

Abstract
Background
Multiple system atrophy (MSA) is marked by abnormal inclusions of α‐synuclein in oligodendrogliocytes, and its etiology remains unknown. Variants in the glucocerebrosidase (GBA) gene have been associated with the other synucleinopathies, dementia with Lewy bodies, and Parkinson's disease. It is unclear whether glucocerebrosidase variants are associated with MSA. The objective of this study was to analyze the frequency of GBA variants among patients who had autopsy‐proven MSA at a brain bank in New York City.
Methods
GBA was fully sequenced in brain tissues from 17 patients with autopsy‐proven MSA for whom there was extractable DNA at the Columbia University New York Brain Bank from 2002 to 2016. To test whether brains from patients with MSA were enriched for GBA variants, the frequency of GBA variants in the MSA brains was compared with that of variants in all brains from patients with pure Alzheimer's disease (AD) at Columbia University for which GBA genotype was available (n = 82).
Results
Of 17 MSA brains, 4 carried GBA variants (23.5%), including 1 that was homozygous for N370S and 1 each that carried heterozygous N370S, T369M, and R496H variants. Among the comparator brains with pure AD, 3 of 82 (3.7%) carried GBA variants (P = 0.0127; Fisher exact test), including 1 each with an N370S homozygous variant, an R496H heterozygous variant, and a T369M heterozygous variant.
Conclusion
A higher frequency of GBA variants was observed among brains from patients who had pathologically diagnosed MSA compared with the frequency of variants in brains from patients who had AD. The results indicate a need for further investigation into the role of glucocerebrosidase and lysosomal dysfunction in the etiology of MSA.
Keywords: Genetics, glucocerebrosidase (GBA), multiple system atrophy (MSA), neuropathologic
Multiple system atrophy (MSA) is an adult‐onset, progressive, neurodegenerative disease characterized by parkinsonism, cerebellar ataxia, autonomic dysfunction, and corticospinal system dysfunction. Neuropathologically, there are α‐synuclein accumulations in oligodendroglial cells that form glial cytoplasmic inclusions.
One proposed mechanism for the development of MSA is disturbance of the elimination pathway for α‐synuclein. Intracellular α‐synuclein can be degraded by either the ubiquitin proteasome system or by autophagy, which is the lysosomal pathway for degrading proteins and organelles. Several studies have demonstrated alteration of the ubiquitin proteasome system and autophagy pathways in models of MSA.1, 2, 3, 4
Glucocerebrosidase, which is encoded by the glucocerebrosidase gene (GBA), is an enzyme involved in the lysosomal degradation of sphingolipids.5 Homozygous mutations of this gene result in Gaucher's disease (GD), which is a rare lysosomal storage disease. Although GD is present across all ethnicities6, it is much more frequent in the Ashkenazi Jewish population.7 People diagnosed with the α‐synucleinopathies Parkinson disease (PD) and dementia with Lewy bodies (DLB) are from 5‐fold to 10‐fold8 and 8‐fold9 more likely, respectively, to carry GBA variants than healthy controls. The prevalence of GBA variants in Alzheimer's disease (AD) has been identified as similar to the prevalence in controls.10 Studies of the association between GBA variants and MSA have been inconclusive.11, 12, 13, 14, 15, 16, 17
In a recent, large study12 that included 969 patients with MSA and 1509 healthy controls from Japan, Europe, and North America, significantly increased odds for the presence of GBA variants were reported among patients who had MSA compared with controls. The strongest association was in the North American cohort, in which the GBA variant prevalence was 2.91% among patients who had MSA compared with 0.34% among controls. Although that study was the best powered, it lacked pathologic confirmation of most of the MSA diagnoses, and it did not report on Jewish ancestry. The objectives of the current study were to assess the prevalence of GBA variants in MSA brains (n = 17) and to compare the variant frequency in MSA brains with that in AD brains (n = 82) collected in the same brain bank. Because there are roughly 1.1 million people who identify as Jewish in New York City alone,18 we were able to investigate the prevalence of GBA variants among pathologically confirmed MSA and AD brains in a population enriched with Jewish people.
Patients and Methods
MSA Brains
All patients had pathologically proven MSA between 2002 and 2016 and had been evaluated at the Columbia Movement Disorders Center at least once before death. Clinical information was extracted from clinical charts and, when available, interview with surviving family members. These findings are summarized in Table 1. Of the 40 MSA brains in the Columbia University brain bank, 17 had tissue available for DNA extraction and GBA gene sequencing. The other 24 brains were autopsied before 2002 or had no frozen tissue available for DNA extraction and reliable sequencing.
Table 1.
Clinical characteristics of all patients who had multiple system atrophy and Alzheimer's disease comparing those with and without glucocerebrosidase gene mutations
| MSA | AD | |||
|---|---|---|---|---|
| Characteristic | Patients with ≥1 GBA mutation, n = 4a | Patients without GBA mutations, n = 13 | Patients with ≥1 GBA mutation, n = 3a | Patients without GBA mutations, n = 79 |
| Age at death: Mean ± SD [range] | 63 ± 2.83 [61–67] | 68.46 ± 8.98 [47–79] | 80. 52 ± 5.68 [77–87] | 81.13 ± 11.15 [49–97] |
| Men: Percentage (no.) | 50 (2) | 15.4 (2) | 33.3 (1) | 43.0 (34) |
| Ashkenazi Jewish descent: Percentage (no.) | 75 (3) | 23.1 (3)b | 33.3 (1) | 17.7 (14) |
| Age of MSA onset: Mean ± SD [range], y | 55 ± 3.2 [52–59] | 61.30 ± 10.14 [41–75] | NA | NA |
| MSA symptom duration: Mean ± SD [range], y | 8 ± 0.8 [7–9] | 7.15 ± 3.31 [3–13] | NA | NA |
| Clinical variant: Percentage (no.) | ||||
| MSC‐C | 50 (2) | 46.2 (6) | NA | NA |
| MSA‐P | 50 (2) | 53.8 (7) | NA | NA |
MSA, multiple system atrophy; AD, Alzheimer's disease; GBA, glucocerebrosidase gene; SD, standard deviation; NA, not applicable; MSA‐C, multiple system atrophy, cerebellar variant; MSA‐P, multiple system atrophy, parkinsonian variant.
One of these patients had a homozygous N370S mutation.
Ethnicity was determined by self‐report in the clinical chart. Three patients had unknown ethnicity, all among non‐GBA carriers, 1 due to adoption.
DNA Extraction and GBA Gene Sequencing
Genomic DNA (50–80 ng) was extracted from frozen brain tissue after proteinase K digestion, phenol chloroform extraction, and ethanol precipitation. Polymerase chain reaction (PCR) was used to amplify fragments suitable for Sanger sequencing using either long‐range PCR or exon‐specific PCR. Most DNA samples were amplified for a 7036 base pair fragment using PrimeSTAR GXL DNA Polymerase (Takara Bio, Mountain View, CA). The 7036 base pair PCR fragment ensured that all GBA exons could be sequenced from 1 PCR fragment without interference from the GBA pseudogene. Samples from a smaller subset of patients for which long‐range PCR was not possible underwent PCR amplification with FastStart Taq DNA polymerase (Roche Diagnostics, Indianapolis, IN) for each exon of GBA. Primer sequences for the single‐exon fragments were also chosen to avoid amplification of the GBA pseudogene and are available upon request. After PCR amplification, both long‐range and exon‐specific PCR fragments were cleaned enzymatically with ExoSAP‐IT (Affymetrix, Santa Clara, CA) according to the manufacturer's instructions. Samples were sequenced in both the forward and reverse directions for the full coding sequence of GBA using ABI BigDye Terminator chemistry (version 1.1; Applied Biosystems by ThermoFisher Scientific, Waltham, MA) and were visualized on an ABI 3730xl DNA analyzer. ABI sequencer files for each sample were compared with a GBA consensus sequence to identify variants using Mutation Surveyor software (SoftGenetics, State College, PA). We also attempted to perform DNA extraction from brains that had only formalin‐fixed tissue banked but were unable to produce DNA for sequencing from those brains.
AD Brains
To compare GBA variant frequency between MSA brains and nonsynucleinopathy‐related brains, we used the brain bank at Columbia University for comparator cases. Because of the rarity of “controls” without neuropathology in this age range, we chose as comparators all brains with a primary neuropathologic diagnosis of AD between 2002 and 2016 that had no secondary diagnosis other than ischemic change or stroke (in particular, with no Lewy body pathology) but that had GBA genotype information available. These brains were collected under the Alzheimer's Disease Research Center protocol, and the genotyping method as well as GBA variants were previously reported.19 Demographic and clinical information, including Ashkenazi Jewish ancestry, were extracted from clinical charts and interviews with surviving family members and treating clinicians. In total, 82 AD brains were included in this study.
Statistical Analysis
Descriptive statistics (mean, standard deviation, and range) are reported for the continuous variables age of onset, age of death, and disease duration. Absolute values with percentages are reported for dichotomous variables. Fisher exact tests were performed to determine statistical significance.
Results
Clinical phenotypes of the MSA brains included 9 with an MSA‐parkinsonian type (MSA‐P) and 8 with an MSA‐cerebellar type (MSA‐C). The mean ± standard deviation age of disease onset in all patients with MSA was 59.94 ± 9.3 years (range, 41–75 years), and the average age at death was 66.8 ± 8.1 years (range, 47–79 years). The average age at death of patients with AD was 81.1 ± 11.0 years (range, 49–97 years). The average duration of disease for patients with MSA from symptom onset to death was 7.6 ± 3.3 years (range, 3–13 years). Among the patients with MSA, there were 6 of Ashkenazi Jewish descent (35.3%), 7 of non‐Jewish Caucasian European descent (41.2%), 1 of Japanese descent (5.9%), and 3 of unknown non‐Jewish ethnicity (17.6%). Among those patients with AD, there were 15 of Ashkenazi Jewish descent (18.3%).
Of the 17 MSA brains tested, 4 (23.5%) had at least 1 known GBA variant, whereas 3 (3.7%) of the 82 AD brains tested had GBA variants (P = 0.0127). The 4 MSA brains with GBA variants are described below. One patient with MSA (Case 4) was homozygous for the N370S mutation but was not diagnosed with GD when alive. The other 3 patients with MSA were heterozygous carriers of either T369M, N370S, or R496H. One patient with AD was homozygous for N370S. The other 2 patients who had AD with variants included 1 T396M heterozygote and 1 R463C heterozygote.
Of the 6 patients with MSA who were of Ashkenazi Jewish descent, as self‐identified in clinical charts, 3 carried GBA variants (50%; 2 with N370S and 1 with R496H), and 3 did not. Only 1 of 15 patients with AD who were of Ashkenazi Jewish descent harbored GBA variants (6.7%) compared with 50% of Jewish patients with MSA (P = 0.0175). Among non‐Jewish patients, 1 of 11 (9.1%) in the MSA cohort and 2 of 67 (3.0%) in the AD cohort had GBA variants (P = 0.46). Table 1 compares clinical characteristics between the patients who had MSA and AD with or without GBA variants, and Table 2 describes the clinical characteristics of patients who had MSA with GBA variants. Figure 1 demonstrates representative pathology from MSA cases with GBA variants.
Table 2.
Clinical and pathologic characteristics of patients who had multiple system atrophy with glucocerebrosidase gene mutations
| Characteristic | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Age at onset, y | 52 | 56 | 59 | 53 |
| Symptom duration, y | 9 | 11 | 8 | 8 |
| Age at death, y | 61 | 63 | 67 | 61 |
| Premortem diagnosis | MSA | MSA | MSA | MSA |
| Family history of neurologic disease | No | Yesa | No | No |
| Ashkenazi Jewish descent | No | Yes | Yes | Yes |
| Genetic results | Heterozygous T369M | Homozygous N370S | Heterozygous N370S | Heterozygous R496H |
| Signs/symptoms of autonomic dysfunction | Orthostasis, urinary symptoms, constipation, erectile dysfunction | Orthostasis, urinary symptoms, constipation | Orthostasis, urinary symptoms, constipation | Orthostasis, urinary symptoms, constipation |
| Early falls (<1 y) | No | No | No | Yes |
| Cognitive signs/symptoms | Yes | Yes | Yes | No |
| RBD | Yes | Yes | No | Yes |
| Signs of parkinsonism | Yes | Yes | Yes | Yes |
| Cerebellar signs | No | No | Yes | Yes |
| Pyramidal signs | No | Yes | Yes | No |
| Pathological diagnosis | MSA‐P | MSA‐P | MSA, mixed | MSA‐C |
| Lewy bodies | None | None | Rare | None |
| Alzheimer disease changes | Tau‐positive neurons and neuropil threads in entorhinal cortex and parahippocampal gyrus | Rare neurofibrillary tangles in entorhinal cortex | Very few neuronal pretangles in amygdala | Rare p‐Tau–labeled neurons and neuropil threads in temporal lobe |
MSA, multiple system atrophy; RBD, rapid eye movement behavior disorder; MSA‐P, multiple system atrophy, parkinsonian variant; MSA‐C, multiple system atrophy, cerebellar variant.
The mother of this patient had a head tremor, and the father had bilateral hand tremors; and 1 paternal uncle and 1 paternal cousin had Parkinson's disease.
Figure 1.
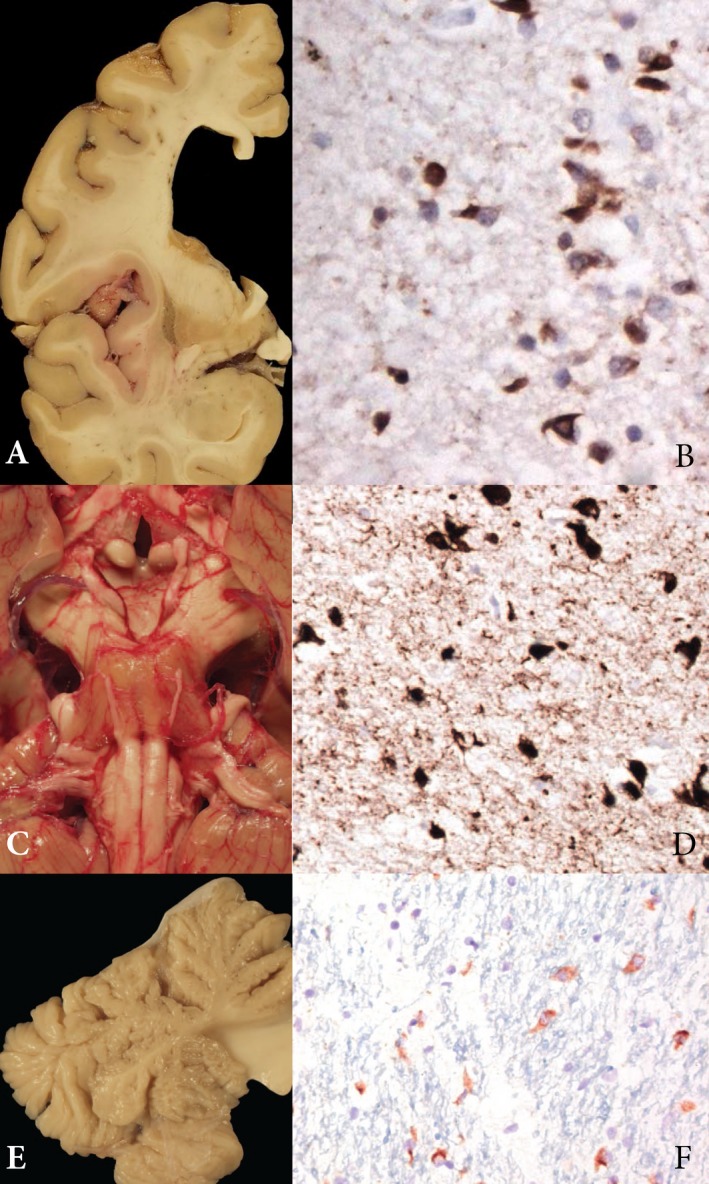
Representative pathology from multiple system atrophy brains with glucocerebrosidase gene (GBA) mutations. (A) Atrophic changes are observed in the lentiform nuclei in Patient 1. (B) α‐Synuclein staining reveals glial cytoplasmic inclusions (GCIs) in the putamen in Patient 1 (original magnification ×630). (C) Severe pontine demyelination and atrophy are observed in Patient 3. (D) Ubiquitin stain reveals abundant GCIs in Patient 3 (original magnification ×400). (E) Cerebellar vermis demonstrates severe atrophy in Patient 4. (F) α‐Synuclein stain reveals abundant GCIs in the cerebellum in Patient 4 (original magnification ×400).
Cases
MSA Case 1
The patient was of western European descent and had no family history of parkinsonism or cerebellar ataxia. Symptoms began at age 52 years with right hand rest tremor, slowness, stiffness, “shuffling” gait with stooped posture, and reduced arm swing. There was no benefit from levodopa (l‐dopa) or dopamine agonists. The patient developed micrographia, falls, freezing of gait, urinary retention requiring intermittent self‐catheterization, sexual dysfunction, orthostatic hypotension, constipation, drooling, dysphagia, rapid eye movement sleep behavior disorder (RBD), and cognitive complaints. The initial diagnosis was normal‐pressure hydrocephalus, and the patient underwent ventriculoperitoneal shunt placement without improvement. On examination at 4 years after onset, the Modified Mini‐Mental State Examination score was 53 of 57,19 and there were square wave jerks without nystagmus, normal reflexes, hypomimia, and symmetric bradykinesia and rigidity with myoclonus. Gait examination revealed stooped posture, difficulty rising from chair, short strides, en‐bloc turning, mild postural instability, and absent arm swing. A brain magnetic resonance imaging (MRI) study demonstrated dilated ventricles and periventricular white matter disease. an 18F‐fluorodeoxyglucose‐positron emission tomography scan demonstrated bilateral reductions in striatal glucose metabolism consistent with an atypical parkinsonian syndrome. The patient died at age 61 years after a 9‐year disease course. Gross pathologic examination revealed mild atherosclerotic disease in the large blood vessels of the brain and severe atrophy in the putamen and caudate. Microscopic evaluation revealed scattered glial cytoplasmic inclusions (GCIs) throughout the cerebral cortex, red nucleus, and basal ganglia. Occasional neurons were labeled with anti‐synuclein antibodies without Lewy bodies. There were τ‐positive staining neurons and neuropils in the temporal lobe, but no neuritic plaques. These findings are consistent with the diagnosis of MSA of the striatonigral type. GBA genotyping revealed a heterozygous T369M variant.
MSA Case 2
The patient was of Ashkenazi Jewish descent and had a family history significant for a mother with life‐long head tremor, a father with bilateral hand tremors, and a paternal uncle and paternal cousin with PD. Symptoms started at age 56 years with hoarse voice and urinary urgency and incontinence. One year later, the patient developed stiffness and discomfort in the left arm, clumsiness in the left hand, breathlessness and stridor, dysphagia, RBD, and balance difficulties with falls. The patient reported constipation, lightheadedness with standing, and cognitive changes without hallucinations. l‐Dopa had a modest effect. On examination 1 year after symptom onset, there was an orthostatic blood pressure drop, and mental status was normal. Movement disorders examination revealed hypometric saccades, hypomimia, no tremor, no dysmetria, and asymmetric bradykinesia. Reflexes were brisk, with equivocal plantar responses. Gait examination revealed stooped posture, normal base, and reduced arm swing. MRI of the brain demonstrated cerebellar atrophy. The patient later developed neck dystonia and postural instability and was wheelchair‐bound 4 years after onset. The patient died 7 years after onset of symptoms. Gross pathologic examination revealed mild global atrophy with marked atrophy of the putamen, globus pallidus, and cerebellum. Microscopic examination revealed scattered GCIs and rare neuronal inclusions, but no Lewy bodies. There was neuronal loss and gliosis throughout the basal ganglia and cerebellum. Rare neurofibrillary tangles were found in the entorhinal cortex without other Alzheimer's pathology. These findings were consistent with a pathologic diagnosis of MSA, mixed type with features of both olivopontocerebellar atrophy and striatonigral degeneration. GBA genotyping revealed a homozygous N370S mutation. The patient had known anemia and Factor VIII and XI deficiency, although GD was not diagnosed during life.
MSA Case 3
The patient was of Ashkenazi Jewish descent and had no family history of movement abnormalities. Symptoms started at age 59 years with balance problems, falls, and a change in golf swing. The patient developed speech changes, reflex blepharospasm, and difficulty with handwriting and typing. By 5 years into the disease, the patient required a walker and 24‐hour assistance. The patient developed constipation, dysphagia, stridor, behavioral symptoms (agitation/anger), severe urinary retention requiring percutaneous catheter, kinetic tremor, painful leg dystonia, and anterocollis. There was no significant response to levodopa. Brain MRI demonstrated cerebellar atrophy with nonspecific T2/fluid‐attenuated inversion recovery (FLAIR) hyperintensities in the pons and cerebellar peduncles. On examination 5 years after onset, the patient had an asymptomatic orthostatic blood pressure drop, normal mental status examination, no nystagmus, absent optokinetic nystagmus, saccadic breakdown of smooth pursuit, hypermetric saccades, and brisk reflexes. There was prominent dysmetria in all extremities, and speech was nasal and scanning. There was hypomimia, bradykinesia on the right, and truncal titubation. The patient was unable to rise from a chair without using the arms. Gait was broad‐based and ataxic, with spontaneous falling. The patient died after a disease duration of 8 years. Gross pathologic examination revealed marked atrophy of the putamen and globus pallidus externa, brainstem, and cerebellum and pallor of the substantia nigra and nucleus coeruleus. On microscopic examination, there were diffuse GCIs in the cerebrum, brainstem, cerebellum, striatum, hypothalamus, and olfactory tract and bulb. There were very few τ‐positive pretangles in the amygdala, without evidence of other Alzheimer's pathology. These pathologic findings were consistent with the diagnosis of MSA, with mixed striatonigral and olivopontocerebellar type. GBA genotyping revealed a heterozygous N370S mutation.
MSA Case 4
The patient was of Ashkenazi Jewish descent with no family history of neurologic disorders. Symptoms began at age 53 years with unsteady gait and falls. Other complaints included numbness in the feet and tingling in the right hand, depression and anxiety, slurred speech, dysphagia, constipation, difficulty with manual dexterity, diplopia, right‐sided ptosis and reflex blepharospasm, RBD, urinary frequency and urgency, and lightheadedness, which later progressed to severe orthostasis with syncope. Brain MRI demonstrated hyperintense FLAIR signal in the cerebellum and cerebellar peduncle. The patient required a walker early and a wheelchair by 5 years. On examination 1 year after onset, the patient experienced a modest orthostatic blood pressure drop, dysarthria, saccadic breakdown of smooth pursuit, and nystagmus. There was dysmetria in the extremities, reduced sensation in the feet, an ataxic gait, and hyperreflexia bilaterally. The patient later developed hypomimia, hypophonia, and bradykinesia on examination. Eight years after symptom onset, the patient developed recurrent abdominal discomfort with vomiting and soon died. Gross pathologic examination demonstrated scattered atherosclerotic plaques and mild, diffuse cerebral atrophy. The brainstem and cerebellum were markedly atrophic. On microscopic examination, GCIs were found within the cortex, subcortical white matter, basal ganglia, pons, and cerebellum. The subcortical white matter showed areas that were pale and vacuolated, which labeled with antibodies against amyloid, likely secondary to a terminal hypoxic‐ischemic episode. There was subtotal loss of the pontine nuclei, myelin of the pontocerebellar fibers, olivocerebellar fibers, and the middle cerebellar peduncle. There were rare, phosphorylated τ‐labeled neurons and neuropil threads in the temporal lobe. It was believed that these findings were consistent with MSA, predominantly the olivopontocerebellar type. GBA sequencing revealed a heterozygous R496H mutation.
Discussion
In this study, we found a higher proportion of GBA variants in MSA brains at the brain bank at Columbia University (23.5%) compared with the proportion among AD brains (3.7%). Seven previous studies have examined the association between GBA variants and MSA11, 12, 13, 14, 15, 16, 17 (Table 3).11, 12, 13, 14, 15, 16, 17, 20 Six studies found no association between GBA variants and MSA. Some of those studies were limited by small sample sizes, lack of pathologic confirmation of MSA diagnosis, methods of genetic testing (full sequencing of GBA vs. genotyping 1 or 2 known mutations), and/or lack of inclusion of ethnic groups known to have a high prevalence of GBA variants (i.e., Ashkenazi Jewish populations) (Table 3). However, the largest clinical study to date identified an association between GBA variants and MSA.
Table 3.
Summary of studies previously reporting no association between multiple system atrophy and glucocerebrosidase gene variants
| Variable | Goker‐Alpan et al., 200617 | Segarane et al., 200914 | Nishioka et al., 201113 | Jamrozik et al., 201011 | Sun et al., 201316 | Srulijes et al., 201315 | Mitsui et al., 201512 |
|---|---|---|---|---|---|---|---|
| Pathologically confirmed? | Yes | Yes | Yes | No | No | No | No |
| No. of patients with MSA | 12 | 108 | 27 | 65 | 54 | 344 | 969 |
| No. of GBA mutation carriers among patients with MSA (%) | 0 (0) | 1 (0.92) | 0 (0) | 0 (0) | 0 (0) | 2 (0.006) with N370S; 4 (0.58) had a nonpathologic variant | 17 (1.8); 2 had homozygous N370S mutations |
| Percentage of GBA mutation carriers among controls | 0.006 Based on previously published data | 1.17 | NA | 0.3 Based on previously published data | 0.0015 | 0 of 325 healthy controls | 0.7 of 1509 healthy controls |
| Method | Whole GBA gene sequencing | Whole GBA gene sequencing | Whole GBA gene sequencing | p.L444p and p.N370S only | p.L444p | p.L444p and p.N370S in 295 with MSA and all controls; whole GBA in 49 with MSA | Whole GBA gene sequencing |
| Country of origin | US | UK | US, Canada, UK | Poland | China | European, no Ashkenazi Jews | Japan, Germany, Italy, US |
MSA, multiple system atrophy; GBA, glucocerebrosidase gene; NA, not applicable.
There are several published reports from this brain bank regarding the frequency of GBA variants in other groups. A previously published study from the Columbia University brain bank reported GBA variants in 6 of 60 patients (10%) who had primary AD pathology (although 2 of those 6 patients had at least some trace of synuclein pathology) and in 1 of 32 (3%) of those without AD or Lewy body pathology, which is significantly lower than the GBA frequency in our MSA cohort (P = 0.0432; Fisher exact test).19 The population allele frequency us 0.0003 for R496H, 0.002210 for N370S, and 0.006571 for T369M.21 Among Ashkenazi Jews, the allele frequencies for R496H and N370S were reportedly 0.0048 and 0.0515, respectively.22
The frequency of GBA variants among Jewish controls in the United States was 7.1%,23 and it was from 16.9% to 18.4% among Jews in New York who had early onset PD.23, 24 In the current study, we identified GBA variants in 3 of 6 patients (50%) with MSA who were of Ashkenazi Jewish descent. In summary, we observed a higher frequency of GBA variants in patients with MSA than in those with AD. Furthermore, the frequency of GBA variants was higher than previously published frequencies both in patients with PD and in healthy controls.
Although the frequency of GBA variants in this study were higher among those of both Ashkenazi Jewish descent and non‐Jewish descent, the association was only statistically significant among Jews. This may be due to power effects, given the lower prevalence of GBA variants in non‐Jewish populations.
Dysfunction of glucocerebrosidase, and probably of the autophagosomal pathway, may represent 1 “hit” in what is likely a pathway that requires multiple “hits” for the development of MSA. Other mechanisms that have been implicated in the development of MSA include mitochondrial dysfunction,25, 26, 27, 28 toxicity of fibrilized α‐synuclein,29 oxidative stress,30 neuroinflammation,31, 32 and even prion‐like propagation of MSA.33 Although the current study makes an argument for the involvement of the pathways for clearance of α‐synuclein in the development of MSA, it also makes clear that other mechanisms also need further investigation.
The major limitation of this study is the small sample size of patients with MSA. The small numbers in our series (n = 17) do make it difficult to draw concrete conclusions from these results alone. Another limitation of this study is that no healthy control brains were analyzed, although information about GBA variant carrier frequencies have been established worldwide, locally, and even within the same brain bank.19, 23, 24, 34, 35, 36 A recent genome‐wide association study in MSA did not reveal candidate loci in the GBA gene.37 However, because there are over 200 known disease‐causing variants in the GBA gene rather than a single haplotype, genome‐wide association studies may not detect association signals even though there is a true association. Similarly, many genome‐wide association studies in PD38, 39, 40, 41, 42, 43 and DLB44 also failed to identify any at‐risk loci within the GBA gene, despite convincing evidence that the association between GBA variants and PD and DLB does exist. The strengths of this study include the pathologic confirmation of MSA and AD diagnoses and the fact that our sample was drawn from a population that had a higher proportion of Ashkenazi Jews compared with the populations in the 7 previous investigations, which was reflected in our sample. Also, complete exon sequencing of GBA was performed, which broadened our ability to detect all coding variants.
Given the robust findings in this study, additional investigation of the correlation between GBA variants and MSA is warranted. Future studies should include a larger international cohort of patients with MSA from populations enriched with people of Ashkenazi Jewish descent.
Author Roles: 1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
M.S.: 1A, 1B, 1C, 2A, 2B, 3A
U.J.K: 1B, 1C, 3B
C.L.: 1C, 3B
E.C.: 1C, 3B
J.P.V.: 1C, 3B
L.C.: 1C, 3B
K.M.: 1C, 3B
M.P.: 1B, 1C, 3B
W.C.N.: 1B, 1C, 3B
W.K.C.: 1B, 1C, 3B
L.S.H.: 1B, 1C, 2B, 3B
R.N.A.: 1A, 1B, 1C, 2B, 3B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: This study was funded by the Parkinson's Disease Foundation and the NIH (NIH/NINDS: K02NS080915; T32 NS07153). Miriam Sklerov has received funding from the National Institute of Health (NIH) (NIH/NINDS T32 NS07153), and the Parkinson Disease Foundation (PDF). Karen Marder has received funding from the NIH, the Michael J. Fox Foundation, the Parkinson Disease Foundation, and the Huntington's Disease Society of America. Roy N. Alcalay receives funding from the NIH (K02NS080915). The authors report no conflicts of interests with relevance to this publication.
Financial disclosures for the previous 12 months: Karen Marder reports fees from Pfizer for an invited lecture; she is a section editor for Springer. Lawrence Honig reports fees for consulting for Bristol‐Myers Squibb, Forum, Fujirebio, Lundbeck, and Eli Lilly Pharmaceuticals; he also received commercial research support from Bristol‐Myers Squibb, Forum, Genentech, Janssen, Lilly, Lunbeck, Pfizer, Roche, TauRx, and vTv. Wendy Chung reports funding from the NIH and the Simons Foundation. Roy Alcalay reports consultation fees from Genzyme/Sanofi and Prophage. The remaining authors report no sources of funding and no conflicts of interest.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Makioka K, Yamazaki T, Takatama M, Nakazato Y, Okamoto K. Activation and alteration of lysosomes in multiple system atrophy. NeuroReport 2012;23:270–276. [DOI] [PubMed] [Google Scholar]
- 2. Stefanova N, Kaufmann WA, Humpel C, Poewe W, Wenning GK. Systemic proteasome inhibition triggers neurodegeneration in a transgenic mouse model expressing human α‐synuclein under oligodendrocyte promoter: implications for multiple system atrophy. Acta Neuropathol 2012;124:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanji K, Odagiri S, Maruyama A, Mori F, Kakita A, Takahashi H, Wakabayashi K. Alteration of autophagosomal proteins in the brain of multiple system atrophy. Neurobiol Dis 2013;49:190–198. [DOI] [PubMed] [Google Scholar]
- 4. Pukaß K, Richter‐Landsberg C. Inhibition of UCH‐L1 in oligodendroglial cells results in microtubule stabilization and prevents alpha‐synuclein aggregate formation by activating the autophagic pathway: implications for multiple system atrophy [serial online]. Front Cell Neurosci 2015;9:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grabowski GA. Gaucher disease. Enzymology, genetics, and treatment. Adv Hum Genet 1993;21:377–441. [PubMed] [Google Scholar]
- 6. Eblan MJ, Nguyen J, Ziegler SG et al. Glucocerebrosidase mutationsare also found in subjects with early‐onset parkinsonism from Venezuela Mov Disord 2006:21:282–283. [DOI] [PubMed] [Google Scholar]
- 7. Beutler E, Grabowski GA. Gaucher disease In: Scriver CR, Beaudet AL, Sly WS, Valle E, eds. The Metabolic and Molecular Bases of Inherited Disease, 8th ed New York, NY: McGraw‐Hill; 2001:3635–3668. [Google Scholar]
- 8. Zhao F, Bi L, Wang W, et al. Mutations of glucocerebrosidase gene and susceptibility to Parkinson's disease: an updated meta‐analysis in a European population. Neuroscience 2016;320:239–246. [DOI] [PubMed] [Google Scholar]
- 9. Nalls MA, Duran R, Lopez G, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 2013;70:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsuang D, Leverenz JB, Lopez OL, et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology 2012;79:1944–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jamrozik Z, Lugowska A, Slawek J, Kwiecinski H. Glucocerebrosidase mutations p. L444P and p.N370S are not associated with multisystem atrophy, progressive supranuclear palsy and corticobasal degeneration in Polish patients. J Neurol 2010;257:459–460. [DOI] [PubMed] [Google Scholar]
- 12. Mitsui J, Matsukawa T, Sasaki H, et al. Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neur 2015;2:417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nishioka K, Ross OA, Vilariño‐Guell C, et al. Glucocerebrosidase mutations in diffuse Lewy body disease. Parkinsonism Relat Disord 2011;17:55–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Segarane B, Li A, Paudel R, et al. Glucocerebrosidase mutations in 108 neuropathologically confirmed cases of multiple system atrophy. Neurology 2009;72:1185–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Srulijes K, Hauser AK, Guella I, et al. No association of GBA mutations and multiple system atrophy. Eur J Neurol 2013;20:e61–e62. [DOI] [PubMed] [Google Scholar]
- 16. Sun QY, Guo JF, Han WW, et al. Genetic association study of glucocerebrosidase gene L444P mutation in essential tremor and multiple system atrophy in mainland China. J Clin Neurosci 2013;20:217–219. [DOI] [PubMed] [Google Scholar]
- 17. Goker‐Alpan O, Giasson BI, Eblan MJ, et al. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology 2006;67:908–910. [DOI] [PubMed] [Google Scholar]
- 18. Pew Research Center . A Portrait of Jewish Americans. Available at: http://www.pewforum.org/2014/10/01/jewish-american-beliefs-attitudesculture-survey/. Accessed October 1, 2016.
- 19. Clark LN, Kartsaklis LA, Wolf Gilbert R, et al. Association of glucocerebrosidase mutations with dementia with Lewy bodies. Arch Neurol 2009;66:578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teng EL, Chui HC. The Modified Mini‐Mental State (3MS) examination. J Clin Psychiatry 1987;48:314–318. [PubMed] [Google Scholar]
- 21. Lek M, Karczewski K, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bronstein S, Karpati M, Peleg L. An update of Gaucher mutations distribution in the Ashkenazi Jewish population: prevalence and country of origin of the mutation R496H. Isr Med Assoc J 2014;16:683–685. [PubMed] [Google Scholar]
- 23. Clark LN, Ross BM, Wang Y, et al. Mutations in the glucocerebrosidase gene are associated with early‐onset Parkinson disease. Neurology 2007;69:1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alcalay RN, Caccappolo E, Mejia‐Santana H, et al. Frequency of known mutations in early‐onset Parkinson disease. Arch Neurol 2010;67:1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kasai T, Tokuda T, Ohmichi T, Ishii R, Tatebe H, Nakagawa M, Mizuno T. Serum levels of coenzyme Q10 in patients with multiple system atrophy [serial online]. PLoS ONE 2016;11:e0147574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Collaboration Multiple‐System Atrophy Research. Mutations in COQ2 in familial and sporadic multiple‐system atrophy. N Engl J Med 2013;369:233–244. [DOI] [PubMed] [Google Scholar]
- 27. Schottlaender LV, Bettencourt C, Kiely AP, et al. Coenzyme Q10 levels are decreased in the cerebellum of multiple‐system atrophy patients [serial online]. PLoS ONE 2016;11:e0149557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ubhi K, Rockenstein E, Kragh C, et al. Widespread microRNA dysregulation in multiple system atrophy—disease‐related alteration in miR‐96. Eur J Neurosci 2014;39:1026–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scholz SW, Houlden H, Schulte C, et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol 2009;65:610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stefanova N, Reindl M, Neumann M, Haass C, Poewe W, Kahle PJ, Wenning GK. Oxidative stress in transgenic mice with oligodendroglial alpha‐synuclein overexpression replicates the neuropathology of multiple system atrophy. Am J Pathol 2005;166:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Radford R, Rcom‐H'cheo‐Gauthier A, Wong MB, et al. The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to α‐synuclein inclusions. Mol Cell Neurosci 2015;65:68–81. [DOI] [PubMed] [Google Scholar]
- 32. Valera E, Masliah E. Immunotherapy for neurodegenerative diseases: focus on α‐synucleinopathies. Pharmacol Ther 2013;138:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prusiner SB, Woerman AL, Mordes DA, et al. Evidence for α‐synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci USA 2015;112:E5308–E5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aharon‐Peretz J, Rosenbaum H, Gershoni‐Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med 2004;351:1972–1977. [DOI] [PubMed] [Google Scholar]
- 35. Sato C, Morgan A, Lang AE, et al. Analysis of the glucocerebrosidase gene in Parkinson's disease. Mov Disord 2005;20:367–370. [DOI] [PubMed] [Google Scholar]
- 36. Toft M, Pielsticker L, Ross OA, Aasly JO, Farrer MJ. Glucocerebrosidase gene mutations and Parkinson disease in the Norwegian population. Neurology 2006;66:415–417. [DOI] [PubMed] [Google Scholar]
- 37. Sailer A, Scholz SW, Nalls MA, et al. A genome‐wide association study in multiple system atrophy. Neurology 2016;87:1591–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. International Parkinson Disease Genomics Consortium ; Nalls MA, Plagnol V. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet 2011;377:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maraganore DM, de Andrade M, Lesnick TG, et al. High‐resolution whole‐genome association study of Parkinson disease. Am J Hum Genet 2005;77:685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pankratz N, Wilk JB, Latourelle JC, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 2009;124:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Simón‐Sánchez J, Schulte C, Bras JM, et al. Genome‐wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 2009;41:1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lill CM, Roehr JT, McQueen MB, et al. Comprehensive research synopsis and systematic meta‐analyses in Parkinson's disease genetics: the PDGene database [serial online]. PLoS Genet 2012;8:e1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Satake W, Nakabayashi Y, Mizuta I, et al. Genome‐wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet 2009;41:1303–1307. [DOI] [PubMed] [Google Scholar]
- 44. Bras J, Guerreiro R, Darwent L, et al. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet 2014;23:6139–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]