

SUMMARY
Antimitotic agents, including Taxol, disrupt microtubule dynamics and cause a protracted mitotic arrest and subsequent cell death. Despite the broad utility of these drugs in breast cancer and other tumor types, clinical response remains variable. Tumor-associated macrophages (TAMs) suppress the duration of Taxol-induced mitotic arrest in breast cancer cells and promote earlier mitotic slippage. This correlates with a decrease in the phosphorylated form of histone H2AX (γH2AX), decreased p53 activation, and reduced cancer cell death in interphase. TAMs promote cancer cell viability following mitotic slippage in a manner sensitive to MAPK/ERK kinase (MEK) inhibition. Acute depletion of major histocompatibility complex class II low (MHCIIlo) TAMs increased Taxol-induced DNA damage and apoptosis in cancer cells, leading to greater efficacy in intervention trials. MEK inhibition blocked the protective capacity of TAMs and phenocopied the effects of TAM depletion on Taxol treatment. TAMs suppress the cytotoxic effects of Taxol, in part through cell non-autonomous modulation of mitotic arrest in cancer cells, and targeting TAM-cancer cell interactions potentiates Taxol efficacy.
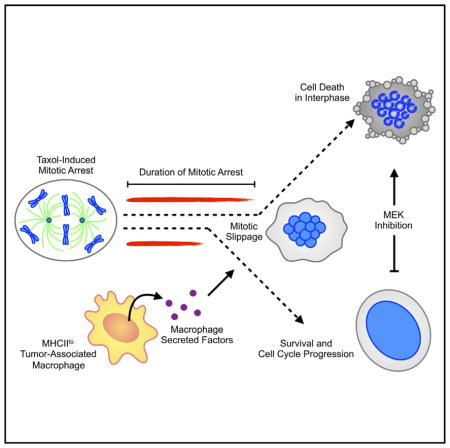
Graphical Abstract
Olson et al. examine how tumor-associated macrophages (TAMs) suppress the duration of Taxol-induced mitotic arrest in breast cancer cells and promote earlier mitotic slippage. TAMs promote cancer cell viability following mitotic slippage through a mechanism that is sensitive to MEK inhibition. Acute depletion of MHCIIlo TAMs in a preclinical breast cancer model increased the ability of Taxol to induce apoptosis and improved therapeutic response.
INTRODUCTION
The microenvironment plays a critical role in regulating tumor development and disease progression (Quail and Joyce, 2013). In the context of chemotherapy treatment, tumor-associated macrophages (TAMs) have emerged as potent regulators of therapeutic response (De Palma and Lewis, 2013; Ruffell and Coussens, 2015). These effector cells can modulate cancer cell survival pathways through the provision of cytokines (Mitchem et al., 2013) and pro-tumorigenic proteases (Shree et al., 2011). Additionally, TAMs can suppress immune-based mechanisms of cytotoxic chemotherapy (DeNardo et al., 2011; Ruffell et al., 2014). Limited research, however, has been conducted into whether microenvironment cells, including TAMs, directly affect the molecular mechanisms by which cytotoxic chemotherapy induces cancer cell damage.
Some intriguing insights into this question have emerged from intravital imaging experiments, showing that antimitotic agents in particular have impaired efficacy against cancer cells in vivo versus what is observed in monoculture in vitro (Orth et al., 2011). Whereas cancer cells propagated in culture arrest for prolonged periods of time following exposure to high doses of antimitotic drugs, often dying during mitosis, when the same cancer cell lines are grown in vivo, they arrest for shorter periods and exit mitosis without dividing in a process termed mitotic slippage (Orth et al., 2011). These results suggest that a microenvironmental component may influence the fate of cancer cells in vivo compared with in vitro. Additionally, the observations that extracellular factors can promote efficient centrosome separation (Mardin et al., 2013) or drive clustering of supernumerary centrosomes (Kwon et al., 2008) suggest a potential role for the microenvironment in regulating mitosis, which has generally been considered a cell-autonomous process. Thus, we sought to evaluate the effect of TAMs on mitotic arrest of cancer cells and their subsequent fate in the context of chemotherapy treatment with Taxol.
RESULTS
TAM Depletion Increases Taxol-Induced DNA Damage Signaling and Cell Death
In order to determine the role of TAMs in the acute response to treatment with the antimitotic agent Taxol, we designed a 1-week trial in which TAMs were depleted with BLZ945, a small-molecule inhibitor of the colony stimulating factor-1 receptor (CSF-1R) (Pyonteck et al., 2013), immediately prior to chemotherapeutic treatment (Figure 1A). FVB/n female mice were orthotopically implanted via mammary fat pad injection with the MMTV-PyMT breast cancer cell line, TS1 (Shree et al., 2011). Following tumor establishment, mice were treated with BLZ945 for 72 hr prior to a single dose of Taxol and continued on BLZ945 in a time course for a further 24–96 hr. It is known that prolonged CSF-1R inhibition (using a chemically distinct small-molecule inhibitor, PLX3397) in combination with Taxol in pre-clinical breast cancer models leads to improved efficacy over time through increased chemotherapy-induced activation of a CD8+ T cell-mediated immune response (DeNardo et al., 2011). For this reason, we restricted our initial analyses to the time points immediately following Taxol treatment, when no significant tumor volume differences were yet observed between Taxol versus Taxol + BLZ945 (Figure 1B). Our preclinical trial design for these initial experiments, therefore, focuses on the acute phase of drug response, enabling precise assessment of the effects of TAM depletion on the cancer cell response to Taxol in vivo through a series of time points.
Figure 1. Depletion of TAMs Increases γH2AX Levels in Response to Taxol Treatment.

(A) Preclinical trial design for combined CSF-1R inhibition and Taxol treatment in an orthotopic mouse model of breast cancer following mammary fad pad (MFP) injection of the TS1 tumor cell line and subsequent tumor establishment.
(B) Plot of tumor volume as a function of time. Treatment groups are vehicle (VEH) 20% Captisol, BLZ945 (BLZ), and Taxol (TAX). Vertical arrow indicates t = 0 when Taxol was administered. The vehicle was administered daily to all animals in the Taxol alone group.
(C) Quantification by image analysis of intratumoral CD68+ cells at 24, 48, and 72 hr after Taxol treatment.
(D–F) Quantitation of the percentage of (D) γH2AX+, (E) cleaved caspase-3+ (CC3+), and (F) Ki67+ cells within the tumor (left) and representative images of tumor sections (right). Scale bars, 50 μm. n = 5 mice in all treatment groups at all time points. All data are presented as mean ± SEM.
(C–F) Data are from whole tumors isolated at the indicated time points according to the trial design in (A).
Student’s t test was used to assess significance between treatment groups: *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S1.
Pretreatment with BLZ945 led to a 47% reduction in TAMs at the time of Taxol administration (day 0) as measured by immunofluorescence staining and quantification of intratumoral CD68+ cells in tissue sections (Figure 1C). By the trial endpoint, BLZ945-treated tumors had 75% fewer CD68+ cells relative to those tumors treated with Taxol alone (Figure 1C). To determine the effects of TAM depletion on the cancer cell response to Taxol treatment, we stained tissue sections for various markers of DNA damage, cell death, and proliferation. A marker of DNA double-strand breaks, the phosphorylated form of histone H2AX (γH2AX), can be used as a means to monitor the effectiveness of cancer therapeutics (Bonner et al., 2008). We found that BLZ945 treatment led to substantially increased levels of γH2AX+ cells at both 24 and 72 hr post-Taxol compared with Taxol alone (Figure 1D). This correlated with significantly elevated levels of cleaved caspase-3+ (CC3+) apoptotic cells at 48 hr after Taxol treatment (Figure 1E). Additionally, BLZ945 significantly reduced proliferation at 24 hr post-Taxol treatment as measured by Ki67 staining (Figure 1F). Collectively, these data indicate that in the context of Taxol treatment, TAM depletion is associated with enhanced DNA damage signaling, increased apoptosis, and suppression of proliferation in cancer cells, suggesting a role for TAMs in modulating the cytotoxic activity of this antimitotic agent.
We next sought to determine whether TAM depletion altered the bioavailability or potency of Taxol within the tumor. Given that the mechanism of action of Taxol involves microtubule stabilization (Schiff and Horwitz, 1980), and stabilized microtubules become acetylated (Schulze et al., 1987), we measured intratu-moral α-tubulin acetylation as a surrogate marker of intratumoral Taxol bioavailability. We observed increased levels of α-tubulin acetylation in tumors following Taxol treatment, with no significant difference compared with tumors additionally treated with BLZ945 (Figure S1A). Moreover, the percentage of phosphohistone H3+ mitotic cells increased similarly in both control and BLZ945-treated mice following Taxol treatment (Figure S1B), showing that the accumulation of mitotic cells is similar in both conditions. These data indicate that macrophages do not interfere with the bioavailability of Taxol nor do they prevent cells from entering mitotic arrest. Rather, TAMs likely directly affect the cytotoxicity of Taxol upon cancer cells.
Because of the complexity of TAM and myeloid cell populations in breast tumors (DeNardo et al., 2011; Franklin et al., 2014), we wanted to determine which specific macrophage subsets are altered in response to acute CSF-1R inhibition. We were particularly interested in the relative depletion of major histocompatibility complex class II high (MHCIIhi) and low (MHCIIlo) macrophage populations following BLZ945 treatment because of their reported phenotypic differences (Franklin et al., 2014) and differential dependency on CSF-1R signaling (Van Overmeire et al., 2016). We observed that following 72 hr of BLZ945 pretreatment there were significant reductions in both MHCIIlo and MHCIIhi populations of CD11b+ F4/80+ macrophages relative to total CD45+ leukocytes (day 0 of the trial; Figures S1C and S1D). However, only the relative proportion of MHCIIlo macrophages remained significantly reduced in the context of Taxol treatment (Figures S1C and S1D), consistent with the critically important role of CSF-1R signaling in this specific TAM population (Van Overmeire et al., 2016). Additionally, there were significant increases in CD4+ and CD8+ T cells in BLZ945-pretreated tumors at the time of Taxol administration (Figures S1E and S1F). This elevation in T cell numbers following CSF-1R inhibition and macrophage depletion is consistent with previous reports (DeNardo et al., 2011; Strachan et al., 2013). Collectively, these data indicate that the depletion of MHCIIlo TAMs within the tumor microenvironment correlates with increased cancer cell DNA damage and apoptosis, and decreased proliferation in cancer cells. Moreover, this suppression of cytotoxic activity is achieved without altering the ability of Taxol to stabilize microtubules and induce mitotic arrest.
Macrophage-Secreted Factors Suppress DNA Damage Signaling and Cell Death following Taxol-Induced Mitotic Arrest
To determine whether TAMs suppress Taxol-induced cytotoxic stress though direct interaction with cancer cells or via another component of the tumor microenvironment, we sought to investigate whether we could model our in vivo findings in a reconstructed in vitro system. Cancer cells are known to respond to antimitotic agents with high levels of intra- and inter-cell line variation (Gascoigne and Taylor, 2008). To this end, we first characterized the TS1 murine breast cancer cell response to Taxol. When we treated TS1 cells with Taxol in monoculture, we observed a loss of 2C DNA content cells by 24 hr, indicating that in response to Taxol treatment, cancer cells were unable to successfully divide and return to a diploid G1 cell state (Figure S2A). This correlated with the emergence at 24 hr of a higher DNA content tetraploid (4C) peak, and at 48 hr of a higher DNA content 8C peak, indicative of tetraploid cells that had re-entered the cell cycle following a failed cell division. In addition, a sub-2C peak indicative of fragmented apoptotic nuclei emerged at 48 hr and increased at 72 hr (Figure S2A). Examination of the nuclear morphology of cells treated for 24 hr with Taxol revealed that the majority of the cells display a multinucleated phenotype (Figure S2B). This phenotype is indicative of mitotic slippage following Taxol-induced mitotic arrest (Panvichian et al., 1998). Thus, we find that the majority of TS1 cancer cells undergo mitotic slippage and become tetraploid within 48 hr following Taxol treatment, and that cell death is typically observed at subsequent time points. This is consistent with intravital microscopy studies that have demonstrated this mechanism of action for maximum tolerated dose Taxol treatment in vivo (Chittajallu et al., 2015; Orth et al., 2011).
We next cultured TS1 tumor cells with bone marrow-derived macrophages (BMDMs) and assessed whether there were any alterations to the DNA content changes in response to Taxol. Notably, we found that co-culture with macrophages substantially reduced the accumulation of sub-2C apoptotic tumor cells at 48 and 72 hr (Figure 2A). This correlated with increased populations of 4C and 8C DNA content cells at 72 hr, indicating that macrophage co-culture promoted the persistence of cancer cells following Taxol-induced mitotic slippage (Figure 2A). Similar to our observations in vivo, in cell culture there was no impact of macrophages on the ability of Taxol to increase the percentage of phosphohistone H3+ mitotic cancer cells as measured by flow cytometry (Figure 2B). By contrast, when γH2AX immunoreactivity was measured in cancer cells following Taxol treatment, we found that co-culture with BMDMs significantly reduced the percentage of γH2AX+ cancer cells throughout the time course analyzed (Figure 2C). Furthermore, we also found that macrophage co-culture transiently reduced the number of cancer cells with phosphorylation of p53 (Figure 2D), an important suppressor of cell growth following failed cytokinesis (Andreassen et al., 2001). These data demonstrate that BMDMs are able to directly suppress the ability of Taxol to induce markers of genotoxic stress, such as γH2AX, in cancer cells.
Figure 2. Macrophages Suppress Genotoxic Stress and Cancer Cell Death.

TS1 tumor cells (TCs) were cultured with Taxol and assayed by flow cytometry at the indicated time points.
(A–D) Flow cytometric analysis of DNA content (n = 4 independent experiments) (A), phospho-H3 immunoreactivity (n = 3 independent experiments) (B), γH2AX immunoreactivity (n = 3 independent experiments) (C), and phospho-p53 immunoreactivity (n = 3 independent experiments) of the TS1 TC line in response to Taxol treatment (50 nM), and the effects of co-culture with bone marrow-derived macrophages (BMDMs) (D).
(E) Comparison of the effects of co-culture with MHCII+ BMDMs on Taxol-induced accumulation of tumor cells with sub-2C DNA content and γH2AX immunoreactivity (n = 4 independent experiments).
(F) Comparison of the effects of direct cell co-culture versus BMDM-conditioned media (CM) supplemented daily [denoted BMDM-CM(+)] on Taxol-induced accumulation of tumor cells with sub-2C DNA content and γH2AX immunoreactivity (n = 4 independent experiments).
(G) Quantification of caspase activation in response to Taxol treatment and the effects of BMDM-CM as measured by western blotting. Representative western blots are shown on the left, and quantification of each caspase is shown on the right (n = 3 independent experiments).
All data are presented as mean ± SEM. Student’s t test was used to assess significance between treatment groups: *p < 0.05; **p < 0.01; ***p < 0.001. See also Figures S2 and S3.
Given the differential depletion of MHCIIlo versus MHCIIhi TAM populations that we observed in response to prolonged CSF-1R inhibition in vivo, we next assessed whether MHCII+ BMDMs were capable of suppressing genotoxic stress and cell death in response to Taxol. MHCII expression was induced on BMDMs by treatment with CSF-2 and interferon γ (IFNγ) for 48 hr prior to co-culture with cancer cells (Figure S2C). Interestingly, MHCII+ BMDMs were unable to suppress cell death or the induction of γH2AX in cancer cells (Figure 2E), indicating that the in vivo effects of BLZ945 treatment on Taxol response are likely due to the specific depletion of the MHCIIlo population of TAMs, rather than simply reducing the number of TAMs overall.
Additionally, we assessed the ability of other stromal cell lines representative of the breast tumor microenvironment for the ability to suppress the cytotoxic effects of Taxol (Figure S2D). We tested two fibroblast cell lines isolated from different stages of MMTV-PyMT tumor development (AdAF, CAF1) (Calvo et al., 2013) and two murine endothelial cell lines (EOMA, C166) (Obeso et al., 1990; Wang et al., 1996). No significant effects were observed upon co-culture of TS1 cancer cells with these different stromal cell types (Figure S2D), further indicating that the robust suppression of γH2AX and cancer cell death is a functional role unique to macrophages and specifically the MHCIIlo population.
We were also interested in determining whether macrophages were similarly capable of reducing γH2AX levels in response to mechanistically distinct chemotherapeutic agents; thus, we treated co-cultures with carboplatin, doxorubicin, etoposide, or gemcitabine. For all chemotherapies tested we observed a reduction in γH2AX levels in cancer cells in the presence of macrophages (Figure S2E), although not all to the same extent as for Taxol treatment (Figure 2C). These observations correlated with increased cancer cell viability in the case of doxorubicin, etoposide, and gemcitabine, but not carboplatin (Figure S2F). Thus, although TAMs can generally reduce DNA damage signaling in cancer cells, the effect on cancer cell fate appears to be specific to the chemotherapeutic agent assessed.
In order to better understand the means by which macrophages suppress cancer cell genotoxic stress in response to chemotherapy, we asked whether the effect could be replicated using macrophage-conditioned media (CM). CM was generated from BMDMs (BMDM-CM) and added to TS1 cells at plating and at the start of the 72 hr experiment (Figure S2G). This partially replicated the reductions in cancer cell death and γH2AX levels observed with direct cell co-culture. However, when the experimental protocol was modified from a single addition of CM at the start of treatment (Figure S2G) to supplementation with fresh CM daily [BMDM-CM(+)], the effects of direct cell co-culture could be fully recapitulated (Figure 2F). These findings indicate that the protective factor(s) is secreted by macrophages, because it is not dependent on direct cell-cell contact between macrophages and cancer cells. Furthermore, cancer cells are apparently dependent upon regular replenishment of this factor(s) from macrophages in order to receive the maximal protective benefit. Previously we have shown that macrophage-secreted cathepsin proteases have an anti-apoptotic effect in the context of chemotherapeutic treatment (Shree et al., 2011). We therefore evaluated whether cathepsin inhibition specifically altered the ability of macrophages to reduce γH2AX levels and nuclear fragmentation, and we found no difference (Figures S3A and S3B). This indicates that there are multiple mechanisms by which macrophages can interfere with chemotherapy efficacy.
We next asked which component of the macrophage-CM was responsible for the protective activity, which is defined herein by reduced γH2AX levels and accumulation of sub-2C cancer cells following Taxol treatment. Fractionation of the macrophage-CM using molecular weight cutoff spin columns determined that the protective activity was specific to the >3 kDa fraction (Figure S3C). Therefore, we reasoned that the macrophage-derived factor(s) is unlikely to be a small-molecule metabolite and is probably proteinaceous in nature. We also treated cancer cells with macrophage-CM and saw no alteration in the ability of Taxol to induce tubulin acetylation (Figure S3D), a marker of microtubule stabilization (Schulze et al., 1987). Thus, as with our in vivo findings, macrophages do not appear to interfere with the ability of Taxol to stabilize cancer cell microtubules, but rather alter the downstream effectors of this treatment.
We next examined whether BMDM-CM alters the activation of apoptotic pathways in response to Taxol treatment by three independent measures: caspase cleavage, ‘‘mitochondrial priming,’’ and MCL1 degradation. We determined that the presence of BMDM-CM reduced the cleavage of caspase-8, -9, and -3, suggesting impaired activation of both initiator and effector caspases (Figure 2G). To assess mitochondrial priming within the cancer cells (Vo et al., 2012), we treated cancer cells at various time points following Taxol addition with the BH3 mimetic ABT-263 to induce mitochondrial depolarization. We observed no effect of macrophage-CM on cancer cell sensitivity to ABT-263 either before or after Taxol treatment (Figure S3E). Additionally, there were no significant effects of macrophage-CM on Taxol-induced MCL1 degradation, or levels of other BCL2 family members (Figure S3F). Therefore, macrophage-CM impairs the ability of Taxol to activate apoptotic caspases, but in a manner that does not make the cells generally resistant to mitochondrial depolarization and apoptosis. Our data thus suggest that the macrophage-supplied activity specifically impairs the ability of mitotic arrest to induce cytotoxic stress and the subsequent initiation of apoptosis in cancer cells.
Macrophages Reduce the Duration of Cancer Cell Mitotic Arrest
To understand how macrophage-secreted factors alter the consequences of Taxol-induced microtubule stabilization and inhibit cancer cell death, we focused on mitotic arrest, the critical intermediate step. To this end, we cultured cancer cells with macrophages and measured the duration of cancer cell mitotic arrest in response to Taxol using live imaging microscopy (Figure 3A). The duration of mitotic arrest was measured as the time between cell rounding and mitotic slippage (Figure 3B). We observed that the vast majority of mitotic arrests ended in mitotic slippage rather than death in mitosis (data not shown). When cancer cells were cultured with macrophages, this resulted in a significant reduction in the duration of mitotic arrest (Figure 3C). To control for any effects of cell density, we plated cancer cells at an increased concentration with no significant impact on the duration of Taxol-induced mitotic arrest (Figure S4A). Additionally, we replicated these findings using the mechanistically distinct inhibitor of the mitotic kinesin Eg5, S-trityl-L-cysteine (STLC) (Figure S4B), demonstrating that macrophage co-culture reduces the duration of mitotic arrest in response to multiple spindle poisons. Given the ability of macrophage-CM to replicate the effects of direct co-culture on γH2AX signaling and cancer cell death, we sought to determine whether macrophage-secreted factors were also responsible for modulating mitotic arrest. We observed that macrophage-CM was indeed sufficient to reduce the duration of Taxol-induced mitotic arrest in cancer cells (Figure 3D). Moreover, fractionation of the CM indicated that the >3 kDa CM fraction was responsible for shortening the duration of mitotic arrest in cancer cells (Figure 3D), consistent with this fraction also reducing levels of γH2AX and apoptosis in Taxol-treated TS1 cells (Figure S3C).
Figure 3. Macrophage-Secreted Factors Reduce the Duration of Taxol-Induced Mitotic Arrest.

TS1 tumor cells were imaged live with or without co-culture with GFP-expressing BMDMs.
(A) Representative images of assays in which cells were imaged by differential interference contrast (DIC) microscopy. BMDMs were identified by GFP fluorescence imaging and excluded from mitotic analyses.
(B) Montage of individual TS1 cells entering mitosis and exiting by mitotic slippage. Numbers indicate time relative to mitotic entry in minutes. Scale bars, 10 μm.
(C) A single representative experiment is plotted measuring the duration of mitotic arrest in TS1 cells treated with Taxol (50 nM) in the presence or absence of BMDMs. In each experiment, at least 30 cells were scored per condition, and this was repeated in three independent experiments.
(D) As in (C), except that tumor cells were additionally treated with complete or fractionated macrophage-CM (<3 kDa or >3 kDa fractions).
(E) Clonogenic assay measuring the effects of BMDM-CM on inhibition of tumor cell colony formation by Taxol. Representative images are shown on the left, and quantification of crystal violet staining is shown on the right (n = 3 independent experiments). Scale bar, 5 mm.
(F) DNA content analysis of tumor cell colonies shown in (E) at day 8 (n = 4 independent experiments). Representative histograms are shown on the left, with quantification of treatment groups on the right.
Data for mitotic arrest duration are presented as mean ± interquartile range. All other data are presented as mean ± SEM. Student’s t test was used to assess significance between treatment groups. *p < 0.05; **p < 0.01; ***p < 0.001. ns, non-significant. See also Figure S4.
The duration of mitotic arrest has been proposed to dictate cell fate in response to microtubule-stabilizing agents (Bekier et al., 2009) and to determine the extent of multinucleation and DNA damage following mitotic slippage (Zhu et al., 2014). In order to uncouple the effects of prolonged mitotic arrest and failed cytokinesis in our experimental system, we used the aurora B kinase inhibitor ZM447439 to abrogate the Taxol-induced mitotic arrest and promote rapid mitotic slippage. Treatment with ZM447439 significantly reduced the induction of γH2AX and p53 activation at 24 hr (Figures S4C and S4D). These data confirm that protracted mitotic arrest is required for Taxol-induced genotoxic stress at the 50 nM concentration used here (Bekier et al., 2009; Zhu et al., 2014). Recent work has suggested that mitotic arrest is not required for response to Taxol, arguing that clinically relevant concentrations of Taxol are actually in the low nanomolar range (Zasadil et al., 2014). We therefore assessed the ability of various concentrations of Taxol to induce accumulation of TS1 cells arrested in mitosis following 8 hr of treatment (Figure S4E). We observed significant increases in phospho-H3+ cells at 6.25 and 12.5 nM with more pronounced increases occurring at concentrations of 25 nM and above (Figure S4E). Indeed, when we examined the DNA content profiles of the cells after 24 hr of treatment, we observed that although at 6.25 nM the profile was unperturbed, at 25 nM the DNA content profile was indicative of failed cell division in most cells (Figure S4F). At 12.5 nM concentrations of Taxol we saw an intermediate phenotype, suggesting mixed mitotic slippage and successful cell division (Figure S4F). We next sought to determine whether macrophages were still protective against the cytotoxic effects of Taxol at these lower concentrations. At both lower concentrations, we observed significant reductions in cancer cell death with BMDM co-culture (Figure S4G). However, the induction of cell death at the 6.25 nM Taxol concentration, where we did not observe mitotic slippage and increased cancer cell ploidy, was minimal.
Given the ability of macrophages to support the survival of cancer cells following Taxol-induced mitotic slippage, we were interested in assessing the long-term fate of these newly tetraploid cells. There are several alternative outcomes for cells following mitotic slippage besides cell death or continued proliferation (Vitale et al., 2011), and although the cells may remain viable, this can occur with a loss of self-renewal capacity if they arrest and become quiescent or senescent. Thus, we sought to examine the effects of macrophage-CM on Taxol treatment in a clonogenic cell survival assay. Cancer cells treated with Taxol in the presence of macrophage-CM formed significantly more colonies than those treated with Taxol alone (Figure 3E). Furthermore, when we assessed the DNA content from the cells in these colonies, we found a greater proportion of tetraploid (4C) cells in the macrophage-CM-treated samples (Figure 3F). When we used lower concentrations of Taxol, we also observed increased clonogenic capacity in cells treated with macrophage-CM (Figure S4H). These data indicate that macrophage-CM promotes the proliferative viability of cancer cells following Taxol-induced mitotic slippage and tetraploidization.
Cancer Cell Mitogen-Activated Protein Kinase Signaling Is Critical for Viability following Taxol Treatment
High levels of mitogen-activated protein kinase (MAPK) have been reported to drive both intrinsic and acquired resistance to taxanes (Cheung et al., 2004; McDaid and Horwitz, 2001; Menendez et al., 2005). Furthermore, inhibitors targeting the MAPK pathway have been shown to potentiate the effects of Taxol in vivo (Holt et al., 2012; McDaid et al., 2005). When we assessed MAPK signaling following Taxol treatment, we observed that macrophage-CM led to a maintenance of extracellular signal-regulated kinase (ERK) 1/2 phosphorylation, whereas the levels in cancer cells treated alone were significantly reduced at 24 and 48 hr (Figure 4A). Treatment of co-cultured macrophages and cancer cells with an MAPK/ERK kinase (MEK) inhibitor (PD0325901) blocked the protective effect of macrophages (Figure 4B). This effect was specific to MAPK pathway inhibition because protein kinase b (AKT) inhibition was insufficient to block the protective effect of macrophages in the context of Taxol treatment (Figures S5A and S5B). Given the reported importance of the epidermal growth factor (EGF)/CSF-1 paracrine loop in TAM-cancer cell communication within the tumor microenvironment (Wyckoff et al., 2004), and the position of EGF receptor (EGFR) upstream of the MAPK pathway (Roberts and Der, 2007), we asked whether this signaling axis was critical to the protective effect of macrophages observed here. Co-cultures of BMDMs and cancer cells were treated with Taxol, with or without the EGFR inhibitor gefitinib (Figure S5C). However, this did not significantly affect Taxol-induced cell death. In order to determine whether cell-autonomous MAPK inhibition in the cancer cells was indeed responsible for reversal of the Taxol-induced cell death phenotype, and to exclude any confounding effect of MAPK inhibition in macrophages, we repeated the assay using macrophage-CM and similarly saw restoration of cell death with MEK inhibition (Figure S5D). MEK inhibition also blocked the ability of macrophage-CM to reduce the duration of Taxol-induced mitotic arrest (Figure S5E). Although MEK inhibition has been reported to enhance death in mitosis in response to prolonged mitotic arrest by microtubule inhibitors (Kawabata et al., 2012), we observed no effect of MEK inhibition alone on Taxol-induced mitotic arrest (Figure S5E), possibly because of the bias of the TS1 cell line toward mitotic slippage. MEK inhibition also restored caspase-3 cleavage in macrophage-CM treated samples (Figure 4C). Thus, targeting the MAPK pathway in combination with Taxol potentiates cancer cell death and blocks the ability of macrophages to modulate mitotic arrest and mediate therapeutic resistance.
Figure 4. Tumor Cell MAPK Signaling Is Critical for Viability following Taxol Treatment.

(A) Analysis of ERK1/2 signaling in tumor cells in response to treatment with Taxol (50 nM) in the presence or absence of BMDM-CM supplemented daily [denoted BMDM-CM(+)]. Representative western blots are shown. Area under the curve (AUC) quantification of band intensities is displayed in the graph below as a ratio of phosphorylated/total ERK1/2 (n = 3 independent experiments).
(B) Flow cytometric analysis of sub-2C DNA content tumor cells in response to Taxol (50 nM) and the MEK inhibitor PD0325901 (500 nM). Tumor cells were treated alone or in co-culture with BMDMs (n = 3 independent experiments).
(C) Analysis of ERK1/2 signaling and caspase-3 activation in tumor cells following 48 hr of treatment with Taxol (50 nM) in the presence or absence of BMDM-CM(+) and the MEK inhibitor PD0325901 (500 nM). Representative western blots are shown. AUC quantification of band intensities is displayed to the right as a ratio of CC3+/GAPDH (n = 3 independent experiments).
(D) Clonogenic assay measuring the effects of the MEK1/2 inhibitor PD0325901 and BMDM-CM on modulating inhibition of tumor cell colony formation by Taxol. Representative images are shown on the left, and quantification of crystal violet staining is shown on the right (n = 4 independent experiments). Scale bar, 5 mm.
(E) Tumor volume plots for the assessment of response to a single dose of Taxol (indicated by vertical dotted line) and acute BLZ945 or PD0325901 treatment. See Figure S5E for schematic of trial design. n = 10 mice in all treatment groups.
(F) Tumor volume plots for the assessment of response to weekly Taxol treatment (indicated by vertical dotted lines) with concurrent PD0325901 or sustained BLZ945 treatment. See Figure S6B for schematic of trial design. n = 10 mice in all treatment groups.
All data are presented as mean ± SEM. Student’s t test was used to assess significance between treatment groups: *p < 0.05; **p < 0.01; ***p < 0.001. See also Figures S5 and S6.
Given the ability of MEK inhibition to restore cell death in the presence of macrophage-CM, we sought to determine the effect of inhibition on clonogenic survival of cancer cells in the context of Taxol treatment. Addition of the MEK inhibitor led to a 53% reduction in outgrowth of cells treated with Taxol and macrophage-CM, to a level comparable with Taxol treatment alone (Figure 4D). Furthermore, when we analyzed the DNA content of the colonies, we observed that the MEK inhibitor reduced the ploidy of the Taxol treatment groups (Figure S5F). Interestingly, we also observed that macrophage-CM significantly blocks the reduction in clonogenic capacity of cancer cells treated with the MEK inhibitor alone. We additionally found that macrophage-CM reversed the reduction in NF-κB p65 activation observed with MEK inhibitor treatment (Figure S5G). Collectively, our data indicate that macrophage-secreted factors can suppress Taxol-induced cytotoxic stress and promote proliferative viability following mitotic slippage in a manner sensitive to MAPK inhibition in the cancer cells (Figure S6A).
Acute CSF-1R and MEK Inhibition Potentiate the Response to Taxol
In order to determine whether targeting the interactions between TAMs and breast cancer cells could modulate the therapeutic response to Taxol in vivo, we designed a trial that combined a single dose of Taxol with acute BLZ945 and/or PD0325901 treatment, followed by trial endpoint analyses 15 days after Taxol administration (Figure S6B). As above, BLZ945 was administered for 3 days to deplete TAMs prior to Taxol treatment, then continued concurrently with Taxol treatment for an additional 3 days. PD0325901 treatment was initiated with Taxol treatment and continued for 3 days (Figure S6B). In this manner, we aimed to confine the inhibitor treatment to the time window when cancer cells are directly responding to Taxol treatment and then monitor subsequent tumor regrowth. Neither BLZ945 nor PD0325901, nor the combination of these two inhibitors, had a significant impact on tumor volumes at endpoint (Figure 4E). In combination with Taxol treatment, however, all acute treatment groups had significantly lower tumor volumes at endpoint compared with Taxol alone (Figure 4E). Notably, no additive effect was observed from the triple combination of BLZ945 + PD0325901 + Taxol treatment versus the single drugs combined with Taxol (Figure 4E).
When we analyzed tissues taken at 72 hr following Taxol treatment, we found that as observed previously for BLZ945 treatment, combined treatment with PD0325901 increased intra-tumoral γH2AX+ and CC3+ cells (Figures S6C and S6D). We next sought to validate these findings in a trial aimed at tumor control where Taxol was administered weekly. Whereas PD0325901 treatment still remained confined to 3 days concurrent with Taxol administration, BLZ945 treatment was now continuous (Figure S6E). As in the earlier trials, we observed a significant reduction in endpoint tumor volume in animals treated with either BLZ945 or PD0325901 in combination with Taxol compared with those treated with Taxol alone (Figure 4F). Interestingly, both inhibitors are essentially interchangeable with respect to potentiation of Taxol, which is consistent with our working model of each inhibitor targeting the same protective interaction of TAMs and cancer cells (Figure S6A).
DISCUSSION
Here, we report that depletion of MHCIIlo TAMs via CSF-1R inhibition increases the levels of cancer cell DNA damage signaling and cell death in response to Taxol treatment in a pre-clinical model of breast cancer. Macrophage-secreted factors are capable of suppressing induction of multiple markers of genotoxic stress and reducing activation of both the intrinsic and the extrinsic apoptotic pathways in response to Taxol-induced mitotic arrest and slippage. This occurs in association with a reduction in the duration of cancer cell mitotic arrest; this is the first example of non-autonomous modulation of mitotic arrest by stromal cells that we are aware of. Macrophages also act on subsequent steps by promoting the clonogenic viability of these newly tetraploid cancer cells in a manner sensitive to MAPK inhibition. Identification of the secreted factor(s) is an active area of research in the laboratory, and we expect this to provide important further insights into the mechanisms by which macrophages confer protection against different chemotherapies.
Previously, the role of the tumor microenvironment in mediating chemoresistance was largely demonstrated to be the result of impaired pharmacokinetics and increased survival signaling (Klemm and Joyce, 2015). Our study and other recent work (Houthuijzen et al., 2014), however, demonstrate the ability of the environment to also modulate the mechanisms through which chemotherapeutic agents induce cytotoxic stress in cancer cells. Intravital microscopy has been particularly informative in evaluating the response to chemotherapy in an intact tumor microenvironment (Nakasone et al., 2012). In this manner, other groups have observed that antimitotic agents lead to shorter mitotic arrest and mitotic slippage in vivo (Chittajallu et al., 2015; Orth et al., 2011). Our data are consistent with these studies and now identify a specific role for TAMs in modulating the mitotic arrest of cancer cells in an intact tumor microenvironment. This paradigm of attenuated response to Taxol treatment in vivo is supported by clinical studies, where serial fine-needle aspirations of breast cancer patients undergoing treatment revealed a greater number of apoptotic cells than cells arrested in mitosis, and responder patients showed a sustained apoptotic response even after levels of mitotic cells had returned to baseline (Symmans et al., 2000). These findings are consistent with mitotic slippage of the cancer cells in vivo, followed by subsequent apoptosis of the tetraploid cells in interphase (Figure S6A), rather than the classical model of death in mitosis following a prolonged arrest. The ability of the tumor microenvironment to support the viability of cancer cells after failed cell division is corroborated by reports that enhanced growth factor signaling can overcome p53-induced G1 arrest (Ganem et al., 2014). The ultimate fate of these cells, however, remains an open question. Tetraploidization has been shown to promote chromosomal instability, tolerance to mitotic errors, and enhanced cancer genome evolution (Dewhurst et al., 2014; Kuznetsova et al., 2015). Aneuploid cells, however, have also been shown to proliferate at slower rates, and thus Taxol-induced tetraploidization might reduce overall tumor growth (Williams et al., 2008).
We have also demonstrated herein that pro-tumorigenic interactions between cancer cells and TAMs can be efficiently targeted via either cellular component, because MEK and CSF-1R inhibitors were interchangeable in potentiating the response to Taxol treatment. Although the microenvironment has been shown to mediate resistance to both conventional and targeted therapeutics (Klemm and Joyce, 2015; Straussman et al., 2012), multiple drug combinations will likely suppress microenvironmental interference. Ultimately, it is becoming clear that the tumor micro-environment can regulate numerous aspects of resistance to therapeutic intervention and in this manner influence cancer cell heterogeneity. Consequently, therapeutic strategies that consider and account for the influence of the microenvironment, including TAMs, will be critical to improve clinical outcomes.
EXPERIMENTAL PROCEDURES
Culture of Established Cell Lines and Drug Treatments
All cell lines were maintained in high-glucose DME media supplemented with 10% fetal bovine serum, 2 mM glutamine, and 100 U/mL penicillin/streptomycin. The TS1 cell line was derived in J.A.J.’s laboratory as previously reported (Shree et al., 2011). CAF1 and AdAF lines were provided by Dr. Erik Sahai (Calvo et al., 2013). EOMA and C166 cell lines were purchased from the ATCC (Obeso et al., 1990; Wang et al., 1996). Where indicated, 50 nM Taxol (Sigma), 50 μM carboplatin (Selleckchem), 300 nM doxorubicin (Selleckchem), 20 μM etoposide (Selleckchem), 400 nM gemcitabine (Selleckchem), 10 μM JPM-OEt (synthesized by the Organic Synthesis Facility at Memorial Sloan Kettering Cancer Center [MSKCC]), 50 μM to 3.2 nM ABT-263 (ApexBio), 10 μM STLC (Tocris), 2.5 μM ZM447439 (Selleckchem), 500 nM PD0325901 (Selleckchem), 500 nM MK2206 (Selleckchem), and 500 nM gefitinib (Selleckchem) were added.
Mice
All animal studies were approved by the Institutional Animal Care and Use Committee of MSKCC under protocol 04-08-022. Wild-type (WT; FVB/n) and CAG-EGFP (FVB/n) mice were purchased from The Jackson Laboratory and bred within the MSKCC animal facility. The TS1 orthotopic model of breast cancer has been reported previously (Shree et al., 2011) and is described fully, along with in vivo drug treatments, in the Supplemental Experimental Procedures.
Immunoprofiling of Tumors
After counting, single-cell suspensions in FACS buffer (1% IgG Free BSA in PBS; Jackson ImmunoResearch) were incubated with 1 μL of Fc block (BD) for every 1 × 106 cells for at least 15 min at 4°C. Cells were then stained with the antibodies for 10 min at 4°C, washed with FACS buffer, and resuspended in FACS buffer containing DAPI (1 μg/mL) for live/dead cell exclusion, unless otherwise noted. Antibodies used for flow cytometry are listed in Table S1. For analysis, samples were run on a BD LSR II (Becton Dickinson), and all subsequent compensation and gating were performed with FlowJo analysis software (Tree Star).
Intracellular Flow Cytometry and DNA Content Analysis
For intracellular flow cytometry and DNA content analyses, cells were plated at a density of 1 × 105 per well in a 12-well plate, either alone, with an equal number of stromal cells, or BMDM CM. For co-cultures only CAG-EGFP BMDMs were used, and other stromal cell lines were fluorescently labeled with CellTracker Green CMFDA Dye (Molecular Probes). In experiments with BMDMs, the media were supplemented with 10 ng/mL recombinant mouse CSF-1 (R&D Systems). In experiments with MHCII+ BMDMs, the media were supplemented with 10 ng/mL recombinant mouse CSF-2 (R&D Systems). Twenty-four hours after plating, cells were treated and then collected at indicated time points by trypsinization. The samples were then fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) and stained overnight at 4°C using primary antibodies at the concentrations listed in Table S2. Allophycocyanin-tagged secondary antibodies (Jackson ImmunoResearch) were used at a 1:300 dilution and incubated for 1 hr at room temperature, followed by resuspension in FACS buffer containing DAPI (5 μg/mL) for DNA content evaluation. For analysis, samples were run on a BD LSR II (Becton Dickinson), and all subsequent compensation and gating were performed with FlowJo analysis software (Tree Star).
Live Imaging of Tumor Cell Response to Antimitotic Agents
For live imaging experiments, tumor cells were plated at a density of 1 × 105 per well in a 12-well glass-bottom plate (In Vitro Scientific), either alone, with an equal number of CAG-EGFP BMDMs, or with BMDM-CM. In experiments with BMDMs, the media were supplemented with 10 ng/mL recombinant mouse CSF-1 (R&D Systems). Twenty-four hours after plating, cells were treated and imaged for 24 hr using a Nikon Eclipse TiE inverted microscope equipped with an environmental chamber maintained at 35°C–37°C and 5% CO2. Mitotic arrest duration in live imaging movies was measured using Nikon Elements Software (Nikon) with at least 30 cells analyzed per treatment condition per experiment.
Additional information on experimental methods can be found in the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Tumor-associated macrophages (TAMs) reduce the duration of Taxol-induced mitotic arrest
TAMs promote cancer cell viability following mitotic slippage
The effects of TAMs on cancer cell response to Taxol are sensitive to MEK inhibition
Depletion of MHCIIlo TAMs increased the ability of Taxol to induce apoptosis in vivo
Acknowledgments
We thank Drs. Leila Akkari and John Maciejowski for critical review of the manuscript. We are grateful to Dr. Robert Benezra for invaluable experimental advice, Dr. Leila Akkari for assistance with oral gavage dosing, Nisarg Shah for excellent technical support, and other members of J.A.J.’s laboratory for insightful comments and discussion. We thank Novartis for supplying BLZ945, and Dr. Erik Sahai for providing the PyMT fibroblast cell lines. This research was supported by the Breast Cancer Research Foundation (J.A.J.), National Cancer Institute fellowship F31CA171384 (to O.C.O.), the Gerstner Sloan Kettering Graduate School (O.C.O.), the Silvian Foundation (O.C.O. and J.A.J.), the Geoffrey Beene Foundation (E.A.F., J.A.J., and O.C.O.), and National Cancer Institute Cancer Center Support Grant P30 CA008748 awarded to MSKCC.
Footnotes
AUTHOR CONTRIBUTIONS
O.C.O. and J.A.J. conceived the study, designed and interpreted experiments, and wrote the manuscript. O.C.O. performed all experiments. H.K. performed the live imaging analyses of mitotic arrest with O.C.O., and D.F.Q. performed orthotopic trials with O.C.O. All authors analyzed data and commented on the manuscript. J.A.J. and E.A.F. supervised the study.
Supplemental Information includes Supplemental Experimental Procedures, six figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.03.038.
References
- Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell. 2001;12:1315–1328. doi: 10.1091/mbc.12.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekier ME, Fischbach R, Lee J, Taylor WR. Length of mitotic arrest induced by microtubule-stabilizing drugs determines cell death after mitotic exit. Mol Cancer Ther. 2009;8:1646–1654. doi: 10.1158/1535-7163.MCT-08-1084. [DOI] [PubMed] [Google Scholar]
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637–646. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HW, Ling MT, Tsao SW, Wong YC, Wang X. Id-1-induced Raf/MEK pathway activation is essential for its protective role against taxol-induced apoptosis in nasopharyngeal carcinoma cells. Carcinogenesis. 2004;25:881–887. doi: 10.1093/carcin/bgh087. [DOI] [PubMed] [Google Scholar]
- Chittajallu DR, Florian S, Kohler RH, Iwamoto Y, Orth JD, Weissleder R, Danuser G, Mitchison TJ. In vivo cell-cycle profiling in xenograft tumors by quantitative intravital microscopy. Nat Methods. 2015;12:577–585. doi: 10.1038/nmeth.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23:277–286. doi: 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Grönroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RL, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014;4:175–185. doi: 10.1158/2159-8290.CD-13-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921–925. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Cornils H, Chiu SY, O’Rourke KP, Arnaud J, Yimlamai D, Théry M, Camargo FD, Pellman D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell. 2014;158:833–848. doi: 10.1016/j.cell.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. Cancer cells display profound intra-and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–122. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Holt SV, Logié A, Odedra R, Heier A, Heaton SP, Alferez D, Davies BR, Wilkinson RW, Smith PD. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br J Cancer. 2012;106:858–866. doi: 10.1038/bjc.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houthuijzen JM, Daenen LG, Roodhart JM, Oosterom I, van Jaarsveld MT, Govaert KM, Smith ME, Sadatmand SJ, Rosing H, Kruse F, et al. Lysophospholipids secreted by splenic macrophages induce chemotherapy resistance via interference with the DNA damage response. Nat Commun. 2014;5:5275. doi: 10.1038/ncomms6275. [DOI] [PubMed] [Google Scholar]
- Kawabata T, Tanimura S, Asai K, Kawasaki R, Matsumaru Y, Kohno M. Up-regulation of pro-apoptotic protein Bim and down-regulation of anti-apoptotic protein Mcl-1 cooperatively mediate enhanced tumor cell death induced by the combination of ERK kinase (MEK) inhibitor and microtubule inhibitor. J Biol Chem. 2012;287:10289–10300. doi: 10.1074/jbc.M111.319426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015;25:198–213. doi: 10.1016/j.tcb.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova AY, Seget K, Moeller GK, de Pagter MS, de Roos JA, Durrbaum M, Kuffer C, Müller S, Zaman GJ, Kloosterman WP, Storchová Z. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle. 2015;14:2810–2820. doi: 10.1080/15384101.2015.1068482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardin BR, Isokane M, Cosenza MR, Krämer A, Ellenberg J, Fry AM, Schiebel E. EGF-induced centrosome separation promotes mitotic progression and cell survival. Dev Cell. 2013;25:229–240. doi: 10.1016/j.devcel.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaid HM, Horwitz SB. Selective potentiation of paclitaxel (taxol)-induced cell death by mitogen-activated protein kinase kinase inhibition in human cancer cell lines. Mol Pharmacol. 2001;60:290–301. doi: 10.1124/mol.60.2.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaid HM, Lopez-Barcons L, Grossman A, Lia M, Keller S, Pé rez-Soler R, Horwitz SB. Enhancement of the therapeutic efficacy of taxol by the mitogen-activated protein kinase kinase inhibitor CI-1040 in nude mice bearing human heterotransplants. Cancer Res. 2005;65:2854–2860. doi: 10.1158/0008-5472.CAN-04-4391. [DOI] [PubMed] [Google Scholar]
- Menendez JA, Vellon L, Mehmi I, Teng PK, Griggs DW, Lupu R. A novel CYR61-triggered ‘CYR61-alphavbeta3 integrin loop’ regulates breast cancer cell survival and chemosensitivity through activation of ERK1/ERK2 MAPK signaling pathway. Oncogene. 2005;24:761–779. doi: 10.1038/sj.onc.1208238. [DOI] [PubMed] [Google Scholar]
- Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeso J, Weber J, Auerbach R. A hemangioendothelioma-derived cell line: its use as a model for the study of endothelial cell biology. Lab Invest. 1990;63:259–269. [PubMed] [Google Scholar]
- Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ. Analysis of mitosis and antimitotic drug responses in tumors by in vivo microscopy and single-cell pharmacodynamics. Cancer Res. 2011;71:4608–4616. doi: 10.1158/0008-5472.CAN-11-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panvichian R, Orth K, Day ML, Day KC, Pilat MJ, Pienta KJ. Paclitaxel-associated multimininucleation is permitted by the inhibition of caspase activation: a potential early step in drug resistance. Cancer Res. 1998;58:4667–4672. [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27:462–472. doi: 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26:623–637. doi: 10.1016/j.ccell.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff PB, Horwitz SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci USA. 1980;77:1561–1565. doi: 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze E, Asai DJ, Bulinski JC, Kirschner M. Posttranslational modification and microtubule stability. J Cell Biol. 1987;105:2167–2177. doi: 10.1083/jcb.105.5.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell-McGuinn KM, Zabor EC, Brogi E, Joyce JA. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25:2465–2479. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, Daniel D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. OncoImmunology. 2013;2:e26968. doi: 10.4161/onci.26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmans WF, Volm MD, Shapiro RL, Perkins AB, Kim AY, Demaria S, Yee HT, McMullen H, Oratz R, Klein P, et al. Paclitaxel-induced apoptosis and mitotic arrest assessed by serial fine-needle aspiration: implications for early prediction of breast cancer response to neoadjuvant treatment. Clin Cancer Res. 2000;6:4610–4617. [PubMed] [Google Scholar]
- Van Overmeire E, Stijlemans B, Heymann F, Keirsse J, Morias Y, Elkrim Y, Brys L, Abels C, Lahmar Q, Ergen C, et al. M-CSF and GM-CSF receptor signaling differentially regulate monocyte maturation and macrophage polarization in the tumor microenvironment. Cancer Res. 2016;76:35–42. doi: 10.1158/0008-5472.CAN-15-0869. [DOI] [PubMed] [Google Scholar]
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–392. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, Deangelo DJ, Frattini MG, Letai A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151:344–355. doi: 10.1016/j.cell.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SJ, Greer P, Auerbach R. Isolation and propagation of yolk-sac-derived endothelial cells from a hypervascular transgenic mouse expressing a gain-of-function fps/fes proto-oncogene. In Vitro Cell Dev Biol Anim. 1996;32:292–299. doi: 10.1007/BF02723062. [DOI] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–7029. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME, Weaver BA. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med. 2014;6:229ra43. doi: 10.1126/scitranslmed.3007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Zhou Y, Shi J. Post-slippage multinucleation renders cytotoxic variation in anti-mitotic drugs that target the microtubules or mitotic spindle. Cell Cycle. 2014;13:1756–1764. doi: 10.4161/cc.28672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.