

Abstract
Known gene mutations account for approximately 50% of the hereditary risk for breast cancer. Moderate and low penetrance variants, discovered by genomic approaches, account for an as yet unknown proportion of the remaining heritability. A truncating mutation c.325C>T:p.Arg109* (R109X) in the ATP-dependent helicase ERCC3 was observed recurrently among exomes sequenced in BRCA negative, breast cancer-affected individuals of Ashkenazi Jewish ancestry. Modeling of the mutation in ERCC3 deficient or CRISPR/Cas9 edited cell lines showed a consistent pattern of reduced expression of the protein and concomitant hypomorphic functionality when challenged with UVC exposure or treatment with the DNA alkylating agent IlludinS. Overexpressing the mutant protein in ERCC3-deficient cells only partially rescued their DNA repair-deficient phenotype. Comparison of frequency of this recurrent mutation in over 6500 chromosomes of breast cancer cases and 6800 Ashkenazi controls showed significant association with breast cancer risk (ORBC=1.53, ORER+=1.73) particularly for the estrogen receptor positive (ER+) subset (P<0.007).
INTRODUCTION
Genetic susceptibility to breast cancer has been shown to be strongly associated with rare coding gene mutations and common non-coding genomic variants (1). Loss of function mutations in well characterized genes, such as those in BRCA1/2 and other members of the homologous recombination pathway, are routinely used for clinical risk assessment in cancer, but account for a small fraction of excess familial risk (2). Common single nucleotide polymorphisms (SNPs) are yet to demonstrate clinical utility, but over 90 SNPs account for over 37% of familial relative risk (3). An as yet to be defined proportion of the remaining familial risk is accounted for by variants of “moderate” risk, including coding or non-coding variants in the DNA repair pathways such as nucleotide excision repair (NER), mismatch repair (MMR) and base excision repair (BER), which play an important role in checkpoint and genomic integrity maintenance (4, 5). In addition to serving as candidate cancer susceptibility genes, members of the NER pathway also play a critical role in DNA damage repair caused by chemotherapeutic agents and radiation exposure. ERCC3 is an ATP dependent DNA helicase that is part of the TFIIH transcription factor complex. In this report, we characterize the functions and demonstrate an association of a recurrent founder mutation in ERCC3 with breast cancer risk.
RESULTS
Identification of a Recurrent ERCC3 R109X Mutation in Ashkenazi Probands with BRCA1/2 Negative Breast Cancer
Exome sequencing was carried out on DNA extracted from peripheral blood/saliva of 46 early onset (<45 years) and 13 familial BRCA wild type breast cancer probands (33 with known ER positive status) of Ashkenazi Jewish ancestry. We filtered for rare protein truncating variants using public data sources such as ESP, 1000genomes and ExAC without TCGA. We further filtered for recurrent mutations in the DNA repair genes with low background mutation load using the smallest RVIS percentage (6) amongst 70 genes, calculated based on the ExAC data. We observed three individuals in the same BRCA wild type kindred (Fig. 1) with the same protein truncating mutation, R109X in ERCC3 (HGMD: c.325C>T: p.R109*; chr2:128050332; rs34295337). Sanger sequencing was used to confirm the next generation sequencing findings (shown in Supplementary Figure 1). Two of the siblings were affected with estrogen receptor positive breast cancer and the third was a male with breast cancer. The same mutation was also observed in two other individuals at relatively young age (<40 years), from unrelated kindreds with multiple cases of breast cancer. Details of the families and cancer incidence is described in Supplementary Table 1. The variant is almost absent in other world populations, while relatively rare in Caucasians in public data sources such as ExAC Consortium (Non-Finnish EUR MAF=0.0008, ESP-EUR MAF=0.0007). Analysis of the structure of the first 300 amino acids of ERCC3 shows that R109 is most likely part of a right handed alpha helix (Supplementary Figure 2). However, no crystal structure is available for high confidence prediction.
Figure 1.
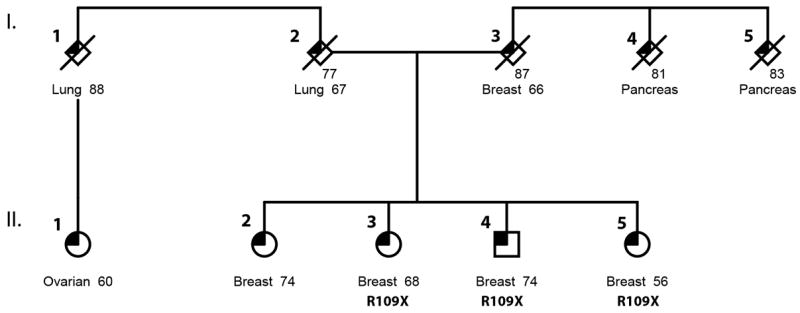
Identification of germline mutations in ERCC3 in a family with multiple breast cancer cases. Sequencing was performed on 3 individuals affected with breast cancer, confirming identification of ERCC3 R109X in all 3 affected siblings. Pathology reports for individuals II-3 and II-5, show both with well-differentiated (low grade) invasive ductal carcinoma diagnosed at Stage IA. One of the tumors was ER+PR+Her2+ (II-3) the other one was ER+PR-Her2- (II-5).
Phenotype Rescue by Complementation Assay
To understand whether this variant has a deleterious effect on the gene function, we carried out a series of in vitro functional assays. Using a previously well characterized ERCC3 deficient cell line derived from an XP/CS patient (7), phenotype rescue after DNA damage was assessed by cell viability assay. The parental XPCS2BA cell line and derived lines stably overexpressing the ERCC3 R109* mutant (R109X), or the wild type (WT) (Fig. 2A & 2B) showed varying degrees of sensitivity to DNA damage inducing agents such as UVC and a fungal sesquiterpene IlludinS. We observed that cells overexpressing ERCC3 R109X showed significantly higher cell death compared to the ERCC3 WT, while the untransfected cells all died upon exposure to higher doses of both IlludinS and UVC (Fig. 2C and 2D). Exposure at 2ng/ml of IlludinS (Fig. 2C), a fungal toxin from the Jack O’lantern mushroom (Omphalotus illudens) that creates DNA damage which blocks transcription with demonstrated antitumor activity (8), showed that 3%, 42% and 77% of the XPCS2BA, R109X and WT cells survived, respectively (P<0.0001). At a dose of 4ng/ml, most of the WT cells survived, demonstrating that the mutant cells were transiently and partially able to withstand UV induced DNA damage, while failing to adequately repair DNA damage to ensure survival. Similarly, following UVC irradiation at a dose of 10J/m2, 72 hours post-exposure, 42% of the R109X cells survived compared to 63% of WT cells (P<0.0001). At a dose of 4ng/ml, these numbers reduced more drastically for the mutant, showing the same trend as the IlludinS exposure experiment. (Fig. 2D).
Figure 2.
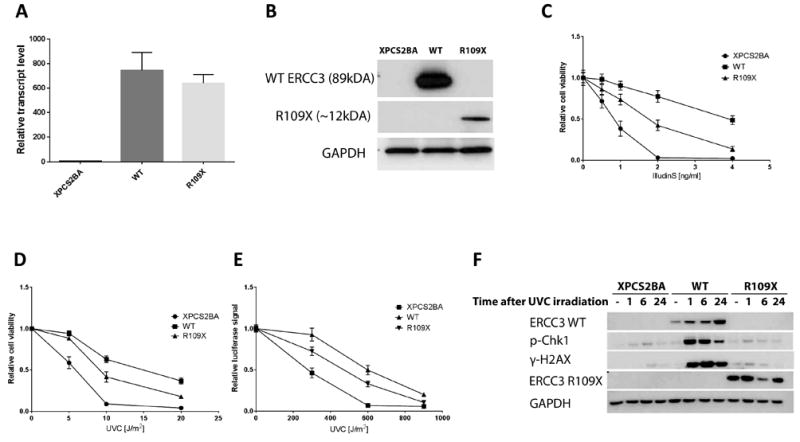
Functional Evaluation of the ERCC3 mutant via overexpression in an ERCC3 deficient cell line. (A) transcript and (B) protein levels in XPCS2BA parental cell line and cell lines stably overexpressing a lentiviral contruct containing wild-type ERCC3 (WT) or the R109X mutant (R109X) cDNA. Expression from the mutant cDNA produces a truncated protein fragment of about 12kDa. (C-D) Relative cell viablilty of XPCS2BA and ERCC3 wild-type and mutant overexpressing cell lines at 72 hours following treatment with increasing doses of IlludinS (C) or UVC (D). (E) Host cell reactivation assay showing reduced DNA repair ability of the mutant as opposed to wild-type ERCC3 overexpressing cell lines. Data represents the mean of three experiments with error bars representing the SD (C) or SEM (D, E). (F) Phosphorylation of H2AX and Chk1 in response to UVC-induced DNA damage. XPCS2BA, WT or R109X cell lines were harvested at different time points following exposure to 20J/m2 UVC and activation of H2AX and Chk1 was assessed by western blotting.
Host Cell Reactivation Assay
Using an exogenous source of DNA, namely a luciferase reporter plasmid, we measured the ability of intact live cells to repair DNA damage. Through measurement of the reporter, the extent of reactivation of the damaged plasmid is quantitated and is a readout of DNA repair activity. Relative luminescence measured after co-transfection of the reporter plasmid (exposed to 600J/m2) showed a 1.5 fold difference between mutant and WT overexpression cell lines, suggesting a markedly reduced reporter reactivation in the R109X (Fig. 2E) (P<0.0001). We observed almost no reactivation in the parental cell line at this dose.
Activation of the DNA Damage Response Pathway following UVC Irradiation
DNA damage response is marked by activation of H2AX (γ-H2AX) and leads to the induction of cell cycle checkpoint initiation by activation of Chk1. To explore the ability of ERCC3 R109X to trigger the checkpoint cascade, in response to DNA damage, we examined γ-H2AX and phosphorylated Chk1 following UVC-mediated DNA damage induction in XPCS2BA cells. UV-induced phosphorylation of H2AX is reduced in cells deficient in the nucleotide excision repair pathway (9). In agreement with this, we find lower levels of activated H2AX in the XPCS2BA cell line compared to cells where WT ERCC3 expression has been restored. In the mutant, we observed lower γ-H2AX levels, almost as low as in the parental cell line. Activation of the checkpoint kinase Chk1 is also strongly reduced in the mutant as compared to the WT overexpressing cells, and similar to the untransfected XPCS2BA (Fig. 2F).
Functional Characterization of Genome Edited ERCC3 Mutants in Human Mammary Epithelial Cells
Overexpression of ERCC3 mutant constructs does recapitulate physiological levels of protein in a germline heterozygous mutation state within cells. Therefore, using CRISPR/Cas9, we engineered several heterozygous mutations in the mammary epithelial cell line HMLE that mimic the site of the originally discovered recurrent mutation in breast cancer individuals (Supplementary Table 2, Supplementary Figure 3). Quantitative real time PCR of ERCC3 transcripts (Exons 1, 2 and Exons 9, 10) showed relative transcript levels reduced by half in these cells when compared to the parental HMLE cell line (Fig. 3A). Western blotting also showed the reduction in total ERCC3 protein levels (Fig. 3B) suggesting that in the CRISPR clones, ERCC3 is transcribed mainly from the remaining WT allele. Since we did not observe any homozygous mutations amongst the surviving CRISPR clones, we are unsure if a homozygous mutation is viable. The ERCC3 CRISPR clones generally showed substantial reduction in relative cell viability 72 hours after exposure to IlludinS (Fig. 3C). At 2ng/ml IlludinS, all the parental HMLE cells survived, while the CRISPR edited cell lines showed significantly reduced survival (P<0.0001). These data suggest that the mutants function at a much lower efficiency compared to WT and hence may be classified as hypomorphs. To demonstrate that the observed effect of IlludinS on the viability of the CRISPR edited HMLE is specific to the resulting ERCC3 deficit, we performed rescue experiments by overexpressing WT ERCC3 in these cells. We observed that the ERCC3 CRISPR cell lines, after stable overexpression of WT ERCC3, showed the same response to IlludinS as the WT HMLE cell line (Fig. 3D), thereby showing complete rescue of the phenotype confirming that this phenotype was indeed a result of the engineered ERCC3 deficiency.
Figure 3.
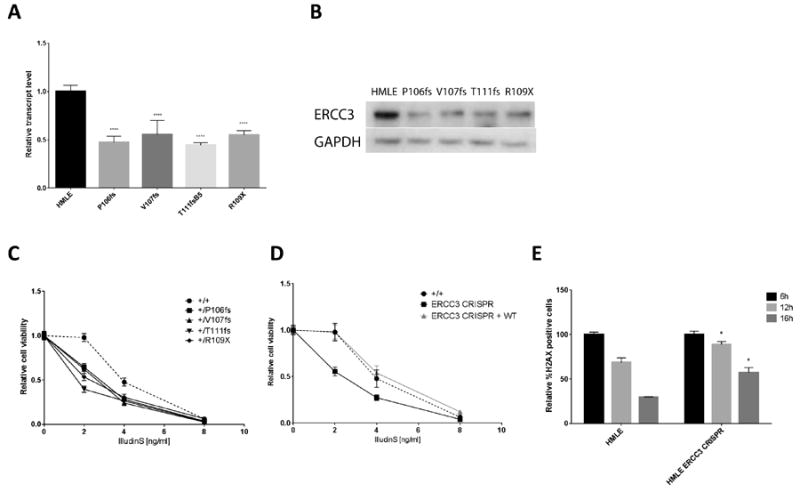
Modeling of ERCC3 R109X by CRISPR/Cas9 in a mammary epithelial cell line and functional analysis. (A) ERCC3 transcript and (B) protein levels in HMLE and CRISPR/Cas9 edited cells lines modeling the heterozygous R109X mutation. (C) Relative cell viability of HMLE control and CRISPR edited cell lines at 72 hours following treatment with increasing doses of IlludinS. Data represents the mean of three experiments with error bars representing the SEM. (D) Relative cell viability of HMLE control and combined CRISPR edited cell lines (with and without re-expression of the wild-type ERCC3) following treatment with IlludinS. (E) Quantification of phosphorylated H2AX by flow cytometry in HMLE control and CRISPR edited cell lines harvested at different time points after DNA damage.
To assess the induction and removal of phosphorylated H2AX in response to DNA damage we performed immunostaining and flow cytometric analysis of γ-H2AX positive cells following treatment with IlludinS. While this led to a similar fold induction of γ-H2AX positive cells in the wild type and CRISPR clones (not shown), the reduction in γ-H2AX was significantly lower in the ERCC3 CRISPR mutants, indicating a less efficient DNA damage repair in these cells (P<0.05) (Fig. 3E).
ERCC3 R109X as a Risk Allele for Breast Cancer in Ashkenazim
Using breast cancer affected individuals and controls from the Memorial Sloan Kettering Cancer Center, the University of Pennsylvania and the Clalit National Israeli Cancer Control Center, we performed a matched case control association of the ERCC3 variant using Taqman genotyping and sequencing across 3286 cases and 2716 controls. A third group of 705 controls were sourced from The Ashkenazi Genome Consortium (TAGC) project [(10) and in preparation] control resource (Table 1A). All control groups showed similar allele frequencies. 101 heterozygote carriers were present in the combined data. A 1.53 fold increased risk was observed for breast cancer (OR=1.53, lower CI= 1.07; P=0.023) after examining over 6500 chromosomes in cases and 6800 chromosomes in controls (Table 1A). A stronger association was observed with the ER positive subtype (OR =1.73, lower CI = 1.19, P=0.007) (Table 1B). The majority of the tumors in the carriers had an ER positive status. These data show that the ERCC3 c.325 T-allele is a moderate risk factor for breast cancer in individuals of Ashkenazi ancestry. “Unphased haplotype analyses from the TAGC heterozygote carriers suggested a founder mutation (Supplementary Figure 4) In the chromosomal region 2q14.3, a 3.4cM long haplotype was observed in the carriers which was significantly longer than in non-carriers (P<10-14).
Table 1.
| A:
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Center | Cases | Controls | Ca-Alt | Ca-Ref | Ctrl-Alt | Ctrl-Ref | P | OR | Lower-CI |
| MSKCC/PENN | 1968 | 1129 | 31 | 3905 | 10 | 2248 | |||
| CLALIT | 1318 | 1587 | 29 | 2607 | 22 | 3152 | |||
| TAGC | 0 | 705 | 0 | 0 | 9 | 1401 | |||
|
| |||||||||
| Total | 3286 | 3421 | 60 | 6512 | 41 | 6801 | 0.023 | 1.53 | 1.07 |
|
| |||||||||
|
B:
| |||||||||
| Center | Cases | Controls | Ca-Alt | Ca-Ref | Ctrl-Alt | Ctrl-Ref | P | OR | Lower-CI |
|
| |||||||||
| MSKCC/PENN | 1288 | 1129 | 23 | 2553 | 10 | 2248 | |||
| CLALIT | 992 | 1587 | 24 | 1960 | 22 | 3152 | |||
| TAGC | 0 | 705 | 0 | 0 | 9 | 1401 | |||
|
| |||||||||
| Total | 2280 | 3421 | 47 | 4513 | 41 | 6801 | 0.007 | 1.73 | 1.19 |
DISCUSSION
Here, we identified a truncating germline mutation in the first putative helicase domain of the DNA repair gene ERCC3, and report a strong genetic association with risk of breast cancer in individuals of Ashkenazi Jewish (AJ) ancestry. The R109X variant, seen in 1.83% of AJ breast cancer individuals and 2.06% in the ER+ subset, confers moderate risk (ORBC=1.53; ORER+=1.73). The same mutation was also observed twice as a somatic event in a breast carcinoma and a soft tissue sarcoma (Supplementary Figure 5A). Mutations within the ERCC3 helicase domain are also often seen in tumors such as melanoma, in addition to non-small cell lung cancer, colorectal, esophagogastric and bladder cancers. In general, ERCC3 is seldom disrupted in somatic tissue by genomic integrity loss such as amplification or deletions (Supplementary Figure 5B). In the LOVD database, the variant was seen in 6/8600 individuals of European ancestry, a similar frequency to public databases. As a core component of the TFIIH basal transcription factor, ERCC3 ATP-dependent DNA helicase has key functions in both RNA transcription by RNA polymerase II and in nucleotide excision repair (NER) following DNA damage (11). It has previously been associated with Mendelian DNA repair disorders such as xeroderma pigmentosum (XP), combined xeroderma pigmentosum/cockayne syndrome(XP/CS) and trichothiodystrophy (TTD) with hallmarks of increased photosensitivity, cancer predisposition and impaired development (12, 13). Hypomorphic mutations in the DNA damage repair gene NBN cause the autosomal recessive chromosomal instability disorder Nijmegen breakage syndrome (NBS) in homozygous individuals, but also have been shown to lead to increased cancer incidence in heterozygous relatives of NBS patients thus providing a plausible example of a hypomorphic mutation that is also a susceptibility gene (14, 15). Similarly, ERCC3 R109X behaves as a hypomorph in our functional assays. In experiments using an ERCC3 deficient cell line, R109X was partially successful at phenotype rescue, with the cells exhibiting intermediate repair capability, suggesting that they are more likely to harbor a second hit following genotoxic events. Importantly, ERCC3 transcript expression was shown to be lower in the proband’s cells compared to a non-mutation carrier (Supplementary Figure 6). Dosage-dependency of key molecular components of the DNA damage repair pathway, such as haploinsufficiency, has been previously demonstrated (16-20), leading to tumorigenesis in vitro and in vivo models of H2AX, BLM and CHEK1.
ERCC3 homozygous knockout mice have been shown to be embryonic lethal, indicating that the gene is necessary for development (21). Additionally, very few ERCC3 mutations have been reported, even amongst XP patients, suggesting the gene is intolerant to common mutational mechanisms. Analysis of gene based mutability from the ESP and ExAC exome data have shown that quantitative metrics that predict gene-conservation and mutation tolerance rank ERCC3 as bearing low background mutational load (22). In light of the rarity of known mutations in the ERCC3, there have thus far been no studies elucidating cancer susceptibility in heterozygous carriers. The only mouse model that has been described modeled the XP/CS hereditary DNA repair deficiency syndromes (21). Heterozygous knockout animals of NER genes XPC and XPE/DDB2 showed increased cancer incidence after exposure to UV irradiation (23, 24), while heterozygous XPC knockout mice also showed elevated frequency of spontaneous mutations as a function of age (25). Also, a mouse model harboring a mutation in the other helicase of the TFIIH complex, XPD, shows a strongly increased cancer incidence in response to UV exposure (26).
This is the first report that shows a specific truncating mutation in ERCC3 conferring increased risk to breast cancer. However, polygenes in DNA repair and/or other pathways, acting epistatically with ERCC3, are likely contributors to and modifiers of the heritable risk of breast cancer, complicating attempts to demonstrate co-segregation of single variants in multiplex kindreds. We have previously demonstrated a modestly elevated breast cancer risk associated with specific mutations of CHEK2 and APC in the Ashkenazi Jewish genetic isolate (27, 28) and other recurring mutations of CHEK2, HOXB13, PALB2, and RAD51C have been variably associated with breast cancer risk in diverse populations (29-32). While more frequent in Ashkenazi Jews, we have observed ERCC3 R109X as well as other ERCC3 mutations in non-Ashkenazi individuals (data not shown), but because of the rarity of these ERCC3 mutations, further population based studies will be required to assess their role as breast cancer susceptibility alleles. While the relative risks of 1.5-2.0 shown here are higher than those associated with most GWAS SNPs, the clinical utility of variants associated with risks in this range remain to be established. Nonetheless, these discoveries provide insight into molecular pathways of breast cancer pathogenesis, and potentially identify therapeutic targets (33-36). The enhanced susceptibility of ERCC3 mutant cells to reagents of the fungal sesquiterpene class, demonstrated here, suggests additional in vivo studies of the therapeutic efficacy versus toxicity of these compounds against breast tumors in an ERCC3 mutant genetic background.
MATERIALS & METHODS
Next Generation Sequencing of Breast Cancer Probands and Controls
One microgram of germline DNA extracted from peripheral blood was used for whole exome capture using the Agilent SureSelect 38Mb paired-end sequencing with the Illumina HiSeq 2000. Sequence reads in the form of FASTQ files were aligned to the human decoy reference (GRCh37) to generate BAM files using BWA v0.7.12. Picard tools was used for quality metric calculation and marking duplicate reads. The Genome Analysis Tool Kit (GATK) version 3.3.0-g37228af was used for variant calling using the haplotype caller algorithm. Variant quality score recalibrated (VQSR) data was used for filtering variants. Variant level and interval level annotations were performed using SNPEff, ANNOVAR, CAVA programs. Downstream analysis consisted of filtering out low quality variant calls and those already reported as common in public databases.
The TAGC (The Ashkenazi Jewish Genome Project) has sequenced the whole genomes of 128 and 577 individuals of AJ ancestry using the Complete Genomics and Illumina X10 sequencing platform at the New York Genome Center, respectively. The paired end libraries were generated using TruSeq DNA Nano kit, sequenced to an average depth of 30 and reads were aligned using a standard pipeline involving BWA version 0.7.8 and GATK version 3.2.2. Extensive QC is performed on all WGS samples, including alignment rates (>97%), median and mean library insert size (>350bp), percent duplication (typically <20%), mean genome coverage (>30x) and uniformity of coverage, TiTv and Het-Hom ratios. An automated concordance check is also performed against the SNP array genotyping data, to ensure against sample swap at any step during the sequencing process, and to further validate the quality of the SNV calls from the sequencing data.
All research participants provided written consent to an IRB-approved research protocol to allow for the collection and use of biospecimens.
Haplotype Analysis
Using only high quality biallelic SNPs, the haplotype lengths carrying the ERCC3 R109X mutation were calculated to either side of the mutation in the TAGC carriers as the length until an opposite homozygous genotype. This is an overestimate, due to the lack of accurate phasing information. The mean length was 3.4cM, estimated by using the Hapmap recombination rates for the region. The mean lengths of haplotypes shared between noncarriers were also computed. The significance of the difference between the distributions of haplotype lengths at the carriers and at 100 random non-carriers was calculated using the Kolmogorov-Smirnov test.
Sanger Sequencing
Sanger Sequencing of the ERCC3 R109X variant was performed using the following primers.
ERCC3_F1: CATGGAGCACCTATGCCTATT
ERCC3_R1: CTGCAACTCATGTTTCCTTGTC
Taqman Genotyping
The allelic discrimination assay C__25963434_10 for genotyping was done using Taqman (Life Technologies, Carlsbad, CA). The assay was run on an ABI HT7900 machine and automatically clustered and manually reviewed. Confirmed heterozygotes were run as positive controls and duplicate concordances checked per plate.
Statistical Analyses
Allele counts were tabulated from the Taqman genotyping after QC. Statistical analysis was performed using R statistical package using the fisher.test module. Since, the truncating genetic variant was hypothesized and shown by functional assays to lead to increased risk associated with reduced DNA repair efficiency, we report one-sided Fisher’s exact test results. Nevertheless, two sided tests for both breast cancer and ER status were also significant at 5% level (BC overall: P=0.036, OR=1.53, 95% CI=1.01-2.34; ER+: P=0.011, OR=1.73, 95% CI=1.11-2.70).
Plasmids
The pENTR221 plasmid containing human ERCC3 ORF was purchased from TransOMIC (Huntsville, USA). The ERCC3 ORF was cloned into the pLX302 lentiviral expression plasmid, a gift from David Root (Addgene # 25896). The ERCC3 R109X mutant was generated from the WT ERCC3 plasmid using the QuickChange II XL Site-Directed Mutagenesis Kit (Agilent). pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene # 48138). Guide RNAs were designed using the CRISPR Design Tool (37). The 24-mer oligonucleotides were synthesized (37)Integrated DNA technologies, Coralville, USA), annealed and cloned into pX458.
Cell Culture and Transfections
The HMLE cell line, established in the laboratory of Robert A. Weinberg at the Whitehead Institute for Biomedical Research, Cambridge, MA, were grown in mammary epithelial growth medium (MEGM) and supplements as recommended by Lonza. The XPCSBA-sv40 cell line was grown in RPMI 1640-HEPES medium (Invitrogen), supplemented with 10% FBS and 1% penicillin-streptomycin and Glutamate. Hek293T cells (ATCC, CRL-3216) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS and 1% penicillin-streptomycin and Glutamate. The HMLE and Hek293T lines were kindly provided by Robert Benezra (MSKCC) and the XPCSBA-sv40 cell line was a generous gift from Jan Hoeijmakers (Rotterdam, Netherlands). The cell lines were not further authenticated. Cell cultures were maintained in a humidified incubator at 37°C in 5% CO2 and tested for mycoplasma on a monthly basis.
Transfections were carried out with the Amaxa® Cell Line Nucleofector® Kit V (Lonza, Walkersville) and Nucleofector IIb device according to the manufacturer’s instructions. Viral vectors, co-transfected with psPAX2 and pseudotyped with VSV-G were produced in Hek293T cells using Lipofectamin2000 transfection reagent. The virus supernatant was concentrated by centrifugation for 90 minutes at 20,000 RPM at 4°C and pellets were dissolved in OptiMEM (Gibco). Transduction of cells with virus supernatant was carried out in the presence of 8μg/ml Polybrene. Stably transfected cell lines were generated by selection with 0.5μg/ml puromycin.
Cells co-transfected with pmaxGFP (Lonza) and the pX458-sgRNA plasmids, containing guide RNAs targeting the ERCC3 locus in close proximity to the ERCC3 mutation site (chr2: 128050332) with or without a repair template (ssODN) containing the specific mutation, were sorted as single cells into 96 well plates based on GFP fluorescence using a BD FACSAria™ cell sorter.
Screening of CRISPR/Cas9 Edited Cell Lines
Single cell clones generated from the transfection mentioned above were expanded and genomic DNA extracted using the QuickExtract DNA Extraction Solution (Epicentre, Madison, WI). The genomic region surrounding the target site was amplified using the following primer sequences:
ERCC3_SURV F, 5’- TGTGGTGTTGGGCAGCTTAT-3’;
ERCC3_SURV R, 5’- ACACTCACTTTGGGCTGCAT-3’;
The purified PCR products were subjected to Sanger sequencing.
Real-time PCR
RNA was extracted 24 hours after transduction using the RNeasy Mini Kit (Qiagen) and reverse transcribed with the ReadyScript cDNA Synthesis Mix (Sigma-Aldrich). Quantitative Real-time PCR analyses were performed on an ABI PRISM 7900HT Sequence Detection System using the Power SYBR® Green PCR Master Mix (Life Technologies) according to the manufacturer’s instructions. Following initial incubation for 10 min at 95°C, amplification was performed for 40 cycles at 95°C for 15 sec and 60°C for 1 min. The RPL32 gene was used as the internal standard. Analysis was performed based on the comparative CT method. Values reported are mean of triplicate experiments. The following primer sequences were used:
RPL32 F, 5’-CATCTCCTTCTCGGCATCA-3’;
RPL32 R, 5’-AACCCTGTTGTCAATGCCTC-3’;
ERCC3 F1, 5’- GTCCGCGAAGATGACAAAATTG-3’;
ERCC3 R1, 5’- AATTCAGGAGACATAGGGCAC-3’;
ERCC3 F3, 5’- ATGGGCAAAAGAGACCGAG-3’;
ERCC3 R3, 5’- CTGACTCATCCACCTGCTTC-3’;
Western Blotting
Protein lysates were prepared in RIPA buffer (Pierce). Samples were run on 4-12% gradient Bis-Tris SDS-PAGE gels (Invitrogen), transferred onto PVDF membranes (Bio-Rad) and probed with antibodies against ERCC3 (ARP37963_P050; 1:2,500; Aviva Systems Biology), phospho-Histone H2A.X (Ser139) (1:1000; Cell Signaling Technology), phospho-Chk1 (Ser345) (1:1000; Cell Signaling Technology) and GAPDH (V-18; 1:400; Santa Cruz Biotechnology). HRP-conjugated secondary antibodies were detected using ECL Prime Western Blotting Detection Reagent (GE Healthcare).
Cell Viability Assays
For viability assessment following treatment with IlludinS (Cayman Chemical), cells were seeded into 96 well plates 24 hours prior to treatment. For post-UV viability assays, cells were exposed to the appropriate doses of UV irradiation and subsequently seeded into 96 well plates. Cell viability was measured after 72 hours using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega).
Host Cell Reactivation Assay
The pIS0 plasmid, derived from the pGL3-control vector containing the Firefly luciferase gene (Promega, Madison, WI) and the pIS2, a derivative from the pRL-SV40 vector (Promega) containing the Renilla luciferase gene (as an internal transfection control) were used for transfection of cells. The pIS0 vector was treated with increasing doses of UVC radiation. These vectors were co-transfected into the XPCS2BA and stable ERCC3 WT and R109X overexpressing cell lines using the Fugene 6 Transfection Reagent (Promega, Madison, WI). Forty-eight hours after DNA transfection, the luciferases’ activity was measured with the Dual-Glo Luciferase Assay System (Promega) and a GloMAX Luminometer (Promega).
Flow cytometry
For flow cytometric analysis, 24h after seeding, cells were either left untreated or were treated with 8ng/ml IlludinS for 1h. Treated cells were subsequently washed with PBS and supplemented with drug-free medium. The cells were harvested at different time points after treatment and fixed with 4% PFA for 10min at RT. Cells were quickly chilled on ice and ice-cold MeOH was added to a final concentration of 90%.
The cells were incubated on ice for 30min and stored at -20°C. For immunostaining the cells were washed twice with PBS/0.5%BSA and incubated with anti phospho-Histone H2A.X (Ser139) Antibody (1:250; Cell Signaling Technology) for 1h at RT, washed again and incubated with Alexa-488 conjugated secondary antibody for 45min at RT. For each condition 20.000 cells were recorded and data from two replicate experiments was analyzed using the FlowJo software (V10.1).
Supplementary Material
Acknowledgments
Additional Contributions: We thank the members of the Clalit National Israeli Cancer Control Center and Department of Community Medicine and Epidemiology, Carmel Medical Center and B Rappaport Faculty of Medicine, Haifa, Israel and the New York Genome Center for the genotyping data. We also thank the members of the Ashkenazi Jewish Genome Project for the control genotypes, Drs. Benezra & Mayr from MSKCC and Dr. Hoeijmakers from Erasmus University for cell lines and plasmids.
GRANT SUPPORT
Funding/Support: This work was supported by the Sharon Corzine Cancer Research Fund, The Robert and Kate Niehaus Center for Inherited Cancer Genomics, the Miele family initiative [K.O.] and the Andrew Sabin Family Fund. This work was also supported by the National Institutes of Health R01CA176785 and the Breast Cancer Research Foundation [F.J.C., K.L.N., M.R., and K.O.], R21-CA139396 [J.V. & Z.S.], Department of Defense W81XWH-13- 1-0338 [K.N.M.]. Also supported by funds from the City of Hope Clinical Cancer Genetics Community Research Network and the Hereditary Cancer Research Registry, supported in part by Award Number RC4CA153828 (PI: J. Weitzel) from the National Cancer Institute and the Office of the Director, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the US Army or the Department of Defense. The work was also supported by the MSKCC Core grant NIH P30CA008748.
Role of the Funder/Sponsor: The funding institutions had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Disclosure of Potential Conflict of Interest: The authors do not have any conflicts of interest to disclose.
References
- 1.Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science. 2014;343:1466–70. doi: 10.1126/science.1251827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Easton DF, Pharoah PD, Antoniou AC, Tischkowitz M, Tavtigian SV, Nathanson KL, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. 2015;372:2243–57. doi: 10.1056/NEJMsr1501341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, Lush MJ, et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet. 2015;47:373–80. doi: 10.1038/ng.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Risinger MA, Groden J. Crosslinks and crosstalk: human cancer syndromes and DNA repair defects. Cancer Cell. 2004;6:539–45. doi: 10.1016/j.ccr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Brosh RM., Jr DNA helicases involved in DNA repair and their roles in cancer. Nat Rev Cancer. 2013;13:542–58. doi: 10.1038/nrc3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vermeulen W, Scott RJ, Rodgers S, Muller HJ, Cole J, Arlett CF, et al. Clinical heterogeneity within xeroderma pigmentosum associated with mutations in the DNA repair and transcription gene ERCC3. American journal of human genetics. 1994;54:191–200. [PMC free article] [PubMed] [Google Scholar]
- 8.Mcmorris TC, Kelner MJ, Wang W, Estes LA, Montoya MA, Taetle R. Structure-Activity-Relationships of Illudins - Analogs with Improved Therapeutic Index. J Org Chem. 1992;57:6876–83. [Google Scholar]
- 9.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9891–6. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carmi S, Hui KY, Kochav E, Liu X, Xue J, Grady F, et al. Sequencing an Ashkenazi reference panel supports population-targeted personal genomics and illuminates Jewish and European origins. Nature communications. 2014;5:4835. doi: 10.1038/ncomms5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaeffer L, Roy R, Humbert S, Moncollin V, Vermeulen W, Hoeijmakers JH, et al. DNA repair helicase: a component of BTF2 (TFIIH) basic transcription factor. Science. 1993;260:58–63. doi: 10.1126/science.8465201. [DOI] [PubMed] [Google Scholar]
- 12.Oh K-S, Khan SG, Jaspers NGJ, Raams A, Ueda T, Lehmann A, et al. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome. Human mutation. 2006;27:1092–103. doi: 10.1002/humu.20392. [DOI] [PubMed] [Google Scholar]
- 13.Singh A, Compe E, Le May N, Egly JM. TFIIH subunit alterations causing xeroderma pigmentosum and trichothiodystrophy specifically disturb several steps during transcription. American journal of human genetics. 2015;96:194–207. doi: 10.1016/j.ajhg.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seemanova E. An increased risk for malignant neoplasms in heterozygotes for a syndrome of microcephaly, normal intelligence, growth retardation, remarkable facies, immunodeficiency and chromosomal instability. Mutation research. 1990;238:321–4. doi: 10.1016/0165-1110(90)90024-6. [DOI] [PubMed] [Google Scholar]
- 15.Digweed M, Reis A, Sperling K. Nijmegen breakage syndrome: consequences of defective DNA double strand break repair. BioEssays : news and reviews in molecular, cellular and developmental biology. 1999;21:649–56. doi: 10.1002/(SICI)1521-1878(199908)21:8<649::AID-BIES4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 16.Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, et al. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–70. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- 17.Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–83. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, Slovek LE, et al. Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002;297:2051–3. doi: 10.1126/science.1074340. [DOI] [PubMed] [Google Scholar]
- 19.Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer cell. 2004;6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 20.Nikkila J, Parplys AC, Pylkas K, Bose M, Huo Y, Borgmann K, et al. Heterozygous mutations in PALB2 cause DNA replication and damage response defects. Nature communications. 2013;4:2578. doi: 10.1038/ncomms3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andressoo JO, Weeda G, de Wit J, Mitchell JR, Beems RB, van Steeg H, et al. An Xpb mouse model for combined xeroderma pigmentosum and cockayne syndrome reveals progeroid features upon further attenuation of DNA repair. Mol Cell Biol. 2009;29:1276–90. doi: 10.1128/MCB.01229-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diderich K, Alanazi M, Hoeijmakers JH. Premature aging and cancer in nucleotide excision repair-disorders. DNA Repair (Amst) 2011;10:772–80. doi: 10.1016/j.dnarep.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itoh T, Cado D, Kamide R, Linn S. DDB2 gene disruption leads to skin tumors and resistance to apoptosis after exposure to ultraviolet light but not a chemical carcinogen. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2052–7. doi: 10.1073/pnas.0306551101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheo DL, Meira LB, Burns DK, Reis AM, Issac T, Friedberg EC. Ultraviolet B radiation-induced skin cancer in mice defective in the Xpc, Trp53, and Apex (HAP1) genes: genotype-specific effects on cancer predisposition and pathology of tumors. Cancer research. 2000;60:1580–4. [PubMed] [Google Scholar]
- 25.Wijnhoven SW, Kool HJ, Mullenders LH, van Zeeland AA, Friedberg EC, van der Horst GT, et al. Age-dependent spontaneous mutagenesis in Xpc mice defective in nucleotide excision repair. Oncogene. 2000;19:5034–7. doi: 10.1038/sj.onc.1203844. [DOI] [PubMed] [Google Scholar]
- 26.Andressoo JO, Mitchell JR, de Wit J, Hoogstraten D, Volker M, Toussaint W, et al. An Xpd mouse model for the combined xeroderma pigmentosum/Cockayne syndrome exhibiting both cancer predisposition and segmental progeria. Cancer cell. 2006;10:121–32. doi: 10.1016/j.ccr.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 27.Shaag A, Walsh T, Renbaum P, Kirchhoff T, Nafa K, Shiovitz S, et al. Functional and genomic approaches reveal an ancient CHEK2 allele associated with breast cancer in the Ashkenazi Jewish population. Human molecular genetics. 2005;14:555–63. doi: 10.1093/hmg/ddi052. [DOI] [PubMed] [Google Scholar]
- 28.Redston M, Nathanson KL, Yuan ZQ, Neuhausen SL, Satagopan J, Wong N, et al. The APCI1307K allele and breast cancer risk. Nat Genet. 1998;20:13–4. doi: 10.1038/1666. [DOI] [PubMed] [Google Scholar]
- 29.Weischer M, Bojesen SE, Ellervik C, Tybjaerg-Hansen A, Nordestgaard BG. CHEK2*1100delC genotyping for clinical assessment of breast cancer risk: meta-analyses of 26,000 patient cases and 27,000 controls. J Clin Oncol. 2008;26:542–8. doi: 10.1200/JCO.2007.12.5922. [DOI] [PubMed] [Google Scholar]
- 30.Alanee S, Couch F, Offit K. Association of a HOXB13 variant with breast cancer. The New England journal of medicine. 2012;367:480–1. doi: 10.1056/NEJMc1205138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erkko H, Xia B, Nikkila J, Schleutker J, Syrjakoski K, Mannermaa A, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–9. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 32.Schnurbein G, Hauke J, Wappenschmidt B, Weber-Lassalle N, Engert S, Hellebrand H, et al. RAD51C deletion screening identifies a recurrent gross deletion in breast cancer and ovarian cancer families. Breast Cancer Res. 2013;15:R120. doi: 10.1186/bcr3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goyal G, Fan T, Silberstein PT. Hereditary cancer syndromes: utilizing DNA repair deficiency as therapeutic target. Fam Cancer. 2016 Jul;15(3):359–66. doi: 10.1007/s10689-016-9883-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sonnenblick A, de Azambuja E, Azim HA, Jr, Piccart M. An update on PARP inhibitors--moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12:27–41. doi: 10.1038/nrclinonc.2014.163. [DOI] [PubMed] [Google Scholar]
- 35.Chen MC, Zhou B, Zhang K, Yuan YC, Un F, Hu S, et al. The Novel Ribonucleotide Reductase Inhibitor COH29 Inhibits DNA Repair In Vitro. Mol Pharmacol. 2015;87:996–1005. doi: 10.1124/mol.114.094987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lok BH, Powell SN. Molecular pathways: understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin Cancer Res. 2012;18:6400–6. doi: 10.1158/1078-0432.CCR-11-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang F. Genome Engineering Resources - Zhang Lab @ MIT. N.p. [01 Apr. 2016];2013 Web. < http:www.genome-engineering.org/>.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.