

Abstract
The processes responsible for cytonuclear discordance frequently remain unclear. Here, we employed an exon capture dataset and demographic methods to test hypotheses generated by species distribution models to examine how contrasting histories of range stability vs. fluctuation have caused cytonuclear concordance and discordance in ground squirrel lineages from the Otospermophilus beecheyi species complex. Previous studies in O. beecheyi revealed three morphologically cryptic and highly divergent mitochondrial DNA lineages (named the Northern, Central, and Southern lineages based on geography) with only the Northern lineage exhibiting concordant divergence for nuclear genes. Here, we showed that these mtDNA lineages likely formed in allopatry during the Pleistocene, but responded differentially to climatic changes that occurred since the last interglacial (~120,000 years ago). We find that the Northern lineage maintained a stable range throughout this period, correlating with genetic distinctiveness among all genetic markers and low migration rates with the other lineages. In contrast, our results suggested that the Southern lineage expanded from Baja California Sur during the Late Pleistocene to overlap and potentially swamp a contracting Central lineage. High rates of intraspecific gene flow between Southern lineage individuals among expansion origin and expansion edge populations largely eroded Central ancestry from autosomal markers. However, male-biased dispersal in this system preserved signals of this past hybridization and introgression event in matrilineal-biased X-chromosome and mtDNA markers. Our results highlight the importance of range stability in maintaining the persistence of phylogeographic lineages, whereas unstable range dynamics can increase the tendency for lineages to merge upon secondary contact.
Keywords: cytonuclear discordance, range expansion, introgression, phylogeography, Pleistocene, Otospermophilus
Introduction
Understanding the spatial and temporal factors that influence the geographic distribution of genetic variation is a central goal in phylogeography (Avise 2000). One common phylogeographic pattern is cytonuclear discordance, or apparent differences in patterns of relative genetic divergence among organelle DNA and nuclear markers (Toews & Brelsford 2012). Several non-mutually exclusive processes have been invoked to explain these phenomena, including simple stochastic variation in coalescent times across loci (Rosenberg 2003), local adaptation of distinct mitochondrial DNA (mtDNA) lineages (e.g., Ribeiro et al. 2011; Pavlova et al. 2013), sex-biased dispersal (e.g., Turmelle et al. 2011; Brandt et al. 2012), and neutral introgression driven by range expansions during past climate oscillations (e.g., Cahill et al. 2013; Chavez et al. 2013). Despite the prevalence of cytonuclear discordance in natural systems, most previous studies have been unable to discern between several competing explanations for the described phylogeographic patterns (reviewed in Toews and Brelsford 2012).
Introgression during range expansions can produce patterns of cytonuclear discordance among closely related species through neutral demographic processes (Currat et al. 2008; Petit & Excoffier 2009; Excoffier et al. 2009), yet this mechanism has been difficult to test explicitly in empirical studies (but see Cahill et al. 2013). Range expansions occur through a series of founder events, ultimately creating clines of decreasing genetic diversity from the expansion origin to the expansion edge (Hewitt 1996; Slatkin & Excoffier 2012; Peter & Slatkin 2013). Range expansions increase the probability of genetic interactions between closely related taxa (species or lineages) through range overlap (Currat et al. 2008); if interbreeding is possible when an expanding taxon collides with a local one, genes from the local taxon are expected to become introgressed into the invading taxon (Currat et al. 2008; Excoffier et al. 2009). Introgression of the invading genome with the local genome occurs in part because individuals from the colonizing taxon will initially exist at low densities, causing heterospecific matings to be more likely than conspecific matings (Currat et al. 2008). However, continued intraspecific gene flow within the invading taxon from the expansion origin to the expansion edge can erode away the signal of introgression from the local taxon, unless the mode of inheritance for particular genetic markers impedes or prevents this process (e.g., sex-linked markers, organelle genomes, Petit and Excoffier 2009; Cahill et al. 2013).
Although past range fluctuations are common in nature and may be responsible for patterns of cytonuclear discordance, testing of this demographic scenario has been hindered by a limited capacity to properly test genetic predictions from range expansion theory. When range expansions are tested using genetic data, many studies test for demographic expansions assuming panmixia (e.g., Tajima’s D, Fu’s Fs, mismatch distributions) and incorporate spatial information afterwards to infer colonization routes (e.g., Rowe et al. 2004; Perktas et al. 2011; Vences 2013) rather than explicitly incorporating spatial information simultaneously. Further, range expansion theory makes explicit predictions about the relative degree of introgression among markers that experience differential levels of drift and intraspecific gene flow across the genome (Currat et al. 2008; Petit & Excoffier 2009). Accordingly, genome scale data is necessary to accurately infer these past demographic processes. Recently, Peter and Slatkin (2013) proposed a method based on a newly developed statistic ψ, the directionality index, to test for evidence of a range expansion against an equilibrium isolation-by-distance model and to locate the origin of expansion. The method utilizes pairwise population comparisons of two dimensional site frequency spectra (2D-SFS, i.e., a summary of allele frequencies between two populations) to (a) detect changes in allele frequencies across geography caused by range expansions and (b) locate the expansion origin by explicitly incorporating spatial data within the same inference framework (Peter & Slatkin 2013). This new method, coupled with advances in sequencing technology that now allow the rapid obtainment of data for 1,000s of loci from populations, enables a robust examination of the impact of range expansions in generating patterns of cytonuclear discordance.
Here, we examine factors that may have led to cytonuclear concordance and discordance within the Otospermophilus beecheyi species complex. O. beecheyi, is a common and abundant ground squirrel in western North America that exhibits male-biased dispersal (Dobson 1979, 1982) and inhabits mesic habitats such as grasslands and agricultural areas (Howell 1938). Previous mtDNA sequencing of this group revealed three highly divergent, morphologically cryptic lineages within O. beecheyi, denoted as the Northern, Central, and Southern lineages based on geography (Álvarez-Castañeda & Cortés-Calva 2011; Phuong et al. 2014). These three lineages are parapatrically distributed, where the Northern lineage ranges from southern Washington to northern California, the Central lineage is found in the Sierra Nevada mountain region, and the Southern lineage extends from northern California to Baja California with an allopatric population in Baja California Sur (Fig. 1a, Phuong et al. 2014). These mtDNA lineages differ by 7–8% in sequence divergence, with the Northern and Southern lineages being more closely related to each other than either is to the Central lineage (Álvarez-Castañeda & Cortés-Calva 2011). In contrast, genetic analyses from a small set of nuclear microsatellites and sequenced loci supported the genetic distinctiveness of the Northern lineage, while the Central and Southern lineages were found to not be evolutionary independent entities (Phuong et al. 2014). Based on these previous results, the Northern lineage was elevated to the species, Otospermophilus douglasii (Phuong et al. 2014), but we refer to it here as the Northern lineage within the O. beecheyi species complex for consistency. While previous work described patterns of divergence in both mtDNA and nuclear markers (Álvarez-Castañeda & Cortés-Calva 2011; Phuong et al. 2014), the genetic data were insufficient to infer past demographic histories. Thus, the processes responsible for cytonuclear discordance in this system have not yet been tested.
Figure 1.

(a) Mitochondrial Bayesian phylogeny inferred from 13 protein coding genes and map of the western United States and Central America depicting the approximate geographic distribution of each lineage with colored shapes representing sampling localities. Labels at nodes represent posterior probabilities. The phylogeny is rooted with Callospermophilus lateralis (not shown). (b) Scatterplot of the first two principal components calculated from SNP data. Circles represent allopatric samples, triangles represent samples from the N/C contact zone, and squares represent samples from the C/S contact zone, as shown in the map in 1a. (c) ADMIXTURE population assignments at K=2 and K=3 from SNP data, with individuals (vertical bars) arranged by lineage and latitude. mtDNA lineage for each individual shown below the ADMIXTURE results. In every graph, samples outside of the contact zones are labeled with a unique number. All individuals at the contact zones are labeled as CS or NC depending on the contact zone the sample originated.
To infer demographic processes that may explain why the Northern lineage exhibits concordance between mtDNA and nuclear markers while the Central and Southern lineages exhibit strong discordance, we employ an exon capture approach. We sequence ~2400 loci that vary in mode of inheritance (i.e., mtDNA, X-linked, autosomal) from individuals among the three lineages of the O. beecheyi species complex and for outgroup comparison, O. variegatus (a closely related species within Otosopermophilus). To generate hypotheses for how past climatic events could have created opportunity for range fluctuation and introgression, we generated distribution models of major mtDNA lineages under current and past climates. We then evaluate the resulting hypotheses against expected patterns of genetic diversity. We also examine the role of selection on the mtDNA as an alternative explanation to patterns of cytonuclear discordance in O. beecheyi.
Methods
Genetic sampling & data collection
We obtained tissue samples from 44 Otospermophilus individuals from several institutions (Fig. 1a, Table S1). Based on tissue availability, we maximized sampling of the geographic extent of each O. beecheyi lineage by including four Northern lineage individuals, six Central lineage individuals, 10 Southern lineage individuals, and three O. variegatus individuals. Each sample represents one individual per locality. We also included four individuals of the Northern lineage and two individuals of the Central lineage from a single region of contact between those two lineages (N/C contact zone, Fig. 1a, Table S1) and six individuals of the Central lineage and nine individuals of the Southern lineage from a locality where both mtDNA haplotypes co-occur (C/S contact zone, Fig. 1a, Table S1). In addition, we included one sample of Callospermophilus lateralis as a distant outgroup with which to polarize ancestry of single nucleotide polymorphisms (SNPs, Table S1). We extracted genomic DNA using Qiagen DNeasy Blood and Tissue kits and prepared index-specific libraries detailed in Meyer and Kircher (2010).
To identify genes for target capture, we performed reciprocal blasts via blastx and tblastn (BLAST+, Altschup et al. 1990) between the annotated Beldingi’s ground squirrel (Urocitellus beldingi) transcriptome (Accession ##, published in this study) and the annotated transcriptome of the Alpine Chipmunk (Tamias alpinus, Bi et al. 2012) and identified loci that were > 7% divergent between the two species to increase the probability of recovering polymorphic loci in O. beecheyi. The U. beldingi transcriptome was sequenced and annotated following RNAseq lab protocols and bioinformatic steps described in Bi et al. (2012). We chose these two species for probe design because of their availability and close phylogenetic affinity to O. beecheyi (Harrison et al. 2003). We ultimately targeted ~1.3 megabases consisting of 2305 nuclear protein coding genes and 13 mitochondrial protein coding genes on an Agilent SureSelect custom 1M-feature microarray. We inferred exon-intron boundaries by comparing U. beldingi transcripts with the genome of the thirteen-lined ground squirrel (Ictidomys tridecemlineatus) using EXONERATE (Slater & Birney 2005), identifying at least 3294 exons that were ≥ 200 bp in length. 249 of these exons were putatively X-linked and had corresponding orthologs on the X-chromosomes of human, mouse, rat, and dog. On the array, we used sequences from the U. beldingi transcriptome as our target sequence because it is more closely related to species within Otospermophilus. Each probe was 60bp in length and, to minimize edge effects, we tiled probes every 1bp for the first and last 6 bp of each locus, while probes were tiled every 4bp for the rest of the locus. We masked probes lying in short repeat or low complexity regions using the program repeatMasker (Smit et al. 2015) and repeated all probes three times on the array.
We pooled 50 libraries (45 made for this study and 5 used in another study) and hybridized this pool on the same array. Before hybridization, pooled libraries were denatured in the presence of excess blocking oligos and an excess 1:1 mixture of Cot-1 DNA isolated from Mus musculus and O. beecheyi. We isolated O. beecheyi Cot-1 DNA using the protocol described in Trifonov et al. (2009). We verified enrichment success using Bioanalyzer traces and through qPCR analysis of pre-capture and post-capture libraries using primers amplifying target and non-target regions, an approach modeled in Hodges et al. (2009) and Bi et al. (2012). We sequenced all individuals on a single Illumina HiSeq 2000 lane with 100bp paired-end reads.
Data filtration & assembly
We trimmed adapters and low quality bases using Trimmomatic (Bolger et al. 2014), merged overlapping paired-end reads using FLASH (Magoč & Salzberg 2011), removed reads with significant homology to human and bacterial (E. coli) genomes using bowtie2 (Langmead & Salzberg 2012), and removed duplicate and low complexity reads using custom Perl scripts. For each individual, we generated raw assemblies with a range of five k-mer values from 21–61 in ABySS (Simpson et al. 2009) and merged assemblies across k-mers using cd-hit (Li & Godzik 2006) and cap3 (Huang & Madan 1999)
To generate a mtDNA protein reference for each lineage, we first annotated mitochondrial proteins for one individual using DOGMA (Dual Organellar GenoMe Annotator, Wyman et al. 2004). Then, we chose one individual from each of the other lineages and performed a reciprocal blast via blastn to create lineage specific mtDNA protein references. Finally, we reconstructed protein coding sequences for each individual by aligning reads to these references using Novoalign (http://novocraft.com) and calling the base at each site directly from read depth. We masked sites that had less than 10X coverage or were not homozygous (minor allele frequency > 20%).
To generate a nuclear gene reference for all samples, we merged all final assemblies from every Otospermohphilus sample using cd-hit and cap3 and performed a reciprocal blast using blastn to identify contigs that matched the targeted exons. To account for chimeric contigs, we performed a self-reciprocal blast with blastn and removed contigs that had > 90% identity to each other. To generate a SNP dataset from the sequenced nuclear exons, we aligned all reads to the nuclear reference using Novoalign for each sample and identified variant sites using SAMtools (Li et al. 2009). To remove potentially paralogous sequences, we used a perl script (SNPcleaner, Bi et al. 2013) to remove contigs out of Hardy-Weinberg Equilibrium (HWE) from samples within each lineage that were outside the contact zones. We did not perform HWE filtering for O. variegatus due to low sample sizes. We kept variant sites with a minimum depth of 3X in at least 40 individuals and generated genotype likelihoods for every site within an individual using ANGSD (Korneliussen et al. 2014). We masked sites with ‘Ns’ when genotype posteriors were below 0.95. The bioinformatic scripts used in this study can be found at https://github.com/CGRL-QB3-UCBerkeley/denovoTargetCapturePopGen.
To assess the success of the capture experiment, we calculated (a) percent targeted bases covered by at least one read, (b) percent of reads aligned to intended targets and (c) sequence coverage for each individual.
mtDNA analysis
We aligned and concatenated 13 mtDNA protein coding genes for all 45 individuals using MUSCLE (Edgar 2004) and inferred a phylogeny using MrBayes (Huelsenbeck & Ronquist 2001) under the partitioning scheme and substitution models estimated by PartitionFinder (Lanfear et al. 2012). We rooted the tree with C. lateralis and we sampled every 10,000 generations over 10 million generations with a 4 million generation burn-in. Further, because selection can explain genealogical discordance between mitochondrial and nuclear genealogies (Ballard & Whitlock 2004; Toews & Brelsford 2012), we tested for evidence of selection on mitochondrial proteins by comparing two site models (M1a and M2a) as implemented in PAML (Yang 2007).
Nuclear population structure analysis
We characterized nuclear genetic structure in two ways: (a) we performed a principal component analysis (PCA) using smartpca from EIGENSOFT v4.2 (http://www.hsph.harvard.edu/alkes-price/software/), and (b) we inferred population assignment in ADMIXTURE, which uses a likelihood approach to estimate ancestry (Alexander et al. 2009). We ran smartpca on all Otospermophilus individuals and only on individuals with Central and Southern lineage mtDNA. We ran ADMIXTURE for K=2 and K=3 populations on individuals within the O. beecheyi species complex. For these analyses, we randomly sampled 1 SNP per gene because both methods assume that SNPs are unlinked.
Quantifying cytonuclear discordance
To quantify the degree of cytonuclear discordance in this system, we calculated sequence divergence (Dxy) for all possible pairwise comparisons across all O. beecheyi mtDNA lineages. We calculated mtDNA sequence divergence (Dxy.mito) using Arlequin (Excoffier et al. 2005), nuclear sequence divergence (Dxy.nuclear) using a perl script, and the ratio between the two values (Dxy.mito / Dxy.nuclear).
Modeling past lineage dynamics
To generate hypotheses for how glacial cycles impacted range dynamics, we modeled distributions across several climate eras using the following grouping of occurrence records: (a) only Northern (b) only Central (c) only Southern (d) Central + Southern and (e) all lineages. We chose to model distributions under varying partitioning strategies because (a) within-species structuring (i.e., subspecies, cryptic lineages) can impact model predictions (Pearman et al. 2010) and (b) we make the assumption that the mtDNA lineages once represented independent evolutionary entities that may have responded differentially to climatic shifts due to being confined to different geographic areas. We partitioned occurrence records from VertNet (vertnet.org; accessed on 30 January 2014) based on the geographic distribution of each mtDNA lineage. We included additional records (Álvarez-Castañeda & Cortés-Calva 2011; Phuong et al. 2014) that were not yet available on VertNet and we removed records if coordinate uncertainties were >10 km or were not reported. To avoid spatial autocorrelation in sampling, we thinned the data by randomly selecting one occurrence record per occupied cell in the bioclimatic layers using a custom R script (R Development Core Team, 2014). The final dataset included 161 unique records for the Northern lineage, 52 for the Central lineage, and 327 for the Southern lineage. We obtained 2.5 arc-minute resolution climate layers from the WorldClim database (Hijmans et al. 2005) under conditions for the present and the Last Glacial Maximum (LGM, ~22,000 years ago) reconstructed under the Community Climate System Model. For the Last InterGlacial (LIG, ~120,000 – 140,000 years ago), we reduced the resolution of the LIG climate layers to 2.5 arc minutes in ARCMAP from the available 30 arc-second resolution layers on the WorldClim database.
We generated species distribution models using Maxent 3.3.1 (Phillips et al. 2006) and parameterized Maxent using 7 of the 19 BIOCLIM variables that were not highly correlated with each other (Pearson correlation coefficient |r| < 0.7, Table S2). To account for sampling bias in occurrence records (VanDerWal et al. 2009), we treated occurrence records of each mtDNA lineage and occurrence records of closely related ground squirrel taxa whose ranges overlap with the focal lineages in this study as pseudo-absences (Table S3, Fig. S1). Occurrence points from closely related taxa were filtered with the same criteria described above. We trained all models on current conditions and projected species distributions onto climates representing each time slice. For each model, we used the following parameters in Maxent (regularization value of 1, convergence threshold of 0.00001, maximum iterations of 500), executed 10 replicates, and generated a consensus distribution map by averaging across all replicates that was outputted in logistic format. We evaluated model performance for the predicted contemporary distribution of each lineage by generating cross-validated Area Under the Curve (AUC) values. The AUC is the probability that the model ranks the presence site higher than the absence site. AUC values range from 0 to 1, with a value of 0.5 indicating that the model has no predictive ability while values closer to 1 implies better models (Swets 1988).
Demographic analyses
To estimate the timing of divergence and the degree of nuclear gene flow between the lineages in this study, we used the program ∂a∂i to fit a three population isolation-with-migration model to the nuclear SNP data (Gutenkunst et al. 2009). ∂a∂i uses a diffusion-based approach to fit an expected demographic model to frequency spectrum data (Gutenkunst et al. 2009). Based on the mtDNA phylogeny (Fig. 1a), we constructed a model where the Central lineage diverges first from an ancestral population, and then the ancestral population splits into the Northern and Southern lineages. After divergence, migration is allowed to occur between lineages and happens continuously throughout their history. Based on previous species tree reconstructions inferred from nuclear sequence data (Phuong et al. 2014), we also constructed a similar isolation-with-migration model, but with the Northern lineage diverging first (Fig. S2). As we are primarily interested in historical, rather than current, introgression, we only included individuals outside of the contact zones for this analysis, except for one Northern lineage individual at the N/C contact zone that clustered with allopatric Northern lineage samples. We removed SNPs that were either (1) nonsynonymous variants, (2) found on X-linked exons, (3) immediately adjacent to each other, or (4) had less than 5 diploid genotypes confidently called per lineage because of assumptions made by the ∂a∂i algorithm (Gutenkunst et al. 2009). We projected down to 10 allele copies per lineage for model fitting and performed the analyses on the folded three dimensional site frequency spectrum (3D-SFS), a summary of shared and private allele frequencies between three populations. We note that formal power analyses have not been conducted with regards to the optimal number of samples for parameter estimation because this is highly dependent on the system and the demographic model. However, results from previous studies imply that 5 diploid individuals are sufficient to estimate relatively ancient events and achieve the goals of this study (McCoy et al. 2014; Robinson et al. 2014). To estimate parameter uncertainties, we used a nonparametric bootstrapping approach, where we generated 100 equal sized datasets by sampling SNPs with replacement from genes in the dataset using a Python script and repeated the ∂a∂i analysis. To convert all parameter estimates from coalescent units to real units, we followed equations and guidelines provided in (Gutenkunst et al. 2009), which incorporates information including effective sequence length, substitution rate, and generation time. We used the average mammalian substitution rate of 2.2E-9 per base per year (Kumar & Subramanian 2002) and assumed the generation time in O. beecheyi to be 1 year (Dobson 1982).
Because our distribution modeling predicted that the Southern lineage expanded its range, we implemented the method described in Peter and Slatkin (2013) in R (R Development Core Team, 2014) to test for range expansion and to locate its origin on combined samples from the Central and Southern lineages (16 individuals), again excluding individuals at the contact zones. The script was provided by B. M. Peter. We included only synonymous nuclear variants and polarized the site frequency spectra with C. lateralis. While 20 diploid individuals are optimal to minimize error of the location of the expansion origin, it is unclear how many samples are necessary to detect range expansions (Peter & Slatkin 2013). Nonetheless, with ~2000 loci and 15–20 geographically dispersed individuals, the method has yielded useful insights into historical range expansions (Potter et al. 2016).
Signatures of introgression due to a recent range expansion should be most evident in markers that have reduced intraspecific gene flow, such as in mtDNA or the X-chromosomes in mammals (Petit & Excoffier 2009; Cahill et al. 2013). Although high intraspecific gene flow can rapidly erode away strong signals of introgression in the autosomes, the reduced levels of intraspecific gene flow experienced by the matrilineal-biased X-chromosomes should retain elevated signatures of past introgression between species, especially if male-biased dispersal is prevalent (Cahill et al. 2013). One prediction from these range dynamics is that we should expect elevated divergence in the X-chromosome relative to autosomes when comparing samples close to the expansion origin vs. the expansion edge because individuals in the recently colonized area will contain X-chromosomes of mixed ancestry (Cahill et al. 2013). Because of relatively high migration rates between the Central and Southern lineages estimated through ∂a∂i and evidence for range expansion from our species distribution modeling and the genetic range expansion test from Peter and Slatkin (2013), we tested for signatures of introgression on the X-chromosome between the Central and Southern lineages. First, we generated the ratio of X-chromosome divergence to autosome divergence (Xdiv/Adiv) by calculating uncorrected pairwise distances from the SNP dataset for female samples from all O. beecheyi lineages using a python script. Then, we categorized the pairwise comparison as either between (a) a sample from the origin vs. a sample outside of the origin, (b) only samples located outside of the origin, or (c) a Northern sample vs. a sample from another lineage. We performed comparisons including three Northern lineage females to determine baseline Xdiv/Adiv values when there is no assumed history of introgression between the two individuals being compared. Based on results from the range expansion analysis, samples from the origin included two females from Baja California Sur, while samples outside the origin consisted of six other females from the Southern and Central lineages, exclusive of individuals from the contact zones. Using R, we performed an analysis of variance (ANOVA) to test for differences between these categories and conducted a post-hoc Tukey HSD test to determine which categories were significantly different from each other.
Results
Sequence capture statistics
We sequenced an average of 6.7 million reads per sample (Table S1). We recovered all mtDNA protein coding genes, sequencing an average of 99.9% of the bases. For each sample, approximately 7% of the reads mapped to the mtDNA protein coding genes, resulting in an average depth of 2495.6X (Table S1). We found corresponding orthologs for all nuclear exons targeted, generating a reference that was roughly 2.5 megabases in length (inclusive of flanking introns). We recovered an average of 97% of the bases with 8.7% of the reads mapping to the targeted exons, generating an average coverage of 17.6X (Table S1). We found high variance in percent reads on target and coverage in our dataset, despite attempts at equimolar pooling before hybridization with the array (Table S1). 354 of the targeted 3294 exons were removed due to being out of HWE. After further data filtering and genotype calling, we discovered 14,117 nuclear SNPs across 2361 loci (exons + flanking introns) that we used for subsequent analyses.
mtDNA analysis
We recovered a mtDNA phylogeny with four divergent lineages corresponding to O. variegatus and the Northern, Central, and Southern lineages within the O. beecheyi complex (Fig. 1a). The topology and support values are consistent with previous studies that inferred relationships using a single mtDNA locus (Fig. 1a, Álvarez-Castañeda and Cortés-Calva 2011; Phuong et al. 2014). In particular, this more extensive dataset confirms that the Central lineage is more divergent from the Northern and Southern lineages than they are from each other. However, even with sequence data from 13 mtDNA protein coding genes, we were unable to infer the earliest diverging mtDNA lineage within Otospermophilus (Bayesian posterior probability = 0.7, Fig. 1a).
Both the nearly neutral model (M1a) and positive selection model (M2a) had equal likelihoods (ℓ = −20668.38), and the positive selection model did not identify any mtDNA sites under positive selection (posterior probability < 0.95, Table S4).
Nuclear structure analysis
The first two PCs explained 31.3% of the variance in the nuclear SNP data (Fig. 1b, Fig. S3). The PCA plot revealed three clusters that correspond to O. variegatus, the Northern lineage, and a mixture of Central and Southern lineage individuals (Fig. 1b). When performing the PCA analysis only on Central and Southern lineage individuals, there was slight separation between Southern lineage individuals, Central lineage individuals, C/S contact zone individuals, and N/C contact zone individuals, but this separation is relatively minor when Northern lineage and O. variegatus individuals were included in the analysis (Fig. 1b, Fig. S4). ADMIXTURE results showed that the Northern lineage was distinct at both K = 2 and K = 3, with evidence of localized hybridization at the N/C contact zone, where all but one individual showed ancestry from a separate population (Fig. 1c). The Central and Southern lineages are indistinguishable from each other at K = 2 (Fig. 1c). At K = 3, the Central and Southern lineages showed evidence of admixture between some allopatrically distributed individuals. All individuals at the contact zone of Central and Southern mtDNA lineages clustered with the Central lineage for nuclear genes (Fig. 1c). These results are consistent with population structure previously inferred from a significantly smaller set of microsatellites and sequenced nuclear loci, such that the Northern lineage is genetically distinct, but the Central and Southern lineages show signs of considerable admixture across large geographic distances (Phuong et al. 2014).
Cytonuclear discordance
Average Dxy.mito was 0.063 and average Dxy.nuclear was 0.00024 (Table 1). When comparing divergence ratios, the comparison between the Central and Southern lineage was a clear outlier (Dxy.mito / Dxy.nuclear = 45.6, Table 1). Divergence ratios for all other lineage comparisons was 24.3 on average (Table 1).
Table 1.
All unique lineage comparisons of mitochondrial sequence divergence (Dxy.mito), nuclear sequence divergence (Dxy.nuclear), and the ratio between mitochondrial and nuclear divergence (Dxy.mito / Dxy.nuclear).
| Lineage 1 | Lineage 2 | Dxy.mito | Dxy.nuclear | Dxy.mito / Dxy.nuclear |
|---|---|---|---|---|
| Central | Northern | 0.0629 | 0.0025 | 25.2 |
| Central | Southern | 0.0638 | 0.0014 | 45.6 |
| Central | O. variegatus | 0.0640 | 0.0026 | 24.6 |
| Northern | Southern | 0.0572 | 0.0025 | 22.9 |
| Northern | O. variegatus | 0.0665 | 0.0029 | 22.9 |
| Southern | O. variegatus | 0.0671 | 0.0026 | 25.8 |
Distribution modeling
We were able to acceptably model the contemporary distribution of each lineage (as reflected by current mtDNA distributions) under current climatic conditions, as indicated by the mean AUC values for the Northern (AUCtest = 0.86, AUCtraining = 0.89), Central (AUCtest = 0.90, AUCtraining = 0.92), and Southern (AUCtest = 0.82, AUCtraining = 0.85) lineages. AUC values decreased when modelling the Central and Southern lineage as one entity (AUCtest = 0.78, AUCtraining = 0.80) or when modelling all lineages as one entity (AUCtest = 0.72, AUCtraining = 0.74). Models for the Northern lineage predicted suitable habitat in areas that closely match its current distribution across all time periods, suggesting range stability over the last 120 thousand years (Fig. 2a). For the Central lineage, models predicted a decline in suitable habitat within the Sierra Nevada (where it is currently distributed) since the LIG (Fig. 2b). The LIG and LGM models for the Central lineages also predicted habitat suitability in areas where it is not currently found today, which may be due to non-climactic variables such as competition or land cover (Fig. 2b). For the Southern lineage, the LIG model predicted a restricted distribution in Baja California and along the coast of California, with habitat suitability increasing across California towards the present (Fig. 2c). This pattern suggests a potential range expansion of the Southern lineage from Baja California northwards into its current distribution in California. The Central + Southern model was similar to the only Southern lineage model, predicting a potential range expansion from Baja California (Fig. S5). When all lineages are modelled as one entity, the LIG model predicted suitable habitat from Baja California and along the coast of the western United States, with increasing suitability eastward towards the present (Fig. S5). We focus on the lineage-specific models for the rest of the study because these provide the hypotheses to be evaluated using the genetic data within Otospermophilus.
Figure 2.

Species distribution models for the (a) Northern lineage, (b) Central lineage, and (c) Southern lineage from climates during the last interglacial (~120–140 thousand years before present (kybp)), the last glacial maximum (~20 kybp), and current conditions. Occurrence points used in distribution models are displayed on current climate maps and are colored black for the Northern lineage, orange for the Central lineage, and blue for the Southern lineage.
Demographic analyses
Demographic modelling in ∂a∂i, conditioned on the mtDNA phylogeny, revealed that the Central lineage diverged from an ancestral Northern and Southern lineage roughly 700,000 years ago, and the Northern and Southern lineages diverged from each other approximately 696,000 years ago (Fig. 3, Table S5). The Southern lineage is inferred to have had the larger effective population size compared with the Northern and Central lineages, which had similar sizes to each other (Fig. 3, Table S5). Crucially, inferred migration rates between the Southern and Central lineages were an order of magnitude higher than between the Northern lineage and either the Central or Southern lineages (Fig. 3, Table S5). Although confidence intervals for the migration rates overlap (Table S5), the migration rate between the Southern and Central lineages was higher than migration rate estimates with the Northern lineage in every bootstrap replicate (Fig. S6). Migration rates were similar when the demographic model was conditioned on the alternative topology based on previous phylogenetic analyses (Phuong et al. 2014, Table S6, Fig. S2, S6)
Figure 3.
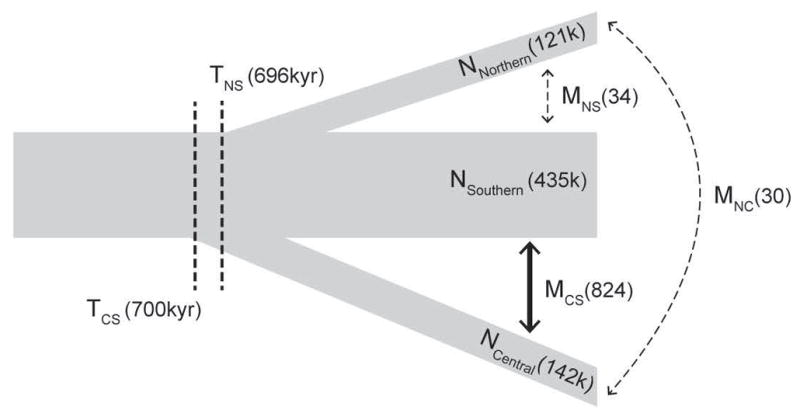
Schematic of the three population isolation-with-migration model with the Central lineage diverging first and the Northern and Southern lineages diverging thereafter. Point estimates reported in parentheses next to each demographic parameter: Divergence times T reported in thousands of years (kyr), effective population sizes N reported in thousands of individuals, and effective migration rates M reported in effective migrants per generation. Confidence intervals for parameter estimates are provided in Table S3.
Using the directionality index, ψ, and the method developed by Peter and Slatkin (2013), we detected strong support for a range expansion (p < 0.0001) and located its origin to Baja California Sur (Fig. 4a) when including all Central and Southern lineage samples as a single population. This result is consistent with predictions from the modeling of potential lineage distributions through time.
Figure 4.

(a) Map of California and Baja California showing probability of origin of the range expansion. Grey represents no probability, and red represents high probability. Points on map represent samples used in the analysis. orange = Central lineage, blue = Southern lineage. (b) Ratio of X-chromosome divergence to autosome divergence when calculating genetic distances between (1) an individual from the origin vs. an individual outside of the origin (Origin vs. “outside origin”), (2) only individuals located outside of the origin (within “outside origin”), or (3) a Northern individual vs. an individual from the Central or Southern lineage (Northern vs. Central + Southern).
Theory predicts that introgression events caused by a range expansions should lead to higher levels of divergence in the X-chromosome relative to the autosomes when comparing individuals from the expansion edge vs. the expansion origin (Cahill et al. 2013). The mean Xdiv/Adiv ratio for comparisons between samples from the expansion origin and expansion edge was 1.48 while comparisons from samples within the colonized area was 1.21 (Fig. 4b). The Xdiv/Adiv ratio when comparisons are made with a Northern lineage individual was 1.1 (Fig. 4b). We found significant differences in these ratios between the three categories (ANOVA, F = 16.37, p < 0.0001, Fig. 4b) and the Tukey HSD test indicated that Xdiv/Adiv was higher in the origin vs. “outside origin” category relative to the other two categories (p < 0.01).
Discussion
Using species distribution models to guide demographic genetic analyses, we provide the following demographic scenario to explain patterns of concordance and discordance among mtDNA and nuclear markers in the O. beecheyi species complex: we propose that the mtDNA lineages represent evolutionary entities that were isolated from each other some time during the Pleistocene. Under this scenario, our demographic modelling placed these splits within the last several hundred thousand years (Fig. 3). In isolation, these lineages accumulated enough divergence to gain genetic distinctiveness in both mtDNA and nuclear DNA, as indicated by concordant genetic structuring in the nuclear genome for 3 out of the 4 mtDNA lineages within Otospermophilus (Fig. 1). Since the LIG, these lineages have responded differentially to climatic fluctuations which have shaped their present day genetic structure. Distribution models for the Northern lineage predicted a stable range since the LIG (Fig. 2a), which correlates with genetic distinction in both mtDNA and nuclear markers (Fig. 1) and a relatively lower rate of migration with the Central and Southern lineages (Fig. 3). These results suggest that the Northern lineage has maintained its evolutionary independence since isolation, despite demonstrated potential to hybridize locally with the adjacent Central lineage (as in the current contact zone, Fig. 1), because its distribution has been more stable across recent climatic shifts.
In contrast, Southern lineage distribution models suggested a recent range expansion from Baja California to its present day distribution in California (Fig. 2c), consistent with our genetic analyses showing statistical support for an expansion from the same region (Fig. 4a). As the Southern lineage expanded northwards across California, it invaded the range of the Central lineage and hybridized with it, causing Southern lineage individuals at the wave front to become massively introgressed by Central lineage nuclear genes (as predicted by range expansion theory, Currat et al. 2008; Petit and Excoffier 2009). High intraspecific gene flow, as evidenced by relatively high migration rates and low levels of genetic divergence among samples classified as Central and Southern individuals (Fig. 1, 3), gradually eroded away significant signals of this hybridization event among the autosomes. Due to strongly male-biased dispersal in this system (Dobson 1979, 1982), erosion of Central ancestry on the X-chromosomes would proceed at a slower pace compared to autosomes while Central ancestry would never be erased from the maternally inherited mtDNA by mating events with male Southern colonizers. We find evidence for both these patterns, including (a) elevated levels of X-chromosome divergence between samples from the expansion origin vs. samples outside the origin (Fig. 4b) and (b) geographically structured and highly divergent Central lineage mtDNA (Fig. 1a). Over time, the Southern lineage largely replaced the nuclear genome of the Central lineage because (a) theoretical simulations predict that the invading taxon will drive the local taxon to extinction if competition exists (Currat et al. 2008; Excoffier et al. 2009), (b) there is little contemporary signature of a genetically distinct Central lineage among autosomes (Fig. 1, Phuong et al. 2014), and (c) distribution models for the Central lineage suggest an apparent gradual decline in habitat suitability since the LIG (Fig. 2b). Further, replacement of the local taxon by the invading taxon is a common pattern across several empirical studies examining similar range dynamics among morphologically distinct taxa (e.g., Vachon 1998; Melo-Ferreira et al. 2005; Cahill et al. 2013) providing support for our scenario here among genetically distinct, but morphologically cryptic lineages.
Why did the Southern lineage invade the Central lineage, but not the Northern? We confirmed patterns from a previous study that hybridization and introgression at the N/C contact zone is geographically limited (Fig. 1, Phuong et al. 2014), yet we also showed that the Northern lineage is able to hybridize with other O. beecheyi lineages. The reasons why hybridization with the Northern lineage is localized and not more widespread are unclear. Reproductive barriers, such as mating preference, hybrid inviability between heterospecific crosses, or ‘high-density blocking’ (i.e., difficulty of secondary dispersers to colonize an already occupied habitat, Waters 2011), may limit the movement of Southern lineage individuals into Northern territory. Future studies examining reproductive isolation in this system may explain why the Southern lineage was not able to invade areas further north.
Several other scenarios could have occurred to generate the cytonuclear discordance pattern exhibited by the Central and Southern lineages. One alternative explanation is that because the nuclear genome evolves much slower than the mtDNA genome, not enough time has passed for these two lineages to gain distinction among nuclear markers. However, the lower nuclear genome substitution rate cannot explain why the Central mtDNA lineage, likely one of the earliest diverging lineage in this complex, remains the only lineage in this complex to not have concomitant genetic distinctiveness among nuclear markers when all the other lineages do (Fig. 1). Indeed, one could argue that the ADMIXTURE K=3 plot provides evidence of nascent divergence between the Central and Southern lineages (Fig. 1c). However, range expansions cause geographic shifts in allele frequencies due to successive founder events which can be detected by clustering algorithms (Peter & Slatkin 2013; Pierce et al. 2014), indicating that the apparent population distinction in the ADMIXTURE K=3 plot may be the result of the range expansion. Alternatively, the apparent distinction of the Central lineage could be remnants of past divergence. While we are currently unable to tease apart these hypotheses, the ADMIXTURE K=3 result remains consistent with the expansion introgression model. One other explanation is that selection on the mtDNA drove divergence among these lineages in the face of nuclear gene flow, as demonstrated in several phylogeographic studies of birds (e.g., Ribeiro et al. 2011; Pavlova et al. 2013). Although we did not find evidence for positive selection in our analyses (Table S4), this does not refute the possibility of selection driving patterns of cytonuclear discordance in this system, such as through asymmetric mating, asymmetric survival of offspring, or lower fitness of male hybrids (Toews & Brelsford 2012). Future studies examining mating behavior and offspring survival may shed light on selective processes amplifying patterns of cytonuclear discordance in this system.
Here, we provide support for a demographic scenario that would otherwise go undetected if only autosomal markers were analyzed, adding to an extensive literature emphasizing the importance of matrilineal-biased markers in providing windows into ancient events in the past histories of organisms (Wilson & Bernatchez 1998; Melo-Ferreira et al. 2005; Currat et al. 2008; Cahill et al. 2013; Streicher et al. 2016). Specifically, we document the existence of a distinct Central lineage that now barely exists, but its past presence can be detected because parts of the Central lineage genome (e.g., X-linked markers, mtDNA) have been ‘fossilized’ (Currat et al. 2008) or captured by the Southern lineage. While most studies that provide convincing evidence for a range expansion and introgression demographic scenario occur in systems where both lineages are still extant (Wilson & Bernatchez 1998; Melo-Ferreira et al. 2005; Cahill et al. 2013), there exists a small handful of cases where similar expansion-introgression events may have driven a once distinct evolutionary entity largely extinct (Bell et al. 2012; Singhal & Moritz 2012; Pereira et al. 2016). Although it is uncertain how often lineages become extinct and have portions of their genomes fossilized in invading colonizers through ‘expansion-introgression’ scenarios in natural systems, range expansions are thought to have occurred in the histories of most species (Excoffier et al. 2009). New demographic methods and sequencing techniques, such as the ones employed in this study, may lead to greater detection of this neutral demographic scenario in explaining patterns of cytonuclear discordance that may otherwise have been attributed to other factors such as selection (e.g., Toews and Brelsford 2012).
Our results highlight the importance of range stability in the maintenance of phylogeographic lineages whereas unstable range dynamics can lead to instances of lineage blending. As suggested in Singhal and Moritz (2013), one implication of this result is that climatically stable regions are therefore more likely to maintain and preserve the accumulation of lineages over time, whereas climatically unstable regions have a greater propensity for lineages to merge or become extinct. More broadly, these results align with ideas concerning the importance of climatic stability in the preservation of biological diversity at several scales of study, ranging from the accumulation of genetic diversity within populations in stable refugial areas to the concentration of species diversity in stable tropical climates (Hewitt 1996; Mittelbach et al. 2007; Carnaval et al. 2009; Sandel et al. 2011). Our study extends this general theme of stability maintaining diversity among genetically divergent and morphologically cryptic, phylogeographic lineages.
Supplementary Material
Acknowledgments
For advice and discussions, we gratefully acknowledge, S Singhal, members of the KC Rowe laboratory, members of the Moritz laboratory, and members of the Ecology and Evolutionary Biology department at the University of California, Los Angeles. For samples, we thank the Burke Museum of Natural History and Culture, the Museum of Southwestern Biology, the Museum of Texas Tech University, the Grinnell Resurvey Project Team, the Deck family, ST Álvarez-Castañeda, TL Morelli, and JL Patton. We thank L Smith for laboratory advice, MCW Lim and DR Wait for assistance with the molecular work, and the Evolutionary Genetics Lab for providing a space to conduct the molecular work. We thank AB Smith for the R script to produce unique occurrence records. For the computational analyses, we thank the Alfaro laboratory for computing time. We thank RC Bell, MCW Lim, S Singhal, three anonymous reviewers from Axios Review, and three other anonymous reviewers from Molecular Ecology for insightful comments on earlier versions of this manuscript. This work was supported by the Barry Goldwater Scholarship, a Museum of Vertebrate Zoology Undergraduate Biodiversity Award, a UC Berkeley Summer Undergraduate Research Fellowship, an NSF GRFP, an NSF GROW fellowship, a Chateaubriand fellowship, a Fulbright Fellowship, and an Edwin M. Pauley Fellowship to MAP, and a grant from the Moore Foundation to CM. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303.
Footnotes
Data accessibility: Raw read data are available at the National Center for Biotechnology Information Sequence Read Archive (Accession Numbers SRX2142140- SRX2142184). Scripts used for data filtering and SNP calling can be found on github (https://github.com/CGRL-QB3-UCBerkeley/denovoTargetCapturePopGen). All other scripts, the U. beldingi transcriptome, and final datasets are available on Dryad (doi:10.5061/dryad.rp011).
Author contributions: MAP KB and CM conceived of the study, MAP and KB conducted the lab work, MAP analyzed the data and wrote the manuscript. All authors read and commented on the manuscript.
References
- Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Research. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschup SF, Gish W, Pennsylvania T, Park U. Basic Local Alignment Search Tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Álvarez-Castañeda ST, Cortés-Calva P. Genetic evaluation of the Baja California rock squirrel Otospermophilus atricapillus (Rodentia: Sciuridae) Zootaxa. 2011;3138:35–51. [Google Scholar]
- Avise J. Phylogeography: The History and Formation of Species. Harvard University Press; 2000. [Google Scholar]
- Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Molecular Ecology. 2004;13:729–744. doi: 10.1046/j.1365-294x.2003.02063.x. [DOI] [PubMed] [Google Scholar]
- Bell RC, MacKenzie JB, Hickerson MJ, et al. Comparative multi-locus phylogeography confirms multiple vicariance events in co-distributed rainforest frogs. Proceedings of the Royal Society B: Biological Sciences. 2012;279:991–999. doi: 10.1098/rspb.2011.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K, Linderoth T, Vanderpool D, et al. Unlocking the vault: Next-generation museum population genomics. Molecular Ecology. 2013;22:6018–6032. doi: 10.1111/mec.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K, Vanderpool D, Singhal S, et al. Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics. 2012;13:403. doi: 10.1186/1471-2164-13-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt AL, Ishida Y, Georgiadis NJ, Roca AL. Forest elephant mitochondrial genomes reveal that elephantid diversification in Africa tracked climate transitions. Molecular Ecology. 2012;21:1175–1189. doi: 10.1111/j.1365-294X.2012.05461.x. [DOI] [PubMed] [Google Scholar]
- Cahill JA, Green RE, Fulton TL, et al. Genomic evidence for island population conversion resolves conflicting theories of polar bear evolution. PLoS Genetics. 2013;9:e1003345. doi: 10.1371/journal.pgen.1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C. Stability predicts genetic diversity in the Brazilian Atlantic Forest hotspot. Science. 2009;323:785–789. doi: 10.1126/science.1166955. [DOI] [PubMed] [Google Scholar]
- Currat M, Ruedi M, Petit RJ, Excoffier L. The hidden side of invasions: massive introgression by local genes. Evolution. 2008;62:1908–20. doi: 10.1111/j.1558-5646.2008.00413.x. [DOI] [PubMed] [Google Scholar]
- Dobson FS. An experimental study of dispersal in the California ground squirrel. Ecology. 1979;60:1103–1109. [Google Scholar]
- Dobson FS. Competition for mates and predominant juvenile male dispersal in mammals. Animal Behaviour. 1982;30:1183–1192. [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Foll M, Petit RJ. Genetic consequences of range expansions. Annual Review of Ecology, Evolution, and Systematics. 2009;40:481–501. [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evolutionary Bioinformatics. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genetics. 2009;5:e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RG, Bogdanowicz SM, Hoffmann RS, Yensen E, Sherman PW. Phylogeny and evolutionary history of the ground squirrels (Rodentia: Marmotinae) Journal of Mammalian Evolution. 2003;10:249–276. [Google Scholar]
- Hewitt GM. Some genetic consequences of ice ages, and their role in divergence and speciation. Biological Journal of the Linnean Society. 1996;58:247–276. [Google Scholar]
- Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A. Very high resolution interpolated climate surfaces for global land areas. International Journal of Climatology. 2005;25:1965–1978. [Google Scholar]
- Hodges E, Rooks M, Xuan Z, et al. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nature protocols. 2009;4:960–974. doi: 10.1038/nprot.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell AH. Revision of the North American ground squirrels with a classification of the North American Sciuridae. North American Fauna. 1938;56:1–256. [Google Scholar]
- Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Research. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, Nielsen R. ANGSD: Analysis of Next Generation Sequencing Data. BMC bioinformatics. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Subramanian S. Mutation rates in mammalian genomes. Proceedings of the National Academy of Sciences. 2002;99:803–8. doi: 10.1073/pnas.022629899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Molecular biology and evolution. 2012;29:1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy RC, Garud NR, Kelley JL, Boggs CL, Petrov DA. Genomic inference accurately predicts the timing and severity of a recent bottleneck in a nonmodel insect population. Molecular Ecology. 2014;23:136–150. doi: 10.1111/mec.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo-Ferreira J, Boursot P, Suchentrunk F, Ferrand N, Alves PC. Invasion from the cold past: extensive introgression of mountain hare (Lepus timidus) mitochondrial DNA into three other hare species in northern Iberia. Molecular ecology. 2005;14:2459–64. doi: 10.1111/j.1365-294X.2005.02599.x. [DOI] [PubMed] [Google Scholar]
- Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor protocols. 2010;2010 doi: 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- Mittelbach GG, Schemske DW, Cornell HV, et al. Evolution and the latitudinal diversity gradient: speciation, extinction and biogeography. Ecology Letters. 2007;10:315–331. doi: 10.1111/j.1461-0248.2007.01020.x. [DOI] [PubMed] [Google Scholar]
- Orozco-Terwengel P, Andreone F, Louis E, Jr, Vences M. Mitochondrial introgressive hybridization following a demographic expansion in the tomato frogs of Madagascar, genus Dyscophus. Molecular Ecology. 2013;22:6074–6090. doi: 10.1111/mec.12558. [DOI] [PubMed] [Google Scholar]
- Pavlova A, Amos JN, Joseph L, et al. Perched at the mito-nuclear crossroads: divergent mitochondrial lineages correlate with environment in the face of ongoing m nuclear gene flow in an Australian bird. Evolution. 2013;67:3412–3428. doi: 10.1111/evo.12107. [DOI] [PubMed] [Google Scholar]
- Pearman PB, D’Amen M, Graham CH, Thuiller W, Zimmermann NE. Within-taxon niche structure: niche conservatism, divergence and predicted effects of climate change. Ecography. 2010;33:990–1003. [Google Scholar]
- Pereira RJ, Martínez-Solano I, Buckley D. Hybridization during altitudinal range shifts: Nuclear introgression leads to extensive cyto-nuclear discordance in the fire salamander. Molecular Ecology. 2016;25:1551–1565. doi: 10.1111/mec.13575. [DOI] [PubMed] [Google Scholar]
- Perktas U, Barrowclough GF, Groth JG. Phylogeography and species limits in the green woodpecker complex (Aves: Picidae): multiple Pleistocene refugia and range expansion across Europe and the Near East. Biological Journal of the Linnean Society. 2011;104:710–723. [Google Scholar]
- Peter BM, Slatkin M. Detecting range expansions from genetic data. Evolution. 2013;67:3274–89. doi: 10.1111/evo.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit RJ, Excoffier L. Gene flow and species delimitation. Trends in Ecology & Evolution. 2009;24:386–393. doi: 10.1016/j.tree.2009.02.011. [DOI] [PubMed] [Google Scholar]
- Phillips SJ, Anderson RP, Schapire RE. Maximum entropy modeling of species geographic distributions. Ecological Modelling. 2006;190:231–259. [Google Scholar]
- Phuong MA, Lim MCW, Wait DR, Rowe KC, Moritz C. Delimiting species in the genus Otospermophilus (Rodentia: Sciuridae), using genetics, ecology, and morphology. Biological Journal of the Linnean Society. 2014;113:1136–1151. [Google Scholar]
- Pierce AA, Zalucki MP, Bangura M, et al. Serial founder effects and genetic differentiation during worldwide range expansion of monarch butterflies. Proceedings of the Royal Society B. 2014;281:20142330. doi: 10.1098/rspb.2014.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter S, Bragg JG, Peter BM, Bi K, Moritz C. Phylogenomics at the tips: Inferring lineages and their demographic history in a tropical lizard, Carlia amax. Molecular Ecology. 2016;25:1367–1380. doi: 10.1111/mec.13546. [DOI] [PubMed] [Google Scholar]
- Ribeiro ÂM, Lloyd P, Bowie RCK. A tight balance between natural selection and gene flow in a southern African arid-zone endemic bird. Evolution. 2011;65:3499–514. doi: 10.1111/j.1558-5646.2011.01397.x. [DOI] [PubMed] [Google Scholar]
- Robinson JD, Coffman AJ, Hickerson MJ, Gutenkunst RN. Sampling strategies for frequency spectrum-based population genomic inference. BMC Evolutionary Biology. 2014;14:254. doi: 10.1186/s12862-014-0254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA. The shapes of neutral gene genealogies in two species: probabilities of monophyly, paraphyly, and polyphyly in a coalescent model. Evolution. 2003;57:1465–1477. doi: 10.1111/j.0014-3820.2003.tb00355.x. [DOI] [PubMed] [Google Scholar]
- Rowe KC, Heske EJ, Brown PW, Paige KN. Surviving the ice: Northern refugia and postglacial colonization. Proceedings of the National Academy of Sciences. 2004;101:10355–10359. doi: 10.1073/pnas.0401338101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandel B, Arge L, Dalsgaard B, et al. The influence of Late Quaternary climate-change velocity on species endemism. Science. 2011;334:660–664. doi: 10.1126/science.1210173. [DOI] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, et al. ABySS: a parallel assembler for short read sequence data. Genome research. 2009;19:1117–23. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal S, Moritz C. Testing hypotheses for genealogical discordance in a rainforest lizard. Molecular Ecology. 2012;21:5059–5072. doi: 10.1111/j.1365-294X.2012.05747.x. [DOI] [PubMed] [Google Scholar]
- Singhal S, Moritz C. Reproductive isolation between phylogeographic lineages scales with divergence. Proceedings of the Royal Society B. 2013;280:1–8. doi: 10.1098/rspb.2013.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater GSC, Birney E. Automated generation of heuristics for biological sequence comparison. BMC bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M, Excoffier L. Serial founder effects during range expansion: a spatial analog of genetic drift. Genetics. 2012;191:171–81. doi: 10.1534/genetics.112.139022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. RepeatMasker Open-4.0 2015 [Google Scholar]
- Streicher JW, McEntee JP, Drzich LC, et al. Genetic surfing, not allopatric divergence, explains spatial sorting of mitochondrial haplotypes in venomous coralsnakes. Evolution. 2016;70:1435–1449. doi: 10.1111/evo.12967. [DOI] [PubMed] [Google Scholar]
- Swets JA. Measuring the accuracy of diagnostic systems. Science. 1988;240:1285–1293. doi: 10.1126/science.3287615. [DOI] [PubMed] [Google Scholar]
- Toews DPL, Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Molecular Ecology. 2012;21:3907–30. doi: 10.1111/j.1365-294X.2012.05664.x. [DOI] [PubMed] [Google Scholar]
- Trifonov VA, Vorobieva NN, Rens W. Fluorescence In Situ Hybridization (FISH) Springer-Verlag; Berlin Heidelberg: 2009. FISH With and Without COT1 DNA; pp. 99–109. [Google Scholar]
- Turmelle AS, Kunz TH, Sorenson MD. A tale of two genomes: Contrasting patterns of phylogeographic structure in a widely distributed bat. Molecular Ecology. 2011;20:357–375. doi: 10.1111/j.1365-294X.2010.04947.x. [DOI] [PubMed] [Google Scholar]
- VanDerWal J, Shoo LP, Graham C, Williams SE. Selecting pseudo-absence data for presence-only distribution modeling: How far should you stray from what you know? Ecological Modelling. 2009;220:589–594. [Google Scholar]
- Waters JM. Competitive exclusion: Phylogeography’s “elephant in the room”? Molecular Ecology. 2011;20:4388–4394. doi: 10.1111/j.1365-294X.2011.05286.x. [DOI] [PubMed] [Google Scholar]
- Wilson CC, Bernatchez L. The ghost of hybrids past: fixation of arctic charr (Salvelinus alpinus) mitochondrial DNA in an introgressed population of lake trout (S. namaycush) Molecular Ecology. 1998;7:127–132. [Google Scholar]
- Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20:3252–5. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular biology and evolution. 2007;24:1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.