.), or viral replication forks (1.3.3.). NOTE: Only Step 1.3.1, 1.3.2, or 1.3.3 should be carried out depending on whether incoming genomes, replicated genomes, or replication forks are to be analyzed in subsequent steps, respectively. NOTE: EdU or f-ara EdU can be substituted for EdC. The toxicity of nucleoside analogs should be monitored and minimized.
- Use prelabeled stocks to carry out infection in Step 1.2 and proceed to Step 2 or 3 within 4 h post infection (hpi) to investigate unreplicated vDNA (Figure 1A). NOTE: Prepare prelabeled virus stocks using the previously described protocol25. Virus stocks must be passed through a G-25 column to remove any residual EdC.
- Label replicating vDNA by adding 5 - 25 µM EdC to the cell culture medium of infected cells for 2 - 4 h (Figure 1B). Before adding, dilute EdC in 1 mL of growth medium.
- To label viral replication forks, after the onset of vDNA replication (≥4 hpi), pulse label replicating vDNA with 5 - 25 µM EdC for 5-20 min. To chase, rinse 3x with chase medium containing 25 - 100 µM 2´deoxycytidine (deoxyC), and then incubate in the presence of chase medium for an additional 20-40 min (Figure 1C). NOTE: Pulse and chase medium should be equilibrated to 37 °C and 5% CO2 before use. NOTE: For pulse chase experiments, it is important to consider the amount of time it takes for nucleosides to enter the cell and be phosphorylated before they can be incorporated into replicating DNA. This must be determined empirically. NOTE: To determine the resolution of pulse chase experiments, calculate the viral replication rate under the experimental conditions used. This can be determined by quantitative PCR of viral genomes during a single step growth time course26. NOTE: For imaging viral genomes proceed to Step 2 and for purification of viral genomes proceed to Step 3.
2. Imaging of EdC labeled DNA (Figure 2A)
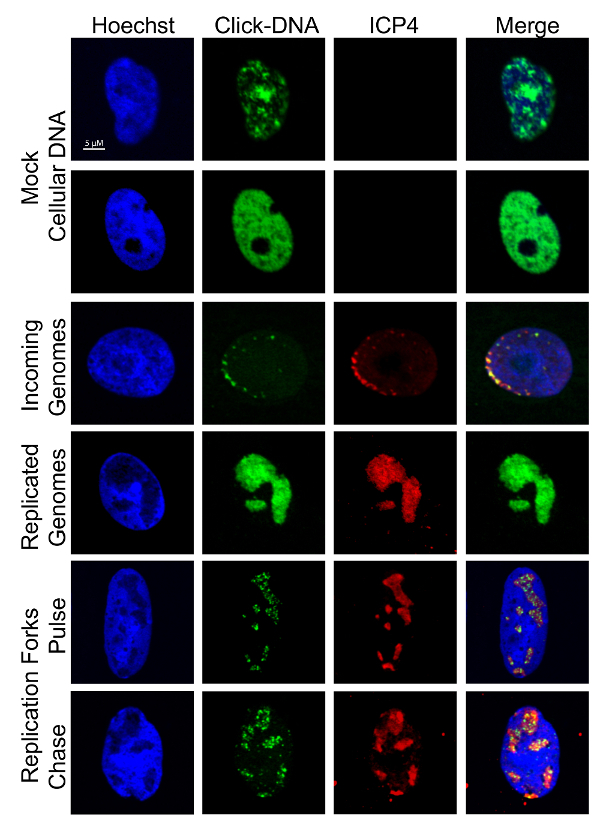
NOTE: It is useful to carry out imaging in tandem with the viral genome purification method to verify that cellular DNA is not labeled in experiments. Imaging can also be used to visualize the nature of labeled vDNA and to validate proteomics data. Examples of cellular and viral DNA staining patterns are shown in Figure 3.
Carry out infections and EdC labeling as indicated in Step 1 except using scaled down conditions on coverslips in a 12-well tissue culture dish containing 1 mL of growth medium.
Fix cells with 1 mL 3.7% paraformaldehyde in 1x PBS for 15 min at RT. After fixation, rinse cells 2x with 1 mL 3% BSA in PBS. Caution: Paraformaldehyde can cause serious or permanent injury. Follow safety precautions. NOTE: The protocol can be paused here and coverslips can be stored at 4 °C in the second wash for up to 3 days.
Permeabilize cells with 1 mL permeabilization buffer [see Table of Materials] for 20 min at room temperature while rocking.
Rinse with 1 mL of 3% BSA and then block with 1 mL 3% BSA in PBS for 30 min at room temperature while rocking.
Add 1 mL of the click reaction cocktail [see Table of Materials] to coverslips to covalently conjugate Alexa Fluor 488 azide to EdC labeled DNA. Incubate at room temperature for 30 min while rocking. Aspirate and rinse two times with 1 mL of PBS.
Stain cellular DNA with 1 mL of a 1:2,000 dilution of Hoechst in PBS for 30 min at room temperature while rocking. Aspirate and rinse two times with 1 mL of PBS.
Stain with primary and secondary antibodies according to standard indirect immunofluorescence protocols25. NOTE: Labeled vDNA colocalizes with ICP8, ICP4, or UL42 and labeled cellular DNA colocalizes with Hoechst stain.
Mount coverslips onto slides and image cells using a fluorescence microscope. NOTE: Slides can be stored at 4 °C for several weeks.
3. Purification of vDNA and Associated Proteins
NOTE: Several aspects of this protocol have been adapted from Leung et al.22 NOTE: All buffers and reagents should be chilled on ice before use and all steps should be carried out on ice unless otherwise indicated.
- Harvest nuclei. NOTE: Before isolation of nuclei, formaldehyde can be used to crosslink proteins to DNA. More stringent wash conditions can then be used during DNA purification on streptavidin-coated beads. For crosslinking and wash conditions see Sirbu et al.21.
- Replace growth medium with 20 mL Nuclei Extraction Buffer (NEB) and incubate for 20 min at 4 °C with occasional rocking. Nuclei will become visible in the microscope.
- Scrape nuclei from the plate using a cell scraper and transfer to a 50 mL conical tube. Centrifuge for 10 min at 2,500 x g at 4 °C to pellet nuclei, discard the supernatant. NOTE: Trypan blue staining can be used to verify the isolation of nuclei from cells.
- Gently dislodge the nuclear pellet in 10 mL PBS, transfer to a 15-mL conical tube, and pellet by centrifuging for 10 min at 2,500 x g at 4 °C. Completely remove PBS. NOTE: Nuclei can be frozen and the protocol can be paused at this point. To do so, gently resuspend the nuclear pellet in 500 µL freezing buffer and incubate the tube in an ethanol/dry ice bath. Store frozen nuclei at -80 °C. Before use, thaw nuclei at room temperature, quickly transfer to ice, add 10 mL PBS, rock gently to mix, pellet by centrifugation for 10 min at 2,500 x g at 4 °C, and completely remove PBS. Freezing nuclei may result in reduced yield.
- Covalently conjugate biotin-azide to EdC labeled vDNA.
- Resuspend the nuclear pellet in 10 mL click reaction mix [see Table of Materials] by gently pipetting up and down 5 times with a 10 mL pipet. NOTE: It is important that the reagents included in the click reaction mix are added in the indicated order.
- Rotate for 1h at 4 °C. While rotating prepare and chill Buffers B1, B2, and B3 [see Table of Materials].
- Pellet nuclei by centrifuging for 10 min at 2,500 x g at 4 °C. Completely remove click reaction mix and wash the pellet by gently resuspending in 10 mL PBS and centrifuging for 10 min at 2,500 x g at 4 °C.
- Gently resuspend the nuclear pellet in 1 mL PBS and transfer to a 1.5 mL microfuge tube using a large bore pipette tip. Pellet nuclei by centrifuging for 10 min at 2,500 x g at 4 °C. Completely remove PBS and flash freeze in liquid nitrogen. NOTE: The protocol can be paused at this point and frozen nuclei can be stored at -80 °C. Freeze thaw helps with subsequent lysis so it is recommended that nuclei be flash frozen even when proceeding to Step 3.3 on the same day.
- Lyse nuclei and fragment DNA.
- Thaw nuclei on ice and resuspend in 500 µL Buffer B1 by pipetting up and down. Incubate on ice for 45 min.
- Sonicate samples 6 times for 30 s each at 40% amplitude using a 3 mm microtip probe. Place samples on ice for 30 s between pulses. After sonication, samples should appear clear, not cloudy. NOTE: Sonication conditions should be optimized for individual sonicators. For detailed instructions on how to optimize sonication conditions refer to Leung et al.22.
- Pellet cell debris by centrifuging at 14,000 x g for 10 min at 4 °C. The pellet size should decrease substantially. Filter the supernatant through 100 µM cell strainer and retain the flow through.
- Add 500 µL Buffer B2 to the filtered supernatant. 900 µL of this sample will be used in Step 3.4.2.
- Take an aliquot of each condition for DNA isolation (50 µL or 1/20th volume) and add 50 µL 2x SDS-bicarb solution [see Table of Materials]. Proceed to the DNA isolation protocol (Step 3.6).
- Take an aliquot of each condition for input protein (50 µL or 1/20th volume) and add 50 µL 2x Laemmli sample buffer, freeze in liquid nitrogen and store at -80 °C. Use in Step 3.5.6.
- Bind biotinylated DNA to streptavidin coated beads.
- Prepare Streptavidin T1 magnetic beads by transferring 300 µL of bead slurry to a 1.5 mL microfuge tube. Prepare one tube of beads per sample. Wash beads 3x with 1 mL Buffer B2 by vortexing to resuspend, applying a magnet to separate the beads, and aspirating the wash buffer. NOTE: Optimized binding conditions result in maximized yield and minimal background binding. It was experimentally determined that Streptavidin T1 magnetic beads have a significantly greater binding capacity than agarose beads, as well as other Streptavidin beads, and less background binding than Streptavidin C1 beads (Figure 4A).
- Add 900 µL of sample from Step 3.3.4. to the washed beads and rotate overnight at 4 °C. Note: Do not vortex beads once sample is added to them. NOTE: If significant amounts of DNA or protein are isolated in the unlabeled negative control, this step can be reduced from overnight to 4 h to reduce background binding.
- Wash beads and elute vDNA and associated proteins. NOTE: It is recommended to use filter tips for the remaining steps.
- Place samples into a magnetic microfuge tube rack, remove supernatant, gently resuspend in 1 mL Buffer B2, rotate at 4 °C for 5 min.
- Repeat Step 3.5.1 three times.
- Place the samples into a magnetic microfuge tube rack, remove supernatant, gently resuspend in 1 mL Buffer B3, and rotate at 4 °C for 5 min.
- Transfer 100 µL bead mixture (1/10th volume) to a new tube for bound DNA isolation. Apply this tube to the magnet, remove supernatant, resuspend beads in 100 µL 1x SDS-bicarb solution and proceed to the DNA isolation protocol (Step 3.6).
- Apply the tube containing the remaining 900 µL bead mixture from Step 3.5.3. to the magnet, remove supernatant, and resuspend beads in 50 µL 2x Laemmli sample buffer to elute protein and DNA-protein complexes.
- Boil samples at 95 °C for 15 min, vortex, quickly spin in the microfuge, and apply magnet. Transfer eluate to a new tube, flash freeze, and store at -80 °C. NOTE: Use cap locks to ensure that tubes do not pop open while boiling. NOTE: The protocol can be paused at this point and samples can be stored at -80 °C for several weeks.
- Analyze protein samples by Coomassie Blue staining, western blotting, or mass spectrometry by standard procedures25. Representative results are shown in Figure 4 and Figure 5. NOTE: To determine if protein yield is sufficient for mass spectrometry, 7.5 µL of each sample is used for analysis by Coomassie Blue staining and 7.5 µL for western blotting of a representative viral genome associated protein (ICP4, ICP8, or UL42). The same volume of lysates from Step 3.3.6. are run alongside to control for input protein levels. The remaining sample (35 µL) is then analyzed by mass spectrometry as described previously25.
- DNA isolation
- Incubate the samples from Steps 3.3.5. and 3.5.4. at 65 °C for 4-16 h. Remove sample from magnetic beads, if necessary.
- Extract samples with 150 µL phenol:chloroform:isoamyl alcohol (PCI) and transfer the aqueous phase to a new tube.
- Extract the remaining PCI with 100 µL Tris-EDTA (TE) and add the aqueous phase to a new tube.
- Extract samples with 200 µL chloroform:isoamyl alcohol (24:1).
- Purify the aqueous phase using the PCR Purification Kit according to the manufacturer's protocol. NOTE: The pH indicator in Buffer PB will turn purple when added to the sample. Add 10 µL 3M NaOAc pH 5.0 to decrease the pH of the sample.
- Determine DNA concentration using a fluorometer. NOTE: This protocol typically yields 100 - 400 ng bead-bound DNA. No DNA should be detected in the eluate of the negative control in which EdC was not added in Step 1.3.
- DNA can be stored in a low DNA binding tube at 4 or -20 °C and can be used for real-time PCR or high throughput sequencing analysis.
Representative Results
The use of click chemistry for the purification of DNA from cells was first accomplished by the iPOND method21. The purpose of iPOND is to purify cellular replication forks for the identification of associated proteins. We have adapted this technique to specifically study vDNA protein interactions during infection. Manipulation of the approach to label viral genomes with EdC (Figure 1), combined with synchronized infections, has allowed for the selective isolation and investigation of separate populations of vDNA. HSV-1 DNA is labeled with EdC (Figure 1) to facilitate covalent attachment to a fluorophore for imaging or biotin for purification (Figure 2). Virus can be prelabeled before infection for the analysis of proteins associated with incoming genomes (Figure 1A) or labeled during DNA replication for the analysis of proteins associated with newly synthesized vDNA (Figure 1B)25. Furthermore, pulse chase analysis can be used to investigate the nature of proteins associated with viral replication forks (Figure 1C)26. DNA can be visualized within cells to provide spatial information about the nature of the labeled DNA and support for identified vDNA-protein interactions (Figure 3). Taken together, the protocols described here allow for the proteomic investigation of multiple aspects of productive infection to provide insight into dynamic changes that occur on HSV-1 genomes. These approaches could be further adapted to investigate latency and reactivation, to examine phenotypes associated with viral mutants, and to study other DNA and RNA viruses.
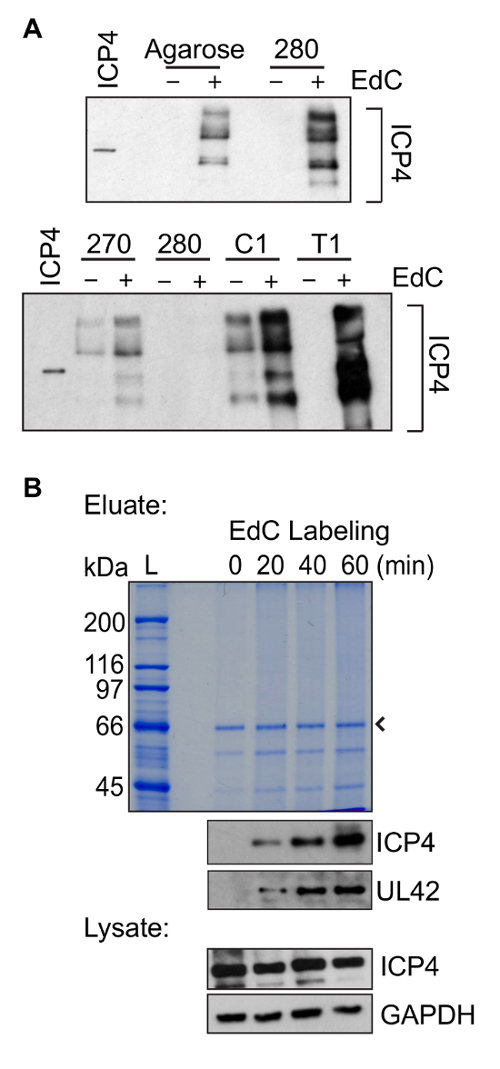
Aspects of this protein purification method were adapted from Leung et al.22. Here a major improvement for the purification of EdC labeled DNA is the use of Streptavidin T1 beads instead of streptavidin-coated agarose beads. To optimize DNA purification, several types of streptavidin-coated beads were tested for nonspecific binding when infection was carried out in the absence of EdC and for maximum protein recovery in the presence of EdC (Figure 4A). For these experiments, western blotting was carried out for the vDNA binding protein ICP4. Purifications carried out on Streptavidin T1 beads resulted in the least amount of background binding in the absence of EdC and the maximum protein recovery in the presence of EdC.
Proteins associated with vDNA can be identified by proteomic methods including western blotting and mass spectrometry. Western blotting can be used to investigate the dynamics of known interactions and mass spectrometry can be used as an unbiased approach to identify novel vDNA associated proteins. An example of protein yield as a function of increasing EdC labeling is shown in Figure 4B. A smear, rather than a clear banding pattern, is usually observed by Coomassie Blue staining. This is likely because there is DNA present in the protein sample or because there is an abundance of protein. It is also important to note that the copper catalyst used in the click reaction can cause partial degradation of protein and nucleic acids28,29. By western blotting, similar levels of ICP4 are typically detected in eluates (Step 3.5.5.) after purification of vDNA that was labeled for 60 min with EdC as that present in the same amount of input sample prepared from nuclear lystates (Step 3.3.6.). This information can be used as a guide to determine whether recovery is sufficient to send the remaining sample for mass spectrometric analysis.
Nano liquid chromatography with tandem mass spectrometry (LC-MS/MS) can be carried out as described previously25 to identify proteins associated with purified vDNA. LC-MS/MS data are analyzed by comparing spectral count (SpC) values between the EdC-labeled experimental sample and the corresponding unlabeled negative control. Proteins are considered to be enriched in the experimental sample based on the following criteria: 1) protein has at least 5 spectral counts (SpC) in the experimental sample, 2) protein is not detected in the control or is enriched over the control by at least four-fold based on dividing SpC values, and 3) the protein is enriched over the control in two or more biological replicates. The normalized spectral abundance factor (NSAF: ![]() ) can be used to account for differences in molecular weight (MW) and total protein yield. This equation determines the relative abundance of individual proteins within a sample and allows for the direct comparison of two different experimental conditions in which different amounts of vDNA are purified (for example, comparing input viral genomes (Figure 1A) to replicated genomes (Figure 1B)).
) can be used to account for differences in molecular weight (MW) and total protein yield. This equation determines the relative abundance of individual proteins within a sample and allows for the direct comparison of two different experimental conditions in which different amounts of vDNA are purified (for example, comparing input viral genomes (Figure 1A) to replicated genomes (Figure 1B)).
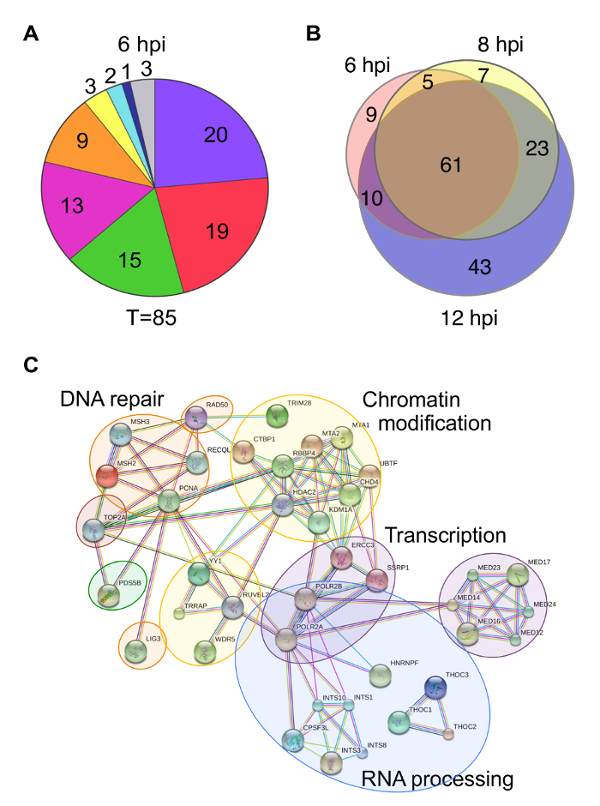
There are multiple ways to present protein enrichment data. Some examples include pie charts, Venn diagrams, and protein interaction networks. Pie charts can be used to illustrate the proportion of proteins identified that are known to function in a specific biological process (Figure 5A). However, problems can arise when one protein has more than one biological function, which is often the case. It is also difficult to compare data across different pie charts. Venn diagrams are useful to demonstrate relationships between proteins identified under different experimental conditions, but do not provide any functional information about the proteins identified (Figure 5B). Protein interaction networks portray a visual illustration of potential interactions between identified proteins and provide information about the types of biological processes that are involved (Figure 5C). Several online resources are available for mapping predicted protein-protein interactions including STRING (Search Tool for the Retrieval of Interacting Genes/proteins)30. Online tools are generally equipped to handle proteomics data from a variety of species and provide valuable information about host proteins involved in viral genome mechanics. However, viral proteins are generally not included in databases, which can complicate analysis. Therefore, it is important to consider the major conclusions one would like to convey when deciding how to present proteomics data.
Figure 1: Approaches to label HSV-1 DNA. (A) To assay the state of incoming virus, resting MRC-5 cells in G0 are infected with prelabeled HSV-1 and genomes are assayed at less than 4 hpi to study unreplicated viral DNA. (B) To assay the state of replicated virus, MRC-5 cells are infected with unlabeled HSV-1 and EdC (orange stars) is added to the growth medium of infected cells and incubated for 2 - 4 h after the onset of viral DNA replication (≥4 hpi). (C) To assay viral replication forks, infected cells are pulse labeled with EdC for 5-20 min. Replication forks can then be chased with deoxyC. EdC labeled DNA is orange. This figure has been modified from Dembowski and DeLuca25. Please click here to view a larger version of this figure.
Figure 2:Procedures described in this paper for analysis of EdC labeled DNA. (A) An outline of the method for imaging EdC labeled DNA. Tagged DNA is represented in green. (B) An outline of the method for the purification and downstream analysis of EdC labeled vDNA. This figure has been modified from Dembowski and DeLuca25. Please click here to view a larger version of this figure.
Figure 3: Visualization of EdC labeled DNA. Cells were infected, labeled, and imaged as described in Figure 1 andFigure 2. Representative images of labeled cellular and viral DNA are shown. Cellular DNA colocalizes with Hoechst stain and vDNA with ICP4. Scale bar: 5 µm.This figure was modified from Dembowski and DeLuca25 and Dembowski et al.26. Please click here to view a larger version of this figure.
Figure 4: Representative protein purification results. (A) Comparison of protein yield when purification steps were carried out using several different types of streptavidin coated beads. Infection was carried out in the presence of EdC (+) to compare protein yield or in the absence of EdC (-) to compare background binding. Infections and EdC labeling were carried out as described in Figure 1B and DNA-protein complexes were purified according to the steps outlined in Figure 2B using comparable amounts of different types of streptavidin coated beads. Top panel: Comparison of protein yield after purification on streptavidin coated agarose beads or Streptavidin M-280. Bottom panel: Comparison of protein yield after purification on Streptavidin M-270, M-280, Streptavidin C1, or Streptavidin T1. Western blotting was carried out with an antibody against the viral protein ICP4. Purified ICP4 is shown in the first lane for comparison. Longer exposure of the bottom panel was required to observe a similar intensity of ICP4 in lanes "280" as in the top panel. (B) Representative protein purification results as a function of time of EdC labeling are shown. Infections and EdC labeling were carried out as described in Figure 1B and DNA-protein complexes were purified according to the steps outlined in Figure 2B using Streptavidin T1 beads. EdC labeling was carried out for 0, 20, 40, or 60 min and Coomassie Blue staining (top) and western blotting (bottom) results are shown. Western blotting was carried out with antibodies specific for ICP4, UL42, or GAPDH. The eluate sample was taken from Step 3.5.5. and lysate from Step 3.3.6. The arrow indicates streptavidin, while L indicates protein ladder. Please click here to view a larger version of this figure.
Figure 5: Examples of different ways to present proteomics data. (A) Pie charts summarize proteins that were identified by mass spectrometry of protein eluates associated with viral genomes purified at 6 hpi. Values indicate the number of proteins identified for each functional category. T indicates the total number of proteins identified. Colors indicate the following categories: purple - RNA processing, red - transcription, green - chromatin remodeling, orange - DNA replication, yellow - nuclear transport, teal - cytoskeleton, dark blue - HSV structural proteins, and gray - other/unknown. (B) Venn diagrams depict the overlap of proteins identified to be associated with viral genomes at 6, 8, or 12 hpi. (C) A STRING map depicts proteins enriched on viral replication forks after a 5 min EdC pulse. Human proteins enriched by 5-fold compared to the unlabeled negative control are shown in the functional interaction map, which was generated using STRING30 with data settings to display only high confidence interactions. Gene names were used to map interactions. Circles indicate proteins that function in the same biological process. This figure was modified from Dembowski and DeLuca25 and Dembowski et al.26. Please click here to view a larger version of this figure.
Discussion
This protocol includes multiple steps which, if not followed carefully, can result in significantly reduced protein yield or contamination with cellular DNA. It is critical that stationary cells are used for all experiments to ensure that cellular DNA is not labeled and purified. This can be confirmed by the absence of cellular DNA polymerases in the protein sample because HSV-1 does not utilize cellular DNA polymerase for genome synthesis. During EdC labeling and nuclei harvesting steps, samples should be staggered to ensure that each is processed equally. During purification steps, care should be taken not to manipulate nuclei by pipetting too much. This can lead to premature lysis of nuclei, as well as sample loss due to sticking to the sides of pipet tips. Furthermore, the sonication step should be optimized for individual sonicators and the amount of streptavidin-coated beads used for biotin binding should be titrated for optimal protein yield.
Several modifications can be made to this protocol and include the use of different viruses or cell types or the addition of a step to crosslink protein to DNA. When choosing a cell type to use for proteomic investigation, it is important to check for the availability of a protein sequence database for that species. A lack of proteomics resources will significantly limit downstream analysis of protein data. Another limitation is the need for a large number of cells, therefore it may be difficult to prepare enough primary cells, such as neurons, for this experiment. This method should be compatible with other DNA or RNA viruses as long as the viral infection does not depend on cellular DNA replication. Furthermore, it would be interesting to explore changes in DNA-protein interactions that occur in HSV-1 mutants. Another modification to this protocol could be the addition of a formaldehyde crosslinking step before isolation of nuclei, which would allow for more stringent wash conditions during purification21. The addition of formaldehyde will not affect the ability to harvest nuclei but may result in decreased protein yield because of difficultly reversing crosslinks during protein elution.
Results from this protocol represent the average state of viral DNA being examined. Therefore, it is difficult to distinguish if complexes are together on the same or separate DNA molecules. vDNA labeling methods described here in combination with high resolution imaging can be used to verify proteomics data and may help to distinguish between different populations of labeled DNA within a cell. Furthermore, ChIP-seq could be used as a follow up method to identify the specific locations of proteins on the viral genome.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We acknowledge Hannah Fox for help in the preparation of this manuscript. This work was supported by the NIH grant R01AI030612.
References
- Knipe DM, Howley PM. Fields virology. 6th. Wolters Kluwer/Lippincott Williams & Wilkins Health; 2013. [Google Scholar]
- Engel EA, Song R, Koyuncu OO, Enquist LW. Investigating the biology of alpha herpesviruses with MS-based proteomics. Proteomics. 2015;15:1943–1956. doi: 10.1002/pmic.201400604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner LM, DeLuca NA. Temporal association of herpes simplex virus ICP4 with cellular complexes functioning at multiple steps in PolII transcription. PloS One. 2013;8:e78242. doi: 10.1371/journal.pone.0078242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TJ, Knipe DM. Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J Virol. 2004;78:5856–5866. doi: 10.1128/JVI.78.11.5856-5866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine-Rodriguez EC, Taylor TJ, Olesky M, Knipe DM. Proteomics of herpes simplex virus infected cell protein 27: association with translation initiation factors. Virology. 2004;330:487–492. doi: 10.1016/j.virol.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Greco TM, Diner BA, Cristea IM. The Impact of Mass Spectrometry-Based Proteomics on Fundamental Discoveries in Virology. Annu Rev Virol. 2014;1:581–604. doi: 10.1146/annurev-virology-031413-085527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conwell SE, White AE, Harper JW, Knipe DM. Identification of TRIM27 as a novel degradation target of herpes simplex virus 1 ICP0. J Virol. 2015;89:220–229. doi: 10.1128/JVI.02635-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suk H, Knipe DM. Proteomic analysis of the herpes simplex virus 1 virion protein 16 transactivator protein in infected cells. Proteomics. 2015;15:1957–1967. doi: 10.1002/pmic.201500020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian N, Bai P, Buchek G, Korza G, Weller SK. Physical interaction between the herpes simplex virus type 1 exonuclease, UL12, and the DNA double-strand break-sensing MRN complex. J Virol. 2010;84:12504–12514. doi: 10.1128/JVI.01506-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath P, Deluca NA. Binding of ICP4, TATA-binding protein, and RNA polymerase II to herpes simplex virus type 1 immediate-early, early, and late promoters in virus-infected cells. J Virol. 2008;82:2339–2349. doi: 10.1128/JVI.02459-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio AL, McAnany PK, Bloom DC. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J Virol. 2006;80:2358–2368. doi: 10.1128/JVI.80.5.2358-2368.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Knipe DM. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J Virol. 2008;82:12030–12038. doi: 10.1128/JVI.01575-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera FJ, Triezenberg SJ. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J Virol. 2004;78:9689–9696. doi: 10.1128/JVI.78.18.9689-9696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent JR, et al. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J Virol. 2004;78:10178–10186. doi: 10.1128/JVI.78.18.10178-10186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Fraser NW. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J Virol. 2008;82:3530–3537. doi: 10.1128/JVI.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catez F, et al. HSV-1 genome subnuclear positioning and associations with host-cell PML-NBs and centromeres regulate LAT locus transcription during latency in neurons. PLoS Pathog. 2012;8:e1002852. doi: 10.1371/journal.ppat.1002852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Murray J, Orr A, Preston CM. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J Virol. 2007;81:10991–11004. doi: 10.1128/JVI.00705-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, et al. Determination of minimum herpes simplex virus type 1 components necessary to localize transcriptionally active DNA to ND10. J Virol. 2003;77:5821–5828. doi: 10.1128/JVI.77.10.5821-5828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishov AM, Maul GG. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J Cell Biol. 1996;134:815–826. doi: 10.1083/jcb.134.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroui MA, et al. Latency Entry of Herpes Simplex Virus 1 Is Determined by the Interaction of Its Genome with the Nuclear Environment. PLoS Pathog. 2016;12:e1005834. doi: 10.1371/journal.ppat.1005834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Cortez D. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat Protoc. 2012;7:594–605. doi: 10.1038/nprot.2012.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KH, Abou El Hassan M, Bremner R. A rapid and efficient method to purify proteins at replication forks under native conditions. BioTechniques. 2013;55:204–206. doi: 10.2144/000114089. [DOI] [PubMed] [Google Scholar]
- Hobbs WE, 2nd, DeLuca NA. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J Virol. 1999;73:8245–8255. doi: 10.1128/jvi.73.10.8245-8255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonte P, Everett RD. Herpes simplex virus type 1 immediate-early protein Vmw110 inhibits progression of cells through mitosis and from G(1) into S phase of the cell cycle. J Virol. 1999;73:9456–9467. doi: 10.1128/jvi.73.11.9456-9467.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembowski JA, DeLuca NA. Selective recruitment of nuclear factors to productively replicating herpes simplex virus genomes. PLoS Pathog. 2015;11:e1004939. doi: 10.1371/journal.ppat.1004939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembowski JA, Dremel SE, DeLuca NA. Replication-Coupled Recruitment of Viral and Cellular Factors to Herpes Simplex Virus Type 1 Replication Forks for the Maintenance and Expression of Viral Genomes. PLoS Pathog. 2017;13:e1006166. doi: 10.1371/journal.ppat.1006166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornberg O, Nyman PO. The dUTPases from herpes simplex virus type 1 and mouse mammary tumour virus are less specific than the Escherichia coli enzyme. J Gen Virol. 1996;77 (Pt 12):3107–3111. doi: 10.1099/0022-1317-77-12-3107. [DOI] [PubMed] [Google Scholar]
- Chiou SH. DNA- and protein-scission activities of ascorbate in the presence of copper ion and a copper-peptide complex. J Biochem. 1983;94:1259–1267. doi: 10.1093/oxfordjournals.jbchem.a134471. [DOI] [PubMed] [Google Scholar]
- Kennedy DC, et al. Cellular consequences of copper complexes used to catalyze bioorthogonal click reactions. J Am Chem Soc. 2011;133:17993–18001. doi: 10.1021/ja2083027. [DOI] [PubMed] [Google Scholar]
- Snel B, Lehmann G, Bork P, Huynen MA. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000;28:3442–3444. doi: 10.1093/nar/28.18.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]