

Abstract
The multidrug resistance (MDR) system actively pumps the antibiotics out of cells causing serious health problems. During the pumping, AcrB (one of the key MDR components), undergoes a series of large-scale and proton-motive conformational changes. Capturing the conformational changes through all-atom simulations is challenging. Here, we implement a hybrid coarse-grained force field to investigate the conformational changes of AcrB in porter domain under different protonation states of Asp407/Asp408 in trans-membrane domain. Our results show that protonation of Asp408 in monomer III (extrusion) stabilizes the asymmetric structure of AcrB; deprotonation of Asp408 induces clear opening of the entrance and closing of the exit leading to the transition from extrusion to access state. The structural changes in porter domain of AcrB are strongly coupled with the proton translocation stoichiometry in trans-membrane domain. Moreover, our simulations support the postulation that the AcrB should adopt the symmetric resting state in a substrate-free situation.
TOC image
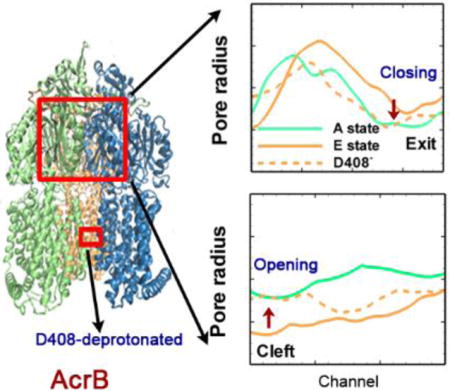
Deprotonation of Asp408 in transmembrane domain induces the opening of the cleft and closing of the exit in porter domain.
Introduction
Multidrug resistance (MDR) represents a serious problem for cancer chemotherapies and treatment of bacterial infectious diseases.1, 2 MDR is usually caused by the overexpression of efflux transporters that actively expel drug or toxin molecules out of the cell. As shown in Figure 1(a), the tripartite efflux system composed of AcrB-AcrA-TolC in Escherichia coli is primarily responsible for the drug resistance and represents the mostly studied MDR system.3–5 As shown, the tripartite system spans across the inner and outer membranes of the cell; AcrB is a member of resistance-nodulation-division (RND) transporter family at the inner membrane and actively pumps drug molecules from the cell to the TolC outer membrane channel,7 AcrA is an adaptor that connects AcrB and TolC.8 Clearly, among the tripartite system AcrB is the key component and plays critical role to the drug export process. AcrB is a proton motive force dependent multidrug efflux pump driven by a substrate/proton antiport mechanism.3, 9–13 As illustrated, the protons are transferred inward to the cytoplasm, creating energy to motivate the outward movement of the substrate molecules. The crystal structure of AcrB was first resolved in 2002,14 as shown in Figure 1(b), it is a homo-trimer. Each monomer can be decomposed into three domains with different functionalities: the trans-membrane (TM) domain responsible for the proton transfer, the porter domain accounting for substrate recognition, binding and extrusion, and the docking domain linking to TolC.15
Figure 1.
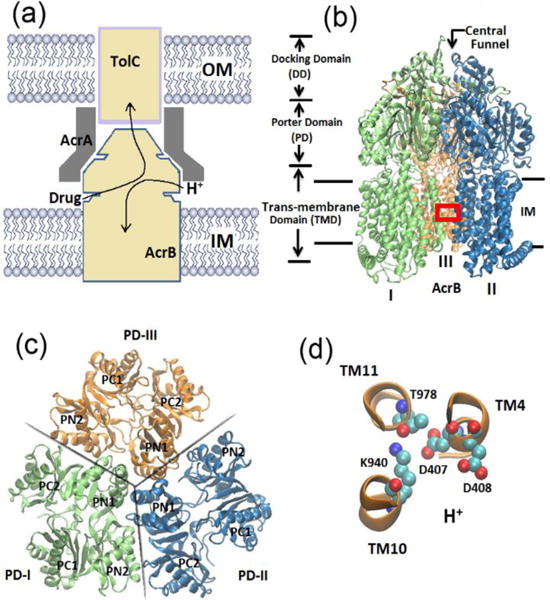
Multidrug resistance (MDR) system. (a) Schematic of the AcrB-AcrA-TolC tripartite efflux system. Arrows indicate the substrate/proton antiport directions. (b) New cartoon representation of the crystal structure of AcrB trimers. Each monomer can be decomposed into trans-membrane domain, porter domain and docking domain. (c) Top view of the porter domains. Each monomer is composed of four subdomains: PC1, PC2, PN1 and PN2. (d) Ball and stick representation of the key residues in TM domain (region indicated by red box in (b)): Asp 407, Asp 408, Lys 940 and Thr978.
Later in 2006 and 2007,10, 16, 17 the X-ray studies showed that the three monomers of AcrB, although similar, exhibits distinct conformations. This asymmetric structure immediately led to the postulation of a three step functionally rotating mechanism for drug export process. Based on this mechanism, each monomer takes one of the three functional states: access (A), binding (B) and extrusion (E). Figure 1(c) shows the cross-sectional view of porter domains for all three monomers, where each monomer consists of four subdomains (PC1, PC2, PN1 and PN2). In monomer I, there is a clear open entrance between PC1 and PC2 subdomain such that substrates can access the vestibule proximal to the aromatic-binding-pocket entrance (A state); in monomer-II, a substrate can bind inside the binding pocket and the conformation is almost similar to monomer-I (B state); and in monomer-III, the vestibule entrance is closed and the exit path between PN1 and PN2 connected from the binding pocket to pore that is linked to TolC channel is open18 (E state). During drug export, the monomers undergo sequential conformational changes from access state to binding then to extrusion and back to access. In this process, the substrate translocation and the associated protein conformational changes primarily occur in the porter domain, while the transduce energy from proton-motive force is in the trans-membrane (TM) domain. Key residues (Asp407, Asp 408, Lys940, and Thr978) of the TM domain are shown in Figure 1(d) along with their α-helices (TM4, TM10 and TM11). It has been shown from site-directed mutagenesis studies19, 20 that these residues were irreplaceable during drug resistance process. Although recent in vivo experiments indirectly supported the aforementioned functionally rotating mechanism,12, 13, 21 the difficulty in in vitro experiments precludes the more direct proof. The molecular picture of the generation of proton driven force at the TM domain, conformational changes at the porter domain, and how they are interconnected are largely unclear.
All-atom molecular simulations are capable of providing the finest spatial/temporal resolution and detailed molecular interaction details. Recently molecular simulations have been performed to investigate the molecular mechanisms involved in drug export of AcrB. Schulz et al.22–24 attempted to address the correlation between conformational cycling and drug extrusion using targeted molecular dynamics. In targeted molecular dynamics, the conformational changes in the protein were mimicked without considering the proton transport and energy transduction. Fischer and Kandt25 studied the water pathways and discussed the possible proton transfer in the TM domain through ~ 50 ns molecular simulations. Feng et al.26 investigated the drug movements in ~20 ns simulations and identified the important roles of Phe617 loop during the drug transport. Vargiu and Nikaido27 studied the multidrug binding properties to the binding pocket of AcrB using all-atom molecular simulations. Most recently Fischer and Kandt28 explored the opening and closing motions in the porter domain of AcrB in ~ 100 ns simulations. Nevertheless, in those studies the large-scale conformational changes in porter domain of AcrB were either facilitated by external means22–24 or mostly overlooked because of the short period of the simulation times from the limit of the all-atom force fields.25, 26, 28 On the other hand, structure-based coarse-grained model has also been adopted to address both the thermodynamics and dynamics of the porter domain of AcrB.29 The multiple-basin model with triple basins representing different states of AcrB has been realized. The results from thermodynamics study showed that the drug dissociation stabilized the symmetric structure of AcrB. The dynamics study indicated strong correlation between conformational changes and the drug export. However, in this coarse-grained model each amino acid in the protein was represented as a bead and only the porter domain of AcrB was considered. Thus, important molecular interaction (salt-bridges/H-bonding) details and the molecular driven force at the TM domain that eventually determine the porter domain dynamics, are missing in such simulations.
In this work, we investigate the conformational changes of AcrB and the associated molecular interactions through atomistic simulations using the hybrid PACE force field originally developed by Han et al.30–35 In PACE, a united-atom-based protein model is coupled with the MARTINI36, 37 water/lipid environment. Through coarse-graining of the environmental molecules (water and lipid), the molecular simulation time can be significantly extended while the molecular details of the protein are still retained. This hybrid force field is particularly suitable for problems involving large-scale conformational changes. The PACE force field has been employed to study the folding and unfolding events in several peptides33 and small proteins34 in microsecond simulations. We have recently implemented the PACE force field to investigate the proton-dependent dynamics and conformational changes of LacY without sugar molecule.38 For the first time, we were able to observe the complete transition from inward-facing to outward-facing conformations of LacY in microsecond molecular simulations. Here we are particularly interested in the effects of the protonation states of two key titratable residues: Asp407 and Asp408 on the molecular interactions at the TM domain, and the resulting large scale conformational changes in the porter domain of AcrB.
Methods
The PACE force field30–35 has been implemented in our simulations. Briefly in PACE the protein atoms are treated with united-atom-based model (each heavy atom represents one site) and coupled with the coarse-grained MARTINI36, 37 water and lipid model (four heavy atoms represent one site). In protein model each heavy atom with the attached hydrogen atoms is generally modeled with one site, but the important hydrogens on backbone and side chain amide groups are also explicitly expressed as dummy atoms.32, 39 The total energy of the system takes into account both the bonded and nonbonded interactions. The bonded interactions account for generic bonded interactions through covalent bonds and interactions involved with rotamers of backbone and side chains. For nonbonded interactions, 12-6 Lennard-Jones (LJ) potentials are used for water-water interactions, water-protein interactions and interactions between nonpolar protein sites. A modified LJ potentials and Coulomb potentials are used to handle hydrogen bonds and electrostatic interactions between polar sites on protein. Details on the modeling development and parameter optimization can be found in Refs.34, 35 It has been reported that using this hybrid PACE force filed, simulations are able to resolve the multiple folding and unfolding events for small peptides, as well as small proteins,33 meanwhile the timescale explored in simulations is significantly increased compared with the all-atom simulations.
All simulations were performed using the modified version of NAMD 2.10.40 PACE hybrid force field were adopted for protein, lipid, water and ion molecules. The simulation models were constructed using CHARMM-GUI41–44 and VMD.45 Figure 2 demonstrates our simulation system, as shown, an asymmetric AcrB trimer with the absence of substrates (PDB ID: 2DHH)16 was initially embedded in a POPE lipid bilayer membrane. The missing parts of AcrB in the original crystal structure are modeled from another AcrB crystal structure (PDB ID: 2J8S).17 The whole system contains 415 lipids after removal of the lipids overlapping with the protein and around 30000 coarse-grained MARTINI waters. Small number of chloride ions was added to neutralize the system. Overall our system contained ~50,000 atoms with dimension of ∼140 × 140 × 160 Å3. The lipid bilayer systems were equilibrated using the standard six-step equilibration process41–44 of gradually turning off the constrains over 280 ps at a temperature of 300 K. In production simulations, periodic boundary conditions were applied in all three directions. The van der Waals interactions were calculated using LJ potential with a cutoff of 12 Å. Production simulations were carried out for ~1.1 μs using NPT (T=300 K, P=1 atm) ensembles. Since the protein force field is close to all-atom model, a time-step of 5 fs was chosen. However, the total number of atoms is significantly reduced compared with all-atom simulations due to the use of MARTINI water and lipids. Graphical software VMD45 has been used for analysis of atomic distances, salt-bridges, H-bonds and taking snap shots.
Figure 2.
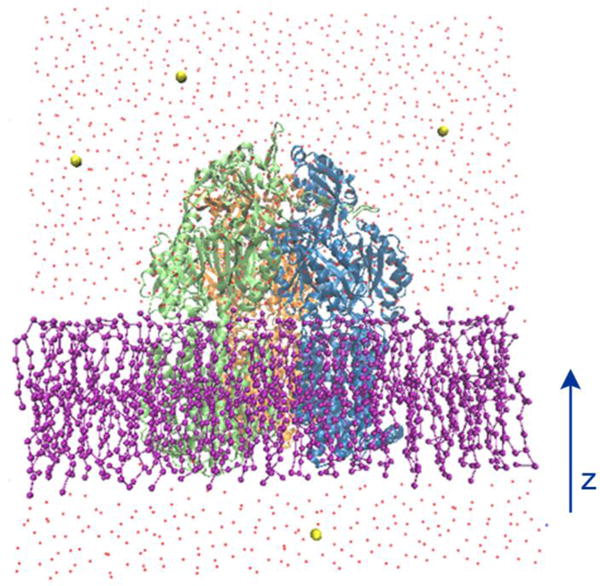
The illustration of the simulation system. AcrB protein (new cartoon representation) is embedded in a POPE lipid bilayer (ball and stick representation). Water molecules are represented by red dots and ions are shown with yellow sphere respectively.
To investigate the large-scale conformational changes in the porter domain of AcrB induced by the different protonation states at TM domain, four model systems have been created in our simulations through varying the protonation state of Asp407 and Asp408 on monomer III while assuming both Asp407 and Asp408 deprotonated on monomer I and II. Model details are presented in Table 1: both Asp407 and Asp408 are deprotonated in the first model; Asp408 is protonated in the second model; Asp407 is protonated in the third model and both Asp407 and Asp408 are protonated in the fourth model. Two independent simulations for each model system were performed for statistical consistency. Since both simulations in each model system produce similar results, for clarity purpose we only present results from one of the realizations in the following sections. The results from the second realization are provided in ESI.
Table 1.
Summary of the simulation model systems. The residues Asp407 and Asp408 in TM domain are kept deprotonated in monomer I and monomer II, but they are changed in monomer III.
| Model System | Monomer I | Monomer II | Monomer III |
|---|---|---|---|
| 1 | D407−/D408− | D407−/D408− | D407−/D408− |
| 2 | D407−/D408− | D407−/D408− | D407−/D408H |
| 3 | D407−/D408− | D407−/D408− | D407H/D408− |
| 4 | D407−/D408− | D407−/D408− | D407H/D408H |
Results
Local Side-chain Dynamics in TM domain and Model Validation
During the drug export process, the AcrB undergoes a series of large-scale conformational changes to accommodate the drug translocation based on the alternating access mechanism. The global protein conformational changes primarily occur in the porter domain, while the driving forces for such changes are from the TM domain where the proton transport happens. It has been recognized that several key residues in the middle of the TM-domain, such as Asp407, Asp408 and Lys940, actively participate the proton transport and play crucial roles during substrate/proton antiport19, 20, 25, 46. According to the asymmetric crystal structure of AcrB, as illustrated in Figure 1, Lys940 is located between Asp407 and Asp408 in monomers I and II, but in monomer III Lys940 is at a different position between Asp408 and Thr978. The different configurations in TM-domain are most likely due to the different protonation states of these residues which may eventually determine the large-scale conformational changes in the porter domain of AcrB.
As listed in Table 1, four model systems have been created by varying of the protonation states of Asp407 and Asp408 in monomer III, while keeping both residues deprotonated in monomers I and II. Through our simulations we first examine the side-chain dynamics and the associated salt-bridge formation/breakage near residues Asp407, Asp408, Lys940 and Thr978 (see Figure 1d) through measuring three distances between: Nζ on Lys940 and Oδ on Asp407, Nζ on Lys940 and Oδ on Asp408, Nζ on Lys940 and Oγ on Thr978. Figure 3a shows the time evaluation of the three distances for 100 ns in monomer II for model system 1, where both Asp408 and Asp407 are deprotonated. The data for monomers I and II are similar, therefore we only discuss the results in monomer II here. As shown, all three distances are steady around the individual values close to the crystal structure within 100 ns. Lys940 forms stable salt-bridges with both Asp408 and Asp407 and resides at a location ~ 7 Å away from Thr978. These data agree with the simulation results from Yamane et al.18 as shown in Figure 3b on exactly the same system but with all-atom force field. However, the side-chain configurations are quite dynamic in monomer III (E state in initial crystal structure) as indicated in Figure 3c. Originally Lys940 is located near Asp407 and Thr978, and is ~ 7 Å away from Asp408 which is consistent with the crystal structure in E state. However, at ~ 10 ns a transition takes place and Lys940 moves away from Thr978 toward Asp408, leading to a configuration similar to that in monomer I and II as shown in Figure 3a. In addition, a stable salt-bridge between Lys940 and Asp407 always exists within 100 ns. The findings are also consistent with the all-atom simulations results from Ref.18 as shown in Figure 3d, in which a similar transition was observed at ~ 50 ns.
Figure 3.
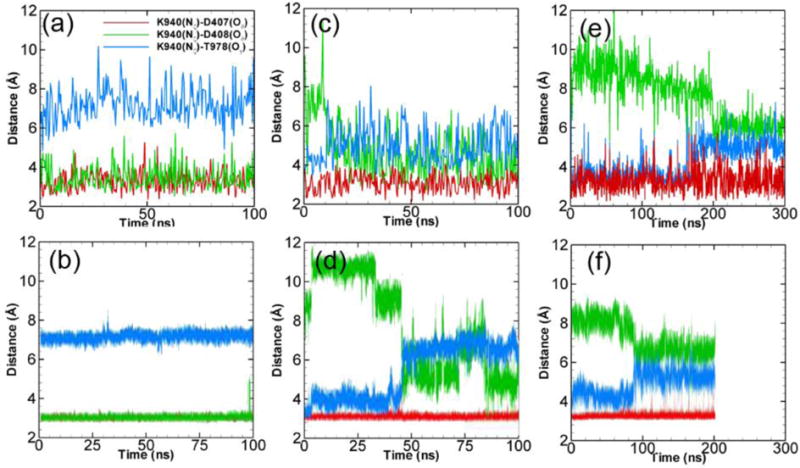
Side chain dynamics at the TM domain. The time evolution of K940-D407 (red), K940-D408 (green) and K940-T978 (blue) distances for monomer II (model 1) (a), monomer III (model 1) (c) and monomer III (model 2) (e). (b), (d) and (f) shows the corresponding results of all-atom simulations from Ref.18 (Simulation results from the second realization can be found in Figure S1 from ESI.)
For model system 2 when the Asp408 on monomer III is protonated, as shown in Figure 3e, Lys940 is always closer to Thr978 than Asp408 within 300 ns simulations. This is consistent with the crystal structure in E state. We also notice a transition at ~ 200 ns with a slight increase in Lys940-Thr978 distance and a slight decrease in Lys940-Asp408 distance. Similar trend was also observed in all-atom simulations18 as shown in Figure 3f but at ~ 90 ns. This has been attributed to the interaction partner switching for the Lys940 amide group from one of the two δ-oxygens on Asp407 interacting with Thr978. Overall, the data show that protonation of Asp408 stabilizes the E state configuration of the monomer III.
Results from model system 3 & 4 are rather dynamic and the early behaviors will not be discussed here. Due to the coarse-graining of the force field and relatively larger time-step, all of our results from PACE exhibit more fluctuations than the results from all-atom simulations. In summary, our simulations of side-chain dynamics in TM domain in early periods indicate that protonation of Asp408 stabilizes configurations of the E state in monomer III, while deprotonation of Asp408 destabilizes the TM domain and induces a transition to A/B state configuration. More importantly, all of our results (including model systems 3 & 4) agree well with the previous all-atom simulation data18 during the early short time period, validating our coarse-grained force fields.
Large-scale Conformation Changes in the Porter Domain
According to the substrate/proton antiport transport mechanism of AcrB, the local configurational changes in the TM domain caused by the proton translocation will lead to the large-scale global movements in the porter domain and eventually the opening/closure of the substrate transport channels to accommodate the substrate export. Based on the crystal structures of AcrB, in each monomer two important binding pockets divided by Phe617-loop (or “switch-loop”) have been identified near the center of porter domain: the outer “proximal binding pocket (proximal BP)” and the inner “distal binding pocket (distal BP)”.47, 48 The proximal BP is connected with several possible entrance channels including vestibule channel,16 cleft channel,10, 17 and a channel from the central cavity.47 These entrance channels and proximal BP are responsible for substrate uptake. An exit channel on the top of the porter domain is connected with the distal BP and obviously responsible for substrate export. Previous simulations with all-atom force fields were able to resolve the side-chain dynamics in the TM domain, but could only capture the initial structural changes in the porter domain due to computational limitation (< 200 ns simulation times). The coarse-grained force fields herein enable us to significantly extend the simulation time to microseconds and directly explore the large-scale conformational changes. In this section, we will focus on the large-scale structural changes in the porter domain of AcrB. We will monitor the conformational changes both at the cleft entrance channel (preferred pathway for larger substrates) and at the exit channel.
Cleft Entrance Channel
Between the cleft entrance and proximal BP, there is a Thr676-loop connecting the PC1 and PC2 subdomains. The Thr676-loop is close to TM domain (see Figure 4a) and adopts different orientations to control the passage/blockage of substrates. In the crystal structure, the Thr676-loops in monomers I and II exhibit configurations that are closer to the TM domain, leading to the opening of the cleft entrance. While in monomer III the Thr676-loop shifts ~ 1 nm towards the switch-loop (or Phe617-loop) closing the entrance and blocking the substrate access. Therefore, as shown in Figure 4a, through our simulations we monitor the opening/closure of the cleft entrance by measuring the center of mass distance between Thr676 and Phe563. Phe563 is located at the TM domain with negligible global movement throughout the simulations.
Figure 4.
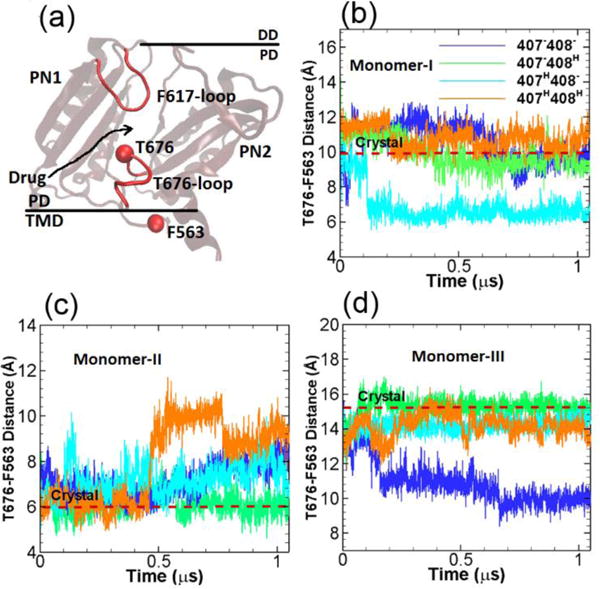
Cleft entrance. (a) Illustration of the relative locations of T676-loop, F617-loop in the porter domain and F563 in the TM domain. The time evolution of the T676-F563 distances in monomer I (b), monomer II (c) and monomer III (d) for the four model systems listed in Table 1. The values of T676-F563 distance in crystal structure (PDB ID: 2DHH) for the three monomers are indicated by red dashed lines for comparison. (Simulation results from the second realization can be found in Figure S2 from ESI.)
Figure 4b–d show the time evolution of the center of mass distances between Thr676 and Phe563 for the four simulation systems (Table 1) with different protonation state and for all three monomers of AcrB respectively. The original distances from the crystal structure are also indicated in the figures by the dashed red lines for reference. As shown, the T676-F563 distance is maximum at ~ 15 Å in monomer III indicating the closure of the entrance channel, while the distances are at 10 and 6 Å in monomers I and II showing the partial opening or opening of the entrance. Among our simulations systems (Table 1), in monomer I (A state in crystal structure) as shown in Figure 4b, the T676-F563 distances are quite stable around the crystal structure value for systems 1, 2 and 4 throughout the simulations. For system 3 the T676-F563 distance decreases to 6 Å at ~ 0.1 μs. In monomer II (B state in crystal structure) as shown in Figure 4c, the T676-F563 distance in system 2 is consistent with the crystal structure value throughout the simulation. A gradual increase of the distances in systems is observed in systems 1 and 3, while the distance is abruptly increased to 10 Å at ~ 0.5 μs in system 4. In monomer III (E state in crystal structure) as shown in Figure 4d, for systems 2, 3 and 4 the T676-F563 distances are similar and fluctuating around the crystal structure value, but for system 1 the distance drops to ~ 10 Å at around 0.2 μs.
Exit Channel
At the exit region of the substrate transport channel around the top of the AcrB, three residues Gln124, Gln125 and Tyr758 form a gate to control the passage/blockage of the substrate.17 Therefore to monitor the opening/closure of the exit channel, we measure the center of mass distances between Gln124 and Tyr758 (see Figure 5a) in our simulations. Figure 5b–d show the time evolution of the Gln124-Tyr758 distances. The red dashed lines are the original distances in the crystal structure, as shown, the Gln124-Tyr758 distances are at ~ 5 Å in monomers I and II when the exit channel is closed, while the exit channel is widely open in monomer III as indicated by the Gln124-Tyr758 distance at ~ 12.5 Å. In monomer I as shown in Figure 5b, the Gln124-Tyr758 distances are stable around the original crystal structure value for systems 1 and 2. In system 3 and 4 the distances are increased indicating the opening the exit channel. In monomer II as shown in Figure 5c, the time evolutions of the Gln124-Tyr758 distances are rather dynamic; Overall the distances are close to the crystal structure value in systems 1 and 2, but are increased to ~ 12 Å and ~ 10 Å in systems 3 and 4 respectively. In monomer III as shown in Figure 5d, for system 1 the Gln124-Tyr758 distance gradually drops to ~ 5 Å closing the exit channel, the distance converges to the crystal structure value ~ 12.5 Å for system 2, while for systems 3 and 4 the distances increase to ~ 16 Å.
Figure 5.
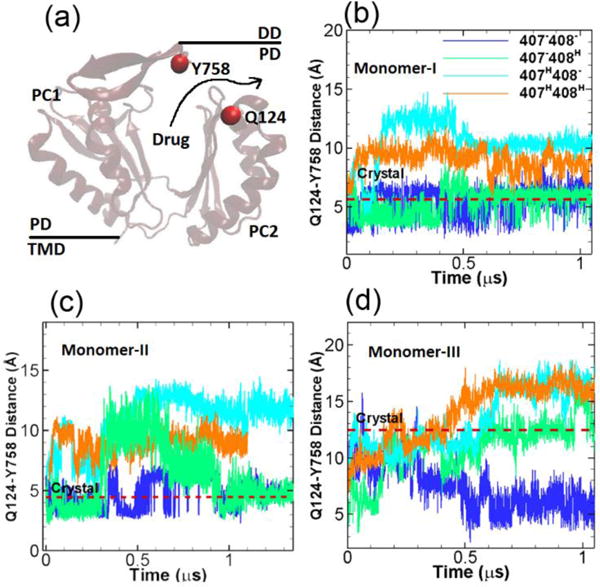
Exit channel. (a) Illustration of the relative locations of Y758 and Q124 at the exit channel. The time evolution of the Q124-Y758 distances in monomer I (b), monomer II (c) and monomer III (d) for the four model systems listed in Table 1. The values of Y758-Q124 distance in crystal structure (PDB ID: 2DHH) for the three monomers are indicated by red dashed lines for comparison. (Simulation results from the second realization can be found in Figure S3 from ESI.)
Channel Radius at the Cleft Entrance and Exit Regions
From the analysis of the four model systems (see Table 1), two model systems (model 1 and model 2) appear remarkably interesting. Starting from the crystal structure of the asymmetric AcrB trimer (PDB ID: 2DHH), we monitored the conformational changes around the cleft entrance and exit regions at the porter domain through measuring the Thr676-Phe563 and Gln124-Tyr758 distances respectively. In model system 1 when both Asp407 and Asp408 are deprotonated in monomer III, the entrance in monomer I stayed open and was consistent with the original crystal structure; the entrance in monomer II slight closed and in monomer III, the conformation near the entrance dramatically changed from the original closed-state (E state) to open-state (A state) (see Figure 4). On the other hand, the exit in monomers I and II stayed tightly closed consistent with the original crystal structures, in monomer III we also observed a dramatic change from open-state (E state) to closed-state (A state) (see Figure 5). For model system 2 when Asp408 was protonated while Asp407 was kept deprotonated, the measurements for all the monomers at both entrance and exit regions showed no changes and converged to the original states of the crystal structures. All the data demonstrated that protonation of Asp408 (model 2) in monomer III assisted in stabilizing the asymmetric structure (ABE) of AcrB, while deprotonation of Asp408 (model 1) caused significant conformational changes and induced a transition from the asymmetric (ABE) to symmetric (AAA) structure. This is also consistent with the measurements of local side-chain dynamics in TM domain from Figure 3.
To further confirm and demonstrate the structural changes during this transition, we measure the radius of cavity along the substrate transport channel from cleft entrance to exit (Figure 6a) for model system 1 at the end of the simulation (~ 1.1 μs). Figure 6b shows the pore radius profile from the cleft entrance to the proximal BP. In the original crystal structure (solid lines), it is clear that both monomers I and II are open at the entrance. Monomer II (~ 4 Å) opens slightly wider than monomer I (~ 3.2 Å). In monomer III, however, the entrance is almost completely closed with pore radius of ~ 1.5 Å. When Asp408 is deprotonated (model system 1), as shown evidently by dashed lines in Figure 6b, the pore radius at the cleft entrance in all three monomers converge to the value of monomer I in crystal structure. On the other hand, along the exit channel as shown in Figure 6c, both monomer I and II are closed and only the monomer III is open in crystal structure. In model system 1 as Asp408 is deprotonated, again the exit channels in all three monomers converge to a situation consistent with monomer I of crystal structure. The pore radius measurements clearly demonstrate the opening movement at the entrance region and closing movement at the exit region of monomer III, and provide strong evidence for the transition from ABE configuration to AAA configuration in model system 1. The channel radius measurements for model system 2 are provided in ESI. As shown in Figure S4 in ESI, when Asp408 is protonated the pore sizes at both cleft entrance and exit are consistent with the original asymmetric crystal structures.
Figure 6.
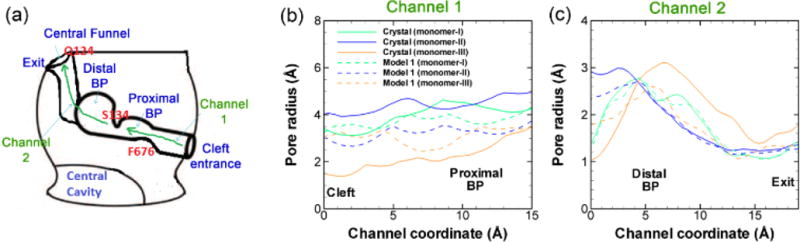
Pore radius profiles. (a) Schematic illustration of the substrate transport channel, from the cleft entrance to proximal binding pocket, and then to the distal binding pocket and finally to the exit central funnel exit. The pore radius profiles calculated by CAVER 2.06 from the cleft entrance to proximal binding pocket (b) and from the distal binding pocket to the exit (c) for all three monomers of the model system 1 at the end of the simulation (~ 1.1 μs). The pore radius profiles from the original crystal structure (PDB ID: 2DHH) are also included as solid lines for comparison. The relative positions of the residues along the substrate channel, F676 and Q124, mentioned in Figure 4 and Figure 5 are illustrated in red text. The channel 1 coordinate is defined from the cleft to S134, and the channel 2 coordinate is defined from the S134 to the exit.
Discussion
It has been postulated that the AcrB adopts symmetric AAA conformation in the absence or low concentration of substrate as the “resting state” with the structural flexibility for substrate intake.49, 50 Recently, Fischer and Kandt28 investigated the opening/closing motions of the porter domain of AcrB with the absence of substrate through ~ 100 ns all-atom molecular simulations. The conformational changes of the porter domain, including the entrance, exit and distal binding pocket regions, were monitored through distance, cross-sectional area and radius of gyration analyses. However, no trend from the original ABE state to AAA resting state was observed in simulations, probably due to the rather short simulation time as pointed out by the authors. On the other hand, Yamane et al.18 performed ~ 200 ns all-atom simulations to explore the functional rotation of AcrB induced by proton-motive force. Similar to our model systems, different protonation states of Asp407 and Asp408 in monomer with E state were examined and the effects on the structural changes in the porter domain were explored. Due to the limited simulation time, only the initial structural changes were captured. Based on the initial changes and the principal component analysis, a transition from asymmetric ABE to symmetric AAA state was proposed when both Asp407 and Asp408 were deprotonated. In this work, we were able to capture the complete conformational transitions in the porter domain due to the benefits of hybrid coarse-grained force field. Through residue distances and pore radius measurements near the entrance and exit regions of the substrate transport channel, we directly illustrated the structural transition from ABE to AAA state when Asp408 was deprotonated. Our results strongly support the hypothesis that without substrates, the AcrB should adopt the resting state (AAA) at appropriate proton translocation stoichiometry.
To address the possible molecular force transmission pathway from the TM domain to the porter domain, we have carefully examined of the simulation results from model system 1 (407−–408−) and compared with results from model system 2 (407−–408H) as well as the crystal structure. As shown in FigureS5 in ESI, the salt-bridges Lys940-Asp407 and Lys940-Asp408 play important roles during the proton translocation. Asp407 and Asp408 are located on TM4 and Lys940 is on TM10. Results show that the protonation of Asp408 actually stabilize the Lys940-Asp407 salt-bridge. Figure 7(a) shows the time evolution of the root-mean-square deviation (RMSD)s for the center of TM10 (transmembrane α-helix 10) calculated relative to the crystal structure. When Asp408 is deprotonated as shown in Figure 7(a), there is a clear transition leading to higher RMSD on TM10 than the case when Asp408 is protonated at ~ 0.5 μs. The promoted dynamics of TM10 will impact the α-helices in vicinity. By comparing all α-helices with the crystal structures in TM domain as shown in Figure 7(c), we observed that the most prominent change, i.e. a clear rotation, took place in TM9. To quantify the rotation, we monitor the time evolution of the angle (θ) formed by TM9 and z-axis. As shown in Figure 7(b), when Asp408 is protonated a stable θ around 40° is observed throughout the simulation. However as Asp408 is deprotonated, a clear angular motion occurs at ~ 0.5 μs changing θ from 40 to 32°. Since TM9 is connected with PC2 subdomain in the porter domain, the global angular motion of TM9 may eventually induces the rotation of PC2 leading to the opening of the cleft entrance and closing of the exit channel. As shown in Figure 1(d), the PC2 subdomain in monomer III is very close to the PN2 subdomain in monomer II. Therefore it is likely that the global motion of PC2 in monomer III will also affect the PN2 in monomer II leading to the structural transitions. Clearly, in addition of TM9 other α-helices also participate the large-scale conformational changes, the complete picture is far more complex and should be further investigated later.
Figure 7.
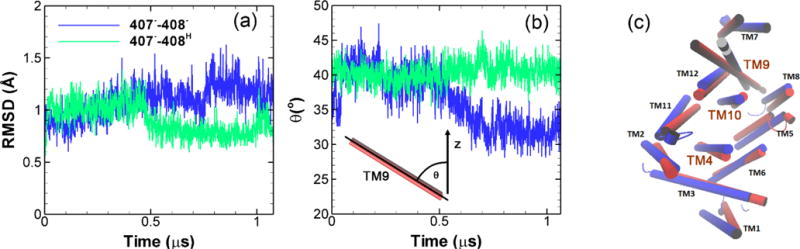
Structural changes in TM domain. (a) Time evolution of root-mean-square deviation (RMSD) relative to the crystal structure for TM10 (transmembrane α-helix 10). (b) Time evolution of the angle (θ) formed by TM9 and z-axis. (c) Cartoon representation of all 12 TM α-helices in monomer III. Red: crystal structure of 2DHH. Blue: final structure from model system 1 (407−–408−).
Conclusions
In this work atomistic simulations have been performed to study the large-scale conformational changes in the porter domain of AcrB driven by the proton-motive force at the TM domain. A hybrid PACE force field, in which the united atom protein model is coupled with MARTINI water/lipid models, was implemented in our simulations. The coarse-graining of the environmental molecules significantly extends the simulation time and enables us to capture the large-scale conformational changes of AcrB in microseconds. As pointed out from Ref. 33, coarse-graining of water/lipid and simplification of the protein-protein/protein-solvent interactions many cause much faster kinetics for protein dynamics than experiments. However, significant insights such as dynamic interactions as well as large-scale protein structural changes can be obtained in such simulations.
Four model systems with the absence of substrates based on the different protonation states of Asp407 and Asp408 in monomer III have been investigated. We first examined the early stage (~ 200 ns) local side-chain dynamics in TM domain through measuring the salt-bridges among Asp407, Asp408, Lys940 and Thr978. The dynamics from our simulations are consistent with the recent all-atom simulation data validating the coarse-grained force field. Then we investigated the large-scale structural changes in the porter domain through residue distances and pore radius measurements at both the entrance and exit regions of the substrate transport channel. Our results indicated that the protonation state of Asp408 was critically important. Protonation of the Asp408 stabilized the asymmetric structures in the porter domain, which is consistent with experiments.46 Deprotonation of Asp408 in the TM domain caused dramatic structural changes on monomer III in the porter domain. Our results clearly showed the opening of the entrance and closure of the exit region of the substrate transport channel, leading to the transition from extrusion (E) to access (A) state. Our data strongly support the postulation that in a substrate-free situation, the AcrB should adopt the symmetric AAA resting state.49, 50 Protonation of Asp407 also caused significant structural changes in the porter domain (Figures 4 and 5 for models 3 and 4) implying its important roles. A substantial decreased function has been observed in experiments with Asp407 mutations.19, 20, 46 Even though the results are incompatible with the existing A,B,E states, transient protonation of Asp407 during proton translocation is possible. Further investigations are needed to clarify the functional roles of the protonation state of Asp407. Finally, our current models do not contain any substrate. Inclusion of the substrate molecules in the simulations and exploration of the coupled substrate and proton transport (substrate/proton antiport) will be interesting and will be addressed in future works.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM122081. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Computational resources were provided in part by the Extreme Science and Engineering Discovery Environment (XSEDE) under grant No. MCB170012.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1.Li XZ, Nikaido H. Drugs. 2004;64:159–204. doi: 10.2165/00003495-200464020-00004. [DOI] [PubMed] [Google Scholar]
- 2.Nikaido H. Annu Rev Biochem. 2009;78:119–146. doi: 10.1146/annurev.biochem.78.082907.145923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zgurskaya HI, Nikaido H. Proc Natl Acad Sci U S A. 1999;96:7190–7195. doi: 10.1073/pnas.96.13.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sulavik MC, Houseweart C, Cramer C, Jiwani N, Murgolo N, Greene J, DiDomenico B, Shaw KJ, Miller GH, Hare R, Shimer G. Antimicrob Agents Chemother. 2001;45:1126–1136. doi: 10.1128/AAC.45.4.1126-1136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tikhonova EB, Zgurskaya HI. J Biol Chem. 2004;279:32116–32124. doi: 10.1074/jbc.M402230200. [DOI] [PubMed] [Google Scholar]
- 6.Petrek M, Otyepka M, Banas P, Kosinova P, Koca J, Damborsky J. BMC Bioinformatics. 2006;7 doi: 10.1186/1471-2105-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paulsen IT, Park JH, Choi PS, Saier MH. FEMS Microbiol Lett. 1997;156:1–8. doi: 10.1111/j.1574-6968.1997.tb12697.x. [DOI] [PubMed] [Google Scholar]
- 8.Dinh T, Paulsen IT, Saier MH. J Bacteriol. 1994;176:3825–3831. doi: 10.1128/jb.176.13.3825-3831.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zgurskaya HI, Nikaido H. Mol Microbiol. 2000;37:219–225. doi: 10.1046/j.1365-2958.2000.01926.x. [DOI] [PubMed] [Google Scholar]
- 10.Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. Science. 2006;313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- 11.Seeger MA, Diederichs K, Eicher T, Brandstaetter L, Schiefner A, Verrey F, Pos KM. Curr Drug Targets. 2008;9:729–749. doi: 10.2174/138945008785747789. [DOI] [PubMed] [Google Scholar]
- 12.Seeger MA, von Ballmoos C, Eicher T, Brandstatter L, Verrey F, Diederichs K, Pos KM. Nat Struct Mol Biol. 2008;15:199–205. doi: 10.1038/nsmb.1379. [DOI] [PubMed] [Google Scholar]
- 13.Takatsuka Y, Nikaido H. J Bacteriol. 2009;191:1729–1737. doi: 10.1128/JB.01441-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- 15.Lomovskaya O, Zgurskaya HI, Nikaido H. Nat Biotechnol. 2002;20:1210–1212. doi: 10.1038/nbt1202-1210. [DOI] [PubMed] [Google Scholar]
- 16.Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Nature. 2006;443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- 17.Sennhauser G, Amstutz P, Briand C, Storchenegger O, Gruetter MG. PLoS Biol. 2007;5:106–113. doi: 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamane T, Murakami S, Ikeguchi M. Biochemistry. 2013;52:7648–7658. doi: 10.1021/bi400119v. [DOI] [PubMed] [Google Scholar]
- 19.Guan L, Nakae T. J Bacteriol. 2001;183:1734–1739. doi: 10.1128/JB.183.5.1734-1739.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takatsuka Y, Nikaido H. J Bacteriol. 2006;188:7284–7289. doi: 10.1128/JB.00683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takatsuka Y, Nikaido H. J Bacteriol. 2007;189:8677–8684. doi: 10.1128/JB.01127-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulz R, Vargiu AV, Collu F, Kleinekathoefer U, Ruggerone P. PLoS Comput Biol. 2010;6:e1000806. doi: 10.1371/journal.pcbi.1000806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vargiu AV, Collu F, Schulz R, Pos KM, Zacharias M, Kleinekathoefer U, Ruggerone P. J Am Chem Soc. 2011;133:10704–10707. doi: 10.1021/ja202666x. [DOI] [PubMed] [Google Scholar]
- 24.Schulz R, Vargiu AV, Ruggerone P, Kleinekathoefer U. J Phys Chem B. 2011;115:8278–8287. doi: 10.1021/jp200996x. [DOI] [PubMed] [Google Scholar]
- 25.Fischer N, Kandt C. Proteins. 2011;79:2871–2885. doi: 10.1002/prot.23122. [DOI] [PubMed] [Google Scholar]
- 26.Feng ZW, Hou TJ, Li YY. Mol Biosyst. 2012;8:2699–2709. doi: 10.1039/c2mb25184a. [DOI] [PubMed] [Google Scholar]
- 27.Vargiu AV, Nikaido H. Proc Natl Acad Sci U S A. 2012;109:20637–20642. doi: 10.1073/pnas.1218348109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer N, Kandt C. BBA-Biomembranes. 2013;1828:632–641. doi: 10.1016/j.bbamem.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 29.Yao XQ, Kenzaki H, Murakami S, Takada S. Nat Commun. 2010;1:117. doi: 10.1038/ncomms1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han W, Wu YD. J Chem Theory Comput. 2007;3:2146–2161. doi: 10.1021/ct700151x. [DOI] [PubMed] [Google Scholar]
- 31.Han W, Wan CK, Wu YD. J Chem Theory Comput. 2008;4:1891–1901. doi: 10.1021/ct800184c. [DOI] [PubMed] [Google Scholar]
- 32.Han W, Wan CK, Jiang F, Wu YD. J Chem Theory Comput. 2010;6:3373–3389. doi: 10.1021/ct1003127. [DOI] [PubMed] [Google Scholar]
- 33.Han W, Wan CK, Wu YD. J Chem Theory Comput. 2010;6:3390–3402. doi: 10.1021/ct100313a. [DOI] [PubMed] [Google Scholar]
- 34.Han W, Schulten K. J Chem Theory Comput. 2012;8:4413–4424. doi: 10.1021/ct300696c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wan CK, Han W, Wu YD. J Chem Theory Comput. 2012;8:300–313. doi: 10.1021/ct2004275. [DOI] [PubMed] [Google Scholar]
- 36.Marrink SJ, de Vries AH, Mark AE. J Phys Chem B. 2004;108:750–760. [Google Scholar]
- 37.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. J Phys Chem B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 38.Jewel Y, Dutta P, Liu J. Proteins. 2016;84:1067–1074. doi: 10.1002/prot.25053. [DOI] [PubMed] [Google Scholar]
- 39.Feenstra KA, Hess B, Berendsen HJC. J Comput Chem. 1999;20:786–798. doi: 10.1002/(SICI)1096-987X(199906)20:8<786::AID-JCC5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 40.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jo S, Kim T, Iyer VG, Im W. J Comput Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 42.Jo S, Lim JB, Klauda JB, Im W. Biophys J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, Qi YF, Lee JM, Monje-Galvan V, Venable RM, Klauda JB, Im W. J Comput Chem. 2014;35:1997–2004. doi: 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qi Y, Cheng X, Han W, Jo S, Schulten K, Im W. J Chem Inf Model. 2014;54:1003–1009. doi: 10.1021/ci500007n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Humphrey W, Dalke A, Schulten K. J Mol Graph Model. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 46.Seeger MA, von Ballmoos C, Verrey F, Pos KM. Biochemistry. 2009;48:5801–5812. doi: 10.1021/bi900446j. [DOI] [PubMed] [Google Scholar]
- 47.Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. Nature. 2011;480:565–569. doi: 10.1038/nature10641. [DOI] [PubMed] [Google Scholar]
- 48.Eicher T, Cha HJ, Seeger MA, Brandstatter L, El-Delik J, Bohnert JA, Kern WV, Verrey F, Grutter MG, Diederichs K, Pos KM. Proc Natl Acad Sci U S A. 2012;109:5687–5692. doi: 10.1073/pnas.1114944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. J Bacteriol. 2006;188:7290–7296. doi: 10.1128/JB.00684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pos KM. Biochimica Et Biophysica Acta-Proteins and Proteomics. 2009;1794:782–793. doi: 10.1016/j.bbapap.2008.12.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.