

Abstract
Brain assembly is hypothesized to begin when pioneer axons extend over non-neuronal cells, forming tracts guiding follower axons. Yet pioneer-neuron identities, their guidance substrates, and their interactions, are not well understood. Here, using time-lapse embryonic imaging, genetics, protein-interaction, and functional studies, we uncover the early events of C. elegans brain assembly. We demonstrate that C. elegans glia are key for assembly initiation, guiding pioneer and follower axons using distinct signals. Pioneer sublateral neurons, with unique growth properties, anatomy, and innervation, cooperate with glia to mediate follower-axon guidance. We further identify a CHIN-1/Chimaerin-KPC-1/Furin double mutant that severely disrupts assembly. CHIN-1/Chimaerin and KPC-1/Furin function non-canonically in glia and pioneer neurons for guidance-cue trafficking. We exploit this bottleneck to define roles for glial Netrin and Semaphorin in pioneer- and follower-axon guidance, respectively, and for glial and pioneer-neuron Flamingo/CELSR in follower-axon navigation. Altogether, our studies reveal previously-unknown glial roles in pioneer-axon guidance, suggesting conserved brain-assembly principles.
Brain formation requires an astonishing orchestration of events, including process fasciculation, axon-substrate interactions, and target-cell recognition. Assembly is thought to initiate when the first axons extend along non-neuronal cells1. Since these events occur in the embryo, where live imaging and functional studies are difficult, identifying pioneer axons and their guidance substrates has been challenging.
Glia physically associate with major axon tracts and also express guidance cues2. Some studies in grasshopper embryos and Drosophila larvae suggest glial roles in axon guidance, while other invertebrate studies rather propose glial roles in axon fasciculation3–7. Moreover, whether glia direct pioneer axon guidance in these systems is not resolved. Similarly, glial contributions to axon guidance in the mammalian central nervous system, and specifically to the guidance of pioneer axons, remain unclear8,9. The requirement of glia for neuronal viability adds additional experimental complexity.
Guidance and cell-adhesion proteins that control axon navigation have been described10. Functional studies of these factors suggest that they account for only select guidance decisions within the hundred-billion-neuron mammalian brain. Thus, unknown components may be necessary to fully explain brain self-assembly. Genetic redundancies may also account for the highly specific axon-guidance defects seen in animals disrupted for broadly-expressed genes11, and may explain why eliminating multiple genes is necessary to elicit guidance defects12.
With unbiased gene-function discovery approaches13, invariant embryonic development14, and glial cells dispensable for neuronal viability15, the nematode Caenorhabditis elegans is a powerful arena in which to decipher cellular and molecular events governing brain assembly. The C. elegans brain neuropil, the nerve ring (NR), consists of >170 sensory, motor, and interneuron processes tightly bundled around the pharynx16, and is enveloped by processes of four CEPsh glia. Axons entering the NR are arranged in bundles (e.g. bilateral amphid, deirid, and sublateral commissures containing 23, 5, and 12 axons, respectively), or occupy non-commissural paths (e.g. the AIY interneuron) (see below). Little is known about NR assembly. Some C. elegans mutants in genes affecting peripheral nerve guidance exhibit partially-penetrant NR navigation defects for some neurons17–19. Other genes including cwn-2/Wnt, functioning in sublateral-commissure neurons, control overall NR placement20. Nonetheless, identities of NR pioneer cells and their functions, and factors affecting overall NR integrity, are not known, and have been the subject of extensive speculation21,22.
Here we demonstrate that C. elegans CEPsh glia promote key early events in NR assembly by directing the extension of a pioneer axon fascicle composed of sublateral-commissure (SubL) neurons. Glia and pioneer neurons extend coalescing processes to generate the NR, and cooperatively direct follower-axon NR entry. We identify a novel mutant, defective in both CHIN-1/Chimaerin and KPC-1/Furin, in which NR entry of >70% of axons is blocked. These proteins cooperate in glia and pioneer neurons for guidance-cue trafficking, and define a choke point we use to identify a network of redundant guidance factors, some specifically required in glia, for NR assembly. Our studies identify pioneer neurons with unique properties and evoke comparisons between CEPsh glia and vertebrate radial glia, defining the initial steps of assembly of an animal brain at single-cell resolution.
Results
CEPsh glia and SubL neurons pioneer the NR and guide its assembly
The C. elegans postembryonic NR is comprised of several neuronal commissures and is surrounded by four CEPsh glial cells (Fig. 1a,b). A corresponding compact and process-rich structure can be seen around the pharynx of 1.5-fold embryos (440 minutes post-fertilization; Supplementary Fig. S1d,h), but not in earlier comma-stage embryos (400 minutes; Supplementary Fig. S1c,g), as observed by electron microscopy (EM) or fluorescence imaging of a ubiquitously-expressed membrane::GFP reporter (Fig. 1c,d). Thus NR assembly initiates between these stages. To study NR formation, we therefore imaged axons of the amphid (AWC, AFD, ASE, AUA), deirid (ADA, BDU), and SubL (SIA, SIB, SMD) commissures, as well as non-commissural neurons (including AIY, BAG) between 360-450 minutes of development. Five bilateral pairs of SubL axons (SIAV, SIAD, SIBV, SIBD, SMDD) enter the NR earlier than any other axons we examined. These emerge ventrally and coalesce in left-right bilateral bundles in bean-stage embryos (Fig. 1e,f), cross the lateral midline (“NR entry”) at comma stage, and meet to form a ring-like structure by the 1.5-fold embryonic stage (Fig. 1g–h). Other axons enter the NR later and at different times. Axons of AIY interneurons (Fig. 1i) and BAG neurons, for example, emerge at the comma stage, only to enter the NR at the 1.5-fold stage (Supplementary Fig. S2e–f). NR entry timing can be sequential within non-pioneer commissures: AWC amphid-commissure axons extend at late bean stage, entering the NR in 1.5-fold embryos (Fig. 1j), while co-commissural AFD and AUA axons emerge at the comma stage, entering the NR only in 2-fold embryos (Supplementary Fig. S2f–h, i, l). ASE amphid-commissure axons and the deirid commissure axons, BDU and ADA, enter the NR even later, after the 2-fold stage (Supplementary Fig. S2j, m–q).
Figure 1. Hierarchical assembly of the embryonic nerve ring.
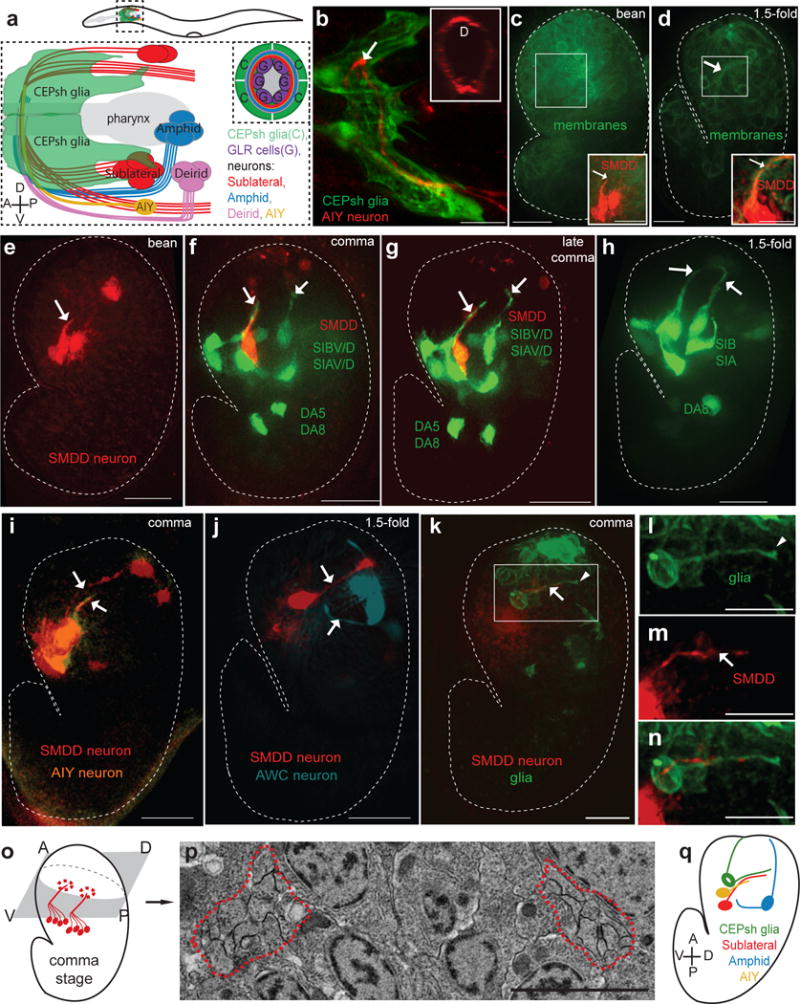
(a–b) The post-embryonic nerve ring is populated by neuronal commissures and enveloped by CEPsh glia. (a) Schematics of lateral view of post-embryonic nerve ring. (b) Imaging of post-embryonic nerve ring in L4 animals. Inset, cross-sectional view. Pttx-3::mCherry (AIY, red), Phlh-17::myristoylated-GFP (CEPsh glia). (c–n) Formation of the embryonic nerve ring starts at late bean stage, with early entry of sublateral commissure axons and CEPsh glia and later entry of other components. (c,d) Phis-72:: myristoylated-GFP (cell membranes). (c–k) Dotted line: embryo outline. D: dorsal, V: ventral, A: anterior, P: posterior. Arrow: axon, arrowhead: CEPsh glia. Scale bars: 10μm. (c-g,i-k,m,n) Pttx-3::mCherry (SMDD, red; AIY, pseudocolored orange in (I). (e–h) Pceh-17::GFP, sublateral neurons SIAV/D, SIBV/D. (j) Phlh-16::GFP (SIA/SIB, red; AWC, pseudocolored blue). (k–n) Pmir-228:: myristoylated-GFP (glia). (b–n) Expression patterns are described in Supplementary Methods and Supplementary Tables S8, S10. l–n: magnified view of boxed region in k. (o,p) A single pair of bilateral bundles are observed in electron micrographs of early comma stage embryos. Schematic (o) and magnification (p) of electron micrograph section. The magnified region corresponds to the region outlined in the blue box of Supplementary Fig. S1g. Red outline: axon bundle. Scale bar: 2μm. (q) Summary of imaging results. CEPsh glia and pioneer neurons enter NR path first, followed by amphid and AIY neurons.
Importantly, processes of ventral CEPsh glia coalesce with SubL axons early (Fig. 1k–n), defining the presumptive NR. Thus, dorsal extension of a ventrally-located process bundle composed, at least in part, of CEPshV glia, SIAV, SIAD, SIBV, SIBD, and SMDD processes is a very early event in NR formation. Supporting this notion, EM of an early comma-stage embryo reveals a single pair of bilateral bundles, each composed of 10 or fewer processes (Fig. 1o,p). Muscle arms, abutting the inner surface of the postembryonic NR16, are not aligned with the early NR tract (Supplementary Fig. S2r–u). Thus, the NR is pioneered by a small, well-defined group of axons and glial processes, with subsequent orderly addition of other axons (Fig. 1q).
To determine whether orderly assembly reflects a functional hierarchy, we ablated early NR entrants by expressing the pro-apoptotic EGL-1/BH3-only protein prior to axon outgrowth and scored cell death by absence of cell-specific markers in larvae (Supplementary Methods). Ablation of either ventral or dorsal CEPsh glia correlates with AIY axon NR entry defects (Fig. 2a–d, Supplementary Table S1), and guidance defects of AWB and AWC co-commissural amphid axons (5/21 animals;23). The SubL bundle enters the NR in CEPsh glia-ablated animals, however, it often de-fasciculates, extending aberrantly anterior to the NR (Fig. 2e–h, Supplementary Table S1). Since SubL neurons enter the NR in CEPsh glia-ablated animals, glial effects on follower-axon guidance are at least partially independent of SubL neurons. Conversely, SIA and SIBV (SubL) neuron ablation correlates with defects in AIY axon (Fig. 2i–l, Supplementary Table S1), and amphid-commissure axon NR entry (25/54 animals), but spares CEPsh glia process growth and NR wrapping (Fig. 2m–p, Supplementary Table S1).
Figure 2. CEPsh glia and SubL axons functionally pioneer the NR.
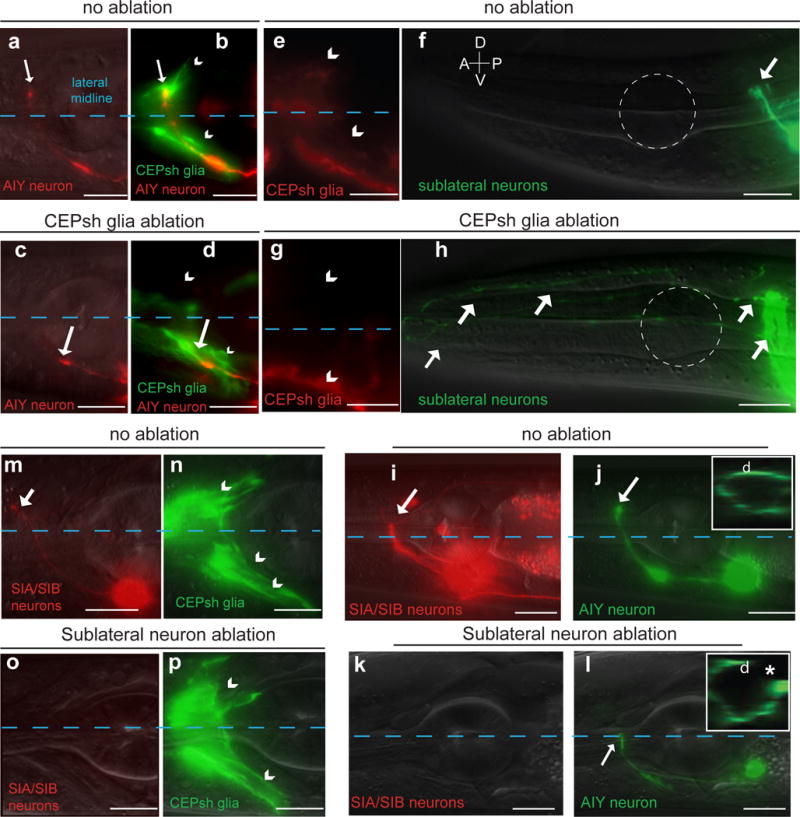
Nerve-ring entry of AIY follower axons and fasciculation of sublateral commissure axons is abnormal in CEPsh-ablated animals. Ablation of neurons of the sublateral commissure results in abnormal AIY nerve-ring entry but spares CEPsh membrane growth. Scale bars: 10 μm. (a–d) Pttx-3::mCherry (AIY), Pmir-228::GFP (glia). (e–h) Pceh-24::GFP (SIA/SIB/SMD), Pptr-10::RFP (glia). Dotted white outline: 1st pharyngeal bulb. (i–p) Pceh-17::RFP (SIA/SIB), Pttx-3::GFP (AIY, green) or Phlh-17:: myristoylated-GFP (CEPsh glia, green). Reporter expression patterns are described in Supplementary Methods and Supplementary Tables S8, S10. Asterisk: NR axon gap.
In summary, the NR is hierarchically assembled: CEPsh glia processes and early-fasciculating SubL axons grow together to mark the presumptive NR. The NR is then populated, in an orderly fashion, by non-commissural, amphid-commissure, and deirid-commissure axons. CEPsh glia promote SubL neuron fasciculation, while CEPsh glia and SubL neurons cooperatively guide follower-axon NR entry.
A NR-assembly-defective mutant reveals cooperative guidance roles for KPC-1/Furin and CHIN-1/Chimaerin
To understand the molecular basis of pioneer-neuron and CEPsh glia functions, we performed a genetic screen, seeking mutants exhibiting AWC/ASE axon NR entry defects reminiscent of CEPsh glia-ablated animals (Supplementary Methods). True-breeding mutants were subjected to whole-genome sequencing (WGS), genetic mapping, and rescue studies. Besides recovering weakly-penetrant mutants in known guidance genes18, we found one mutant, exhibiting uncoordinated movement and highly-penetrant ASE/AWC axon NR entry defects (Fig. 3a–b,o), with no lesion in known C. elegans guidance genes.
Figure 3. NR axon entry is disrupted in kpc-1; chin-1 mutants.
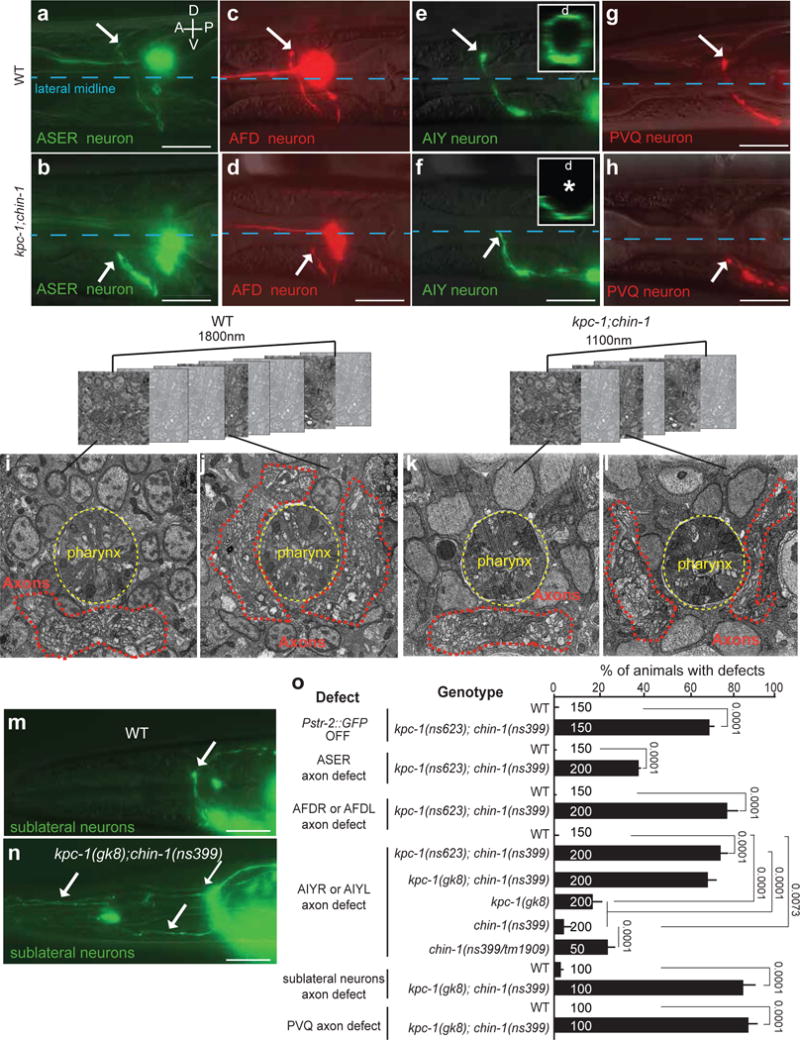
(a–h, m–o) Nerve-ring guidance of axons of different neuron subtypes, and in different commissures, is abnormal in kpc-1; chin-1 mutants. Asterisk, arrow, scale bars: 10μm. D: dorsal, V: ventral, A: anterior, P: posterior. (a,b) Pgcy-5::GFP. (c,d) Pttx-1::RFP. (e,f) Pttx-3::GFP. (b,d,f) kpc-1(ns623); chin-1(ns399). (g,h) Pnpr-11::RFP. (i–l) Nerve-ring structure of L1 animals is abnormal in kpc-1; chin-1 mutants compared to wild-type animals. FIBSEM images of WT (i,j) and mutant (k,l) NR region of age-matched L1 animals. Dotted red line: axons. (h,k,l,n) kpc-1(gk8); chin-1(ns399). (m,n,o) Fasciculation of sublateral commissure neurons is abnormal kpc-1; chin-1 L1 mutant animals compared to wild-type animals. Pceh-24::GFP. (a–h, m–n) Reporter expression patterns are described in Supplementary Methods and Supplementary Tables S8, S10. (o) Numbers inside bars: total animals scored per genotype, n=4 independent scoring experiments. Mean +/− Error bars: SEM. Numbers above bars of significance, p values from Fisher’s exact test. ns: non significant.
NR entry defects in this mutant are prominent (>70%) in all follower neurons examined, including another amphid-commissure neuron (AFD), and the non-commissural interneurons AIY and PVQ (Fig. 3c–h,o). Defective axons often fail to extend dorsally, resulting in NR gaps when viewed in cross section (Fig. 3e,f). This is accompanied by loss of str-2 expression in AWC neurons (Fig. 3o), which depends on bilateral axon contacts24. Focused Ion Beam Scanning Electron Microscopy (FIB-SEM) reconstructions of newly-hatched larvae revealed that ventral aspects of wild-type and mutant NRs contain roughly similar axon numbers, but lateral and dorsal aspects of the mutant NR have markedly fewer processes (Fig. 3i–l; Supplementary Movies S1,S2): ventral, lateral, and dorsal NR segments span >840 nm, >660 nm and >300 nm in the anteroposterior axis in the wild type, but <750 nm, <200 nm and <150 nm in the mutant, respectively. Thus, mutant NR assembly is severely disrupted.
The mutant axon-extension defects are NR-entry specific and do not reflect outgrowth initiation defects. Mutant axons of the tail neuron PVQ have highly-penetrant NR entry defect, but properly extend and navigate along the ventral nerve cord (VNC) prior to NR-entry (Supplementary Fig. S3a–d). Furthermore, the VNC and dorso-ventral motor-neuron commissures, major mid-body axon bundles, as well as positions of neuron and glia cell soma appear grossly normal in mutants (Fig. 3e–j).
Unlike follower neurons, SubL pioneers in the mutant enter the NR, but exhibit fasciculation defects, reminiscent of CEPsh glia-ablated animals (Fig. 3m–o). CEPsh glia morphology, however, appears unaffected in mutant larvae (Supplementary Fig. S3k–l). Therefore, the mutant we identified specifically affects NR entry of various neuronal classes, likely by altering CEPsh glia-pioneer neuron communication.
Genomic and genetic characterization of this mutant revealed, surprisingly, that two lesions are causal for the highly-penetrant NR entry defect. One mutation, kpc-1(ns623), results in a S579T change in the KPC-1 protein, a Kex2/subtilisin-like proprotein convertase homologous to vertebrate Furin. The other mutation, chin-1(ns399), results in a G273E change in the CHIN-1 protein, a GTPase activating protein (GAP) homologous to vertebrate Chimaerins (Supplementary Fig. S4a,b). kpc-1(ns623) is likely a strong loss-of-function allele, as kpc-1(gk8) null mutants25, have similar NR defects when combined with chin-1(ns399) (Fig. 3o). Mutations in the other C. elegans proprotein convertases, AEX-5, EGL-3, and BLI-426, neither affect AIY axon NR entry, nor enhance chin-1 or kpc-1 mutant defects (Supplementary Table S2). chin-1(ns399) is likely not a null allele, as chin-1(tm1909) deletion homozygotes are dead and chin-1(ns399)/chin-1(tm1909) trans-heterozygotes have stronger NR entry defects than chin-1(ns399) animals (Fig. 3o). Our analysis suggests chin-1(ns399) is a dominant negative allele (see below and Fig. 6g).
Figure 6. KPC-1/CHIN-1 control guidance-cue trafficking.
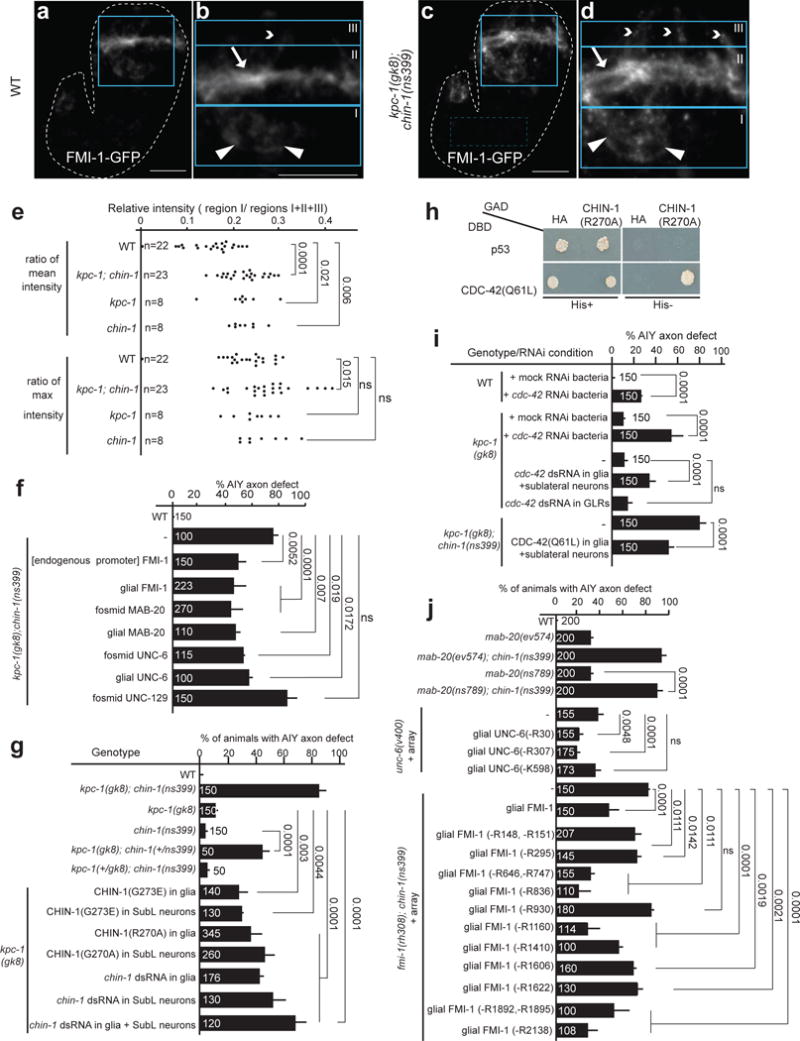
(a–e) FMI-1-GFP localizes to the NR bundle of 1.5-fold wild-type embryos but is detected abnormally outside the NR in kpc-1; chin-1 mutants. (a,c) 1.5-fold embryos expressing Pfmi-1::FMI-1-GFP and magnified view of their head regions (b,d). (b,d) Blue boxes outline regions of interest I (cell bodies, triangles), II (NR, arrows), and III (dendrites, arrowheads). Scale bar: 10 um. (e) FMI-1-GFP ectopic signal is quantified as described in Supplementary Methods (see also Supplementary Fig. S7). Number of animals analyzed appears in the graph (n). Numbers above bars of significance, p values from t-test test (GraphPad). ns: non-significant. t ratios for group comparisons of wild-type and kpc-1; chin-1, wild-type and kpc-1, wild-type and chin-1 are 4.95, 2.44, 2.95, respectively, for mean intensity ratio; and 2.55, 0.74 and 1.50, respectively, for max intensity ratio. Number of degrees of freedom equals the number of pairs minus 1.
(f) Overexpression of UNC-6, MAB-20, FMI-1 but not UNC-129 can partially restore the AIY axon defects of kpc-1; chin-1 mutant animals. (g) chin-1(ns399) encodes a dominant-negative protein similar to CHIN-1 harboring an Arginine finger-mutation. (h) CHIN-1 specifically binds CDC-42 in yeast-two-hybrid assays. p53-DBD and HA-GAD: negative controls, CDC-42(Q61L)-DBD and CHIN-1(R270A)-GAD: positive interactors. His: histidine. (i) cdc-42 RNAi results in abnormal AIY axon nerve-ring entry. Mock RNAi: L4440 vertor. (j) MAB-20, UNC-6 and FMI-1 predicted furin motifs are important for AIY axon nerve-ring entry. (f,g; I,j) cDNA expression is driven by Pmir-228 (glia), Pceh-17 (SubL neurons), Pgly-18 (GLR cells) or endogenous regulatory regions (Online methods). (j) mab-20(ns789): CRISPR allele. (-Rn) or (-Kn): extrachromosomal array of unc-6 or fmi-1 cDNA with single amino-acid deletions perturbing predicted Furin-recognition motifs (Supplementary Fig. S6, Supplementary Table S5). Numbers inside bars: total animals scored per genotype, n=4 independent scoring experiments or number of transgenic lines in rescue experiments. Mean +/− Error bars: SEM. Numbers above bars, exact p values by Fisher’s exact test. ns: non-significant.
Unlike the highly-penetrant NR-entry defects of kpc-1; chin-1 double mutants, those of kpc-1 or chin-1 single mutants are very weak (Fig. 3o). Thus, the kpc-1 and chin-1 genes are functionally redundant and the double mutant kpc-1; chin-1 defines a new class of axon-guidance mutants acting synergistically to assemble the C. elegans brain neuropil.
KPC-1 and CHIN-1 function in CEPsh glia and pioneer neurons during NR assembly onset
To uncover the origin of the kpc-1; chin-1 mutant NR defects, we performed time-lapse imaging during NR assembly. AIY or SubL pioneer axons initiate outgrowth at the bean-stage of wild-type and kpc-1; chin-1 double-mutant embryos. However, extension into the presumptive NR is delayed in the double mutant and, occasionally, in kpc-1 or chin-1 single mutants (Fig. 4a–j, Supplementary Fig. S5). Large growth cones are observed in mutant but not wild-type embryos, suggesting that mutant axons are competent for growth cone initiation, but navigate slowly (Fig. 4g). This is in line with the idea that large growth cones correlate with pausing at choice points27. Furthermore, expression of either kpc-1 or chin-1 cDNA rescues AIY axon defects of kpc-1; chin-1 mutants only when induced at the end of gastrulation (ball stage: 320 min, Supplementary Figure S1) or the “bean” embryonic stage using a heat-shock-responsive promoter (Fig. 4k). Thus, KPC-1 and CHIN-1 are required for proper guidance of axons into the NR prior to or at the time of pioneer neuron entry.
Figure 4. KPC-1 and CHIN-1 act in NR pioneers at the onset of NR assembly.
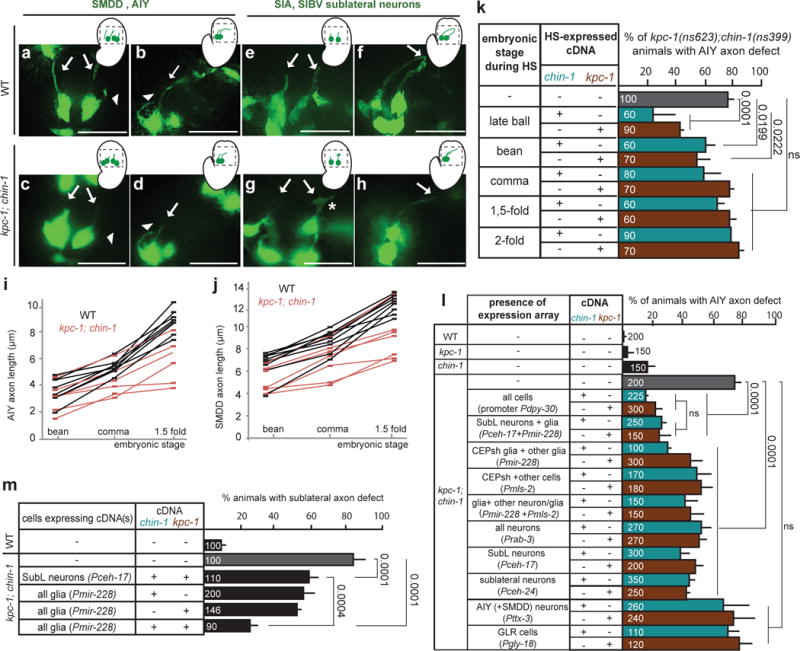
(a–j) Extension of AIY axons and SubL commissure axons into the presumptive nerve-ring is delayed in kpc-1; chin-1 mutants compared to wild-type embryos. Histograms and scale bars, as in Fig. 3. (a–h) Head region (outlined in schematics) of bean or 1.5 fold embryos expressing Pttx-3::GFP (a-d) or Pceh-17::GFP (e-h). Arrows: SubL axons, arrowhead: AIY axon. Asterisk: growth cone. (i–j) Squares: individual axon measurement at given embryonic stage. Lines track individuals across stages. Number of animals analyzed: (i) n=7 for WT, n=6 for chin-1; kpc-1 mutants, (j) n=8 for WT, n=7 for chin-1; kpc-1 mutants. (k) CHIN-1 and KPC-1 expression is necessary prior to the embryonic comma stage for proper nerve-ring assembly. HS: Heat-Shock driving chin-1 or kpc-1 cDNA expression. (l,m) CHIN-1 and KPC-1, acting non-cell-autonomously from SubL neurons and glia, can rescue chin-1; kpc-1 mutant defects of follower axons. Furthermore, CHIN-1 and KPC-1 acting non-cell-autonomously from glia can rescue chin-1; kpc-1 mutant defects of pioneer SubL axons. Rescue of mutant defects by cDNA expression using indicated promoters (Expression patterns are described in Supplementary Methods and Supplementary Tables S8, S10). The following alleles are used unless otherwise indicated: kpc-1(gk8), chin-1(ns399). (k–m) Numbers inside bars: total animals scored per genotype, n=3 independent scoring experiments or number of transgenic lines in rescue experiments. Mean +/− Error bars: SEM. Numbers above bars of significance, p values from Fisher’s exact test. ns: non significant.
KPC-1 and CHIN-1 are broadly expressed before and during NR assembly (Supplementary Fig. S4c,d), consistent with previous studies25,28. However, providing either kpc-1 or chin-1 cDNA in all glia, or in some SubL neurons, partially restores AIY axon NR entry in kpc-1; chin-1 mutants. Moreover, combined cDNA expression in SubL neurons and glia, but not pan-neuronal expression, elicits rescue as efficiently as ubiquitous expression (Fig. 4l). Importantly, kpc-1 or chin-1 cDNA expression in AIY neurons or neighboring GLR cells (Fig. 1a) neither rescues AIY axon defects (Fig. 4l), nor enhances partial rescue obtained by SubL-neuron or glia expression (Supplementary Table S3). Furthermore, each cDNA gives partial rescue when driven separately or in combination by mls-2 or mir-228 promoters, with overlapping expression in CEPsh, CEPso, OLQsh, OLQso, and ADEsh glia (Supplementary Methods). Non-CEPsh glia are positioned >5-10 μm from the NR during NR formation, and none of their processes are NR-associated14. Moreover, mosaic analysis indicates that rescue of AIY axon defects correlates with kpc-1 or chin-1 expression in CEPsh glia, but not in AIY, GLRs, muscle, pharynx, or intestine (Supplementary Table S3).
Although KPC-1 and CHIN-1 function non-cell autonomously to guide AIY axons, we wondered whether they function cell autonomously in SubL neurons. Axon guidance defects of these pioneers are partially rescued by combined KPC-1 and CHIN-1 expression within this commissure, or by glial expression of either KPC-1 or CHIN-1. Yet, significantly better rescue is achieved by expressing KPC-1 and CHIN-1 together in glia (Fig. 4m).
Together, our data suggest that KPC-1 and CHIN-1 cooperate in CEPsh glia, during the onset of NR assembly, to guide SubL pioneer neurons. Both proteins function synergistically in glia and SubL neurons to direct follower axons. These results uncover previously-unknown roles for Chimaerin and Furin in glia, and demonstrate that a specific genetic background can expose the layers of cellular and molecular redundancies controlling NR assembly initiation.
Glia guide pioneer and follower axons using different signals
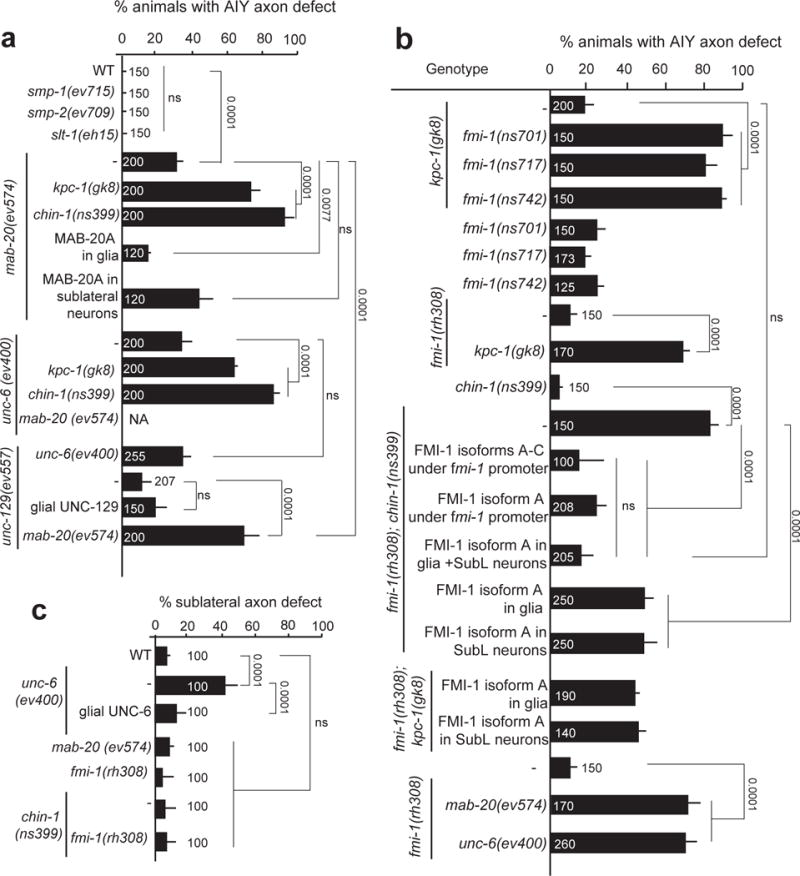
The synergy of KPC-1 and CHIN-1 on NR assembly suggests that redundant factors guiding NR assembly could be identified by seeking mutations that enhance the weak NR defects of either kpc-1 or chin-1 single mutants. Indeed, using candidate gene approaches we found that null mutations in mab-20/Semaphorin29 or unc-6/Netrin21 result in AIY axon NR entry defects, strongly enhanced by kpc-1 or chin-1 lesions (Fig. 5a). Disrupting Semaphorins SMP-1/2 or SLT-1/Slit has no effect on AIY axon NR entry (Fig. 5a). Remarkably, SubL axon navigation is impaired in unc-6 mutants but not in mab-20 mutants. UNC-6 expression was reported in bean-stage CEPsh glia21. Moreover, glia expression of UNC-6 can rescue SubL axon guidance defects of unc-6 mutants (Fig. 5c). mab-20 mRNA is enriched in CEPsh glia (2.5x over all other cells, M. Katz and S.S., pers. comm.), and we found that mab-20 cDNA expression of isoform A in embryonic glia, but not SubL neurons, restores AIY axon NR entry (Fig. 5a). Thus, both MAB-20 and UNC-6 likely function from glia to regulate NR assembly, through distinct mechanisms of follower- and pioneer-axon navigation. UNC-129/TGFß, a muscle-secreted UNC-6/Netrin regulator17, also acts in NR assembly through the Netrin pathway: unc-129 mutations enhance AIY defects of mab-20 mutants, but not of unc-6 mutants (Fig. 5a). This result corroborates a role for glial Netrin signaling in NR assembly.
Figure 5. Glia direct pioneer and follower-axon guidance using distinct signaling pathways.
(a, c) UNC-6/Netrin and MAB-20/Semaphorin regulate nerve-ring assembly. UNC-6 guides primarily pioneer SubL axons while MAB-20 specifically guides follower axons and both act from glia. (b,c) FMI-1/Flamingo/CELSR can act cooperatively from SubL commissure neurons and glia to drive nerve-ring assembly, by specifically guiding follower axons. (a–c) cDNA expression of MAB, 20, UNC-6 and FMI-1 is driven by Pmir-228 (glia), Pceh-17 or Pceh-24 (SubL neurons), or endogenous regulatory regions. Expression patterns are described in Supplementary Methods and Supplementary Tables S8, S10. Numbers inside bars: total animals scored per genotype, n=4 independent scoring experiments or number of transgenic lines in rescue experiments. Mean +/− Error bars: SEM. Numbers above bars, p values from Fisher’s exact test. ns: non significant.
From genetic enhancer screens, in which we mutagenized kpc-1 or chin-1 single mutants, we isolated 21 independent strains with robust AIY axon NR entry defects (>60% penetrance). In addition to new kpc-1 alleles, we isolated 3 mutations in the gene fmi-1, encoding the C. elegans Flamingo/CELSR homolog These fmi-1 alleles (ns701, ns717, ns742; see Supplementary Fig. S6a, and Supplementary Methods), cause weak AIY axon entry defects on their own and strongly enhance the defective phenotype of the kpc-1(gk8) mutation. (Fig. 5b).
FMI-1/CELSR is a transmembrane protein with extracellular cadherin, EGF, and laminin interaction domains (Supplementary Fig. S6a). It is broadly expressed in the C. elegans nervous system during embryogenesis, and promotes axon fasciculation in the VNC30, but a role in NR assembly had not been described. fmi-1 mRNA is enriched in CEPsh glia (13x over all other cells; Menachem Katz and S.S., pers. comm.). The previously-isolated fmi-1(rh308) mutation (Q725Ochre) also displays weak AIY axon NR entry defects and strongly enhances kpc-1 or chin-1 mutations (Fig. 5b). These defects are fully rescued with genomic DNA (generating 3 FMI-1 isoforms, Supplementary Fig. S6a) as well as with fmi-1 regulatory sequences fused to isoform A cDNA (Fig. 5b). A functional FMI-1- GFP protein partially rescues AIY axon defects of fmi-1; chin-1 or fmi-1; kpc-1 mutants when expressed using glial or SubL neuron promoters. Combined expression of fmi-1 cDNA in glia and SubL neurons allows complete rescue, similar to the one achieved by expression under fmi-1 regulatory sequences. Interestingly, fmi-1 or fmi-1; chin-1 mutants are not defective for SubL axon fasciculation (Fig. 5c). Thus, FMI-1, together with KPC-1 and/or CHIN-1, promotes follower axon guidance during NR assembly, and acts from SubL neurons and glia, a surprising result given that other CELSR homologs had been previously suggested to function only from neurons31,32.
Overall, glia appear to differentially guide pioneer and follower axons. Glial UNC-6/Netrin controls pioneer-axon guidance, which can affect follower-axon extension, while MAB-20/Semaphorin and FMI-1/CELSR act in glia for follower-axon and not pioneer-axon guidance (see also below).
KPC-1 and CHIN-1 control glial guidance-cue trafficking
To understand how kpc-1, and chin-1 affect NR axon guidance, we examined FMI-1-GFP subcellular localization. FMI-1-GFP localizes to the NR bundle of 1.5-fold wild-type embryos (Fig. 6a–b). In kpc-1; chin-1 mutants, or kpc-1 or chin-1 single mutants, however, GFP is detected more strongly outside the NR, in cell bodies, dendrites, and proximal axon segments (Fig. 6c–e, Supplementary Fig. S7). FMI-GFP mislocalization is unlikely to be a result of axon morphology defects, as similar mislocalization occurs in chin-1 and kpc-1 single mutants, yet these animals have largely normal neuron structures (Fig. 3o, Supplementary Fig. S5c–d). Thus, KPC-1 and CHIN-1 are required for NR trafficking of FMI-1/Flamingo. This suggests a previously-unknown function for Chimaerins in trafficking, and sheds light on how CELSR proteins are localized.
The partial mislocalization of FMI-1-GFP, as well as the incompletely penetrant defects of the kpc-1; chin-1 double mutant suggest that some trafficking of guidance cues does occur in this strain. If so, then overexpression of guidance cues, allowing more guidance factors to be trafficked, should partially rescue kpc-1; chin-1 double mutants. Indeed, we found that AIY NR entry in kpc-1; chin-1 mutants is partially restored by overexpression of FMI-1, MAB-20, or UNC-6 using endogenous or glia-specific regulatory sequences (Fig. 6f). This suggests that trafficking of MAB-20 and UNC-6 from glia may also require KPC-1/CHIN-1. By contrast, kpc-1; chin-1 mutants are not rescued by overexpression of UNC-129 (Fig. 6f). UNC-129 does not act in glia, as its expression in glia cannot rescue unc-129 mutant NR defects (Fig. 5a). Thus, its inability to rescue kpc-1; chin-1 mutants under its endogenous promoter supports the specificity of our glial overexpression assay. Thus, contrary to previous studies33, CHIN-1 acts upstream, and not downstream, of guidance cues for NR assembly.
In summary, KPC-1/Furin and CHIN-1/Chimaerin function upstream of at least three redundant guidance factors acting specifically in glia (MAB-20/Sema, UNC-6/Netrin) or cooperatively in glia and SubL pioneers (FMI-1/Flamingo), to control NR assembly. These guidance factors also regulate amphid-commissure follower axon navigation, in addition to AIY-axon NR-entry (Supplementary Table S4).
CHIN-1/Chimaerin functions with CDC-42/GTPase
To understand CHIN-1/Chimaerin function, we characterized the chin-1(ns399) mutation. This G273E substitution affects a conserved Arginine finger motif (ArgF, R270), mutations in which allow GTPase binding, but not hydrolytic activity34. We found that NR axon entry defects in kpc-1(null) mutants are enhanced by a heterozygous chin-1(ns399)/+ lesion (Fig. 6g). Glia or SubL-neuron overexpression of CHIN-1(G273E) or CHIN-1(R270A), has similar effects, as does reduction of chin-1 gene function using a hairpin-RNAi in embryonic glia, SubL neurons or both (Fig. 6g). Therefore, chin-1(ns399) is a dominant-negative allele that may block activity of small GTPases.
Through a candidate yeast two-hybrid screen (Supplementary Methods), we identified CDC-42 as a major CHIN-1 interactor (Fig. 6h). Chimaerins bind CDC42 and modulate its GTPase activity35, and CDC42 can regulate trafficking36. NR axon entry defects of follower AIY axons in kpc-1(null) mutants are strongly enhanced by RNAi induced by bacteria-fed cdc-42 dsRNA or by expression of a cdc-42 hairpin in glia and sublateral neurons, but not in neighboring GLR cells (Fig. 6i). This suggests that CDC-42 functions non cell-autonomously from glia and sublateral neurons for follower axon guidance. Given that CHIN-1(G273E) likely represents a dominant negative GTPase activating protein, we reasoned that expression of a constitutively active CDC-42(Q61L) could sequester dominant-negative CHIN-1(G273E), allowing endogenous CDC-42 to bind downstream effectors. In line with this model, expression of CDC-42(Q61L) in NR pioneers partially rescues AIY axon NR defects of kpc-1; chin-1 mutants (Fig. 6i). Thus, CHIN-1 acts through the Rho-family GTPase CDC-42 to control NR assembly.
Predicted KPC-1/Furin cleavage sites in glial guidance cues are required for NR assembly
Furins are implicated in secretory-pathway trafficking37 as well as in Semaphorin and cadherin processing38,39. We identified putative Furin cleavage sites in MAB-20/Sema, UNC-6/Netrin and FMI-1/Flamingo proteins using an RXXR consensus motif, or cleavage sites predicted by the ProP1.0 algorithm (Supplementary Fig. S6a–c, Supplementary Table S5, Supplementary Methods). A single predicted site in MAB-20 fulfills both criteria and is conserved in vertebrate Semaphorin homologs. Mutation of the conserved arginine (R247H) using CRISPR/Cas9 results in AIY axon defects similar to those of mab-20(ev574) null mutants, and enhances defects of single chin-1 mutants (Fig. 6j). The ProP1.0 algorithm also identified three predicted motifs in the UNC-6/Netrin protein that are not conserved and do not conform to the RXXR consensus. unc-6 proteins expressed in glia, and carrying R30 or R37, but not K598 lesions, can rescue AIY entry defects of unc-6 mutants (Figure 6, S6, Supplementary Table S5).
FMI-1 has fifteen predicted furin motifs, six are ProP1.0-predicted cleavage sites not conforming to the RXXR consensus, and nine are RXXR motifs (Figure S6, Supplementary Table S5). None of these are highly conserved. Nonetheless, fmi-1 cDNAs altering amino acids R151, R295, R930, R1410, R1606, or R1622, do not robustly rescue AIY NR entry defects in fmi-1; chin-1 mutants (Fig. 6j). Thus, these MAB-20, UNC-6 and FMI-1 predicted furin motifs, likely recognized by KPC-1, are important in vivo for NR assembly.
Discussion
Our studies support a model for hierarchical assembly of the C. elegans NR (Supplementary Fig. S8). At the onset of embryonic morphogenesis, axons of SubL neurons fasciculate and navigate anteriorly towards ventral CEPsh glia. Dorsally-directed processes of these glia may define a turning point for SubL axons, which coalesce with glial processes to define the NR tract. Based on our EM studies, NR pioneer bundles may contain 2-4 additional processes that we have not yet identified. These may include axons and/or processes of the CEPshD glia. These two dorsal CEPsh glia extend ventrally-directed processes enveloping the adult NR. Although tracking of individual CEPshD processes was challenging, these processes are present before late morphogenesis and their presence is functionally important for follower and pioneer axon guidance (Fig. 2).
CEPsh glia drive SubL axon navigation and then cooperate with these pioneer neurons to promote NR entry of follower axons, which otherwise do not properly enter the NR, and either stop or grow ectopically. This synergy is mediated by glia-secreted UNC-6/Netrin, likely acting primarily for pioneer-axon guidance, and glia-secreted MAB-20/Semaphorin acting specifically for follower-axon guidance. The fasciculation protein FMI-1/CELSR, acts from glia and SubL neurons, perhaps creating a common guidance substrate, which then directs follower axons. Follower axons enter the NR at different times, and do so independently or in commissures that build over time.
SubL pioneers have unique characteristics: we demonstrate that they have CEPsh glia-independent growth potential, unlike follower neurons, and form the earliest fasciculating commissure entering the nerve ring. These neurons are important for follower-axon NR entry, and have previously been implicated in NR positioning20. The NR guidance and positioning activities might reflect distinct temporal roles for SubL commissure components. Adult anatomical studies reveal that SubL neurons occupy central neuropil positions, and are sparsely connected. Indeed, SIA/SIB exhibit no chemical synaptic output onto other NR neurons16 (C. elegans Neural Network wormweb). Thus, NR pioneers represent a distinct neuronal class. Moreover, within the SubL fascicle, SIA/SIB neurons are sufficient for mutant rescue, as these are the neurons common to two different drivers we use (Fig. 4I, Supplementary Tables S3,S10), and appear to grow slightly earlier than SMD neurons (Fig. 1f, Supplementary Fig. S2b–d), suggesting these may be the key components of the SubL bundle.
Our studies also suggest that molecular and cellular redundancies may have previously hindered identification of NR assembly cues. Key to our dissection of the process was the identification of the kpc-1; chin-1 double mutant, which revealed a bottleneck that was used to expose the activities of multiple guidance factors. Importantly, while KPC-1/Furin and CHIN-1/Chimaerin are broadly expressed, and are implicated in many cellular processes25,28,40, the dominant-negative chin-1 mutation we uncovered, when accompanied by kpc-1 null lesions, has remarkably specific synergy in NR assembly. We suggest that a similar mutant combination may uncover novel guidance molecules and interactions in other organisms as well.
Our studies also provide new insights into Chimaerin and Furin functions in the nervous system. Furins are known to cleave guidance cues38,41; however, functional evidence of roles for these proteases in glia are not well-documented. Murine β2-Chimaerin mediates Sema3F-dependent axon pruning42 while human α2-chimaerin mutations cause Duane’s retraction syndrome 2 (DURS2-DRS), and the protein has been suggested to act cell-autonomously in neurons, downstream of a Semaphorin/Plexin pathway for oculomotor nerve guidance33. Intriguingly, however, loss of murine α2-chimaerin causes aberrant axon midline crossing11 and dominant α2-chimaerin lesions cause axon stalling and defasciculation43. The similarities between these abnormalities and the NR defects we observe raise the possibility that CHIN-1 may function non-autonomously upstream of guidance cues in pioneer neurons and/or glia in vertebrates as well.
We reveal key roles for C. elegans CEPsh glia in regulating NR assembly. Although, CEPsh glia have not been demonstrated to be neural progenitors like vertebrate radial glia44, our work, nonetheless, suggests intriguing similarities between these cells in their development, gene expression, and roles in CNS formation. Like embryonic CEPsh glia, vertebrate radial glia extend thin bipolar processes that associate early in development with neurons and which demarcate CNS domains45. Radial glia of the optic tectum express Semaphorins and Furin41,46, as do CEPsh glia, and may be the first source of Netrin1 expression in the embryonic telencephalon47. Importantly, radial glia pial processes were recently reported to be the functionally-relevant sites of Netrin1 activity during commissural-axon guidance in the vertebrate spinal cord48,49. The same guidance molecule in embryonic CEPsh glia drives NR pioneer neuron guidance. Finally, radial glia morphologically transform into astrocytes, glial cells with elaborate membranes that extend fine projections approximating synapses44,50. Similarly, embryonic CEPsh glia develop into adult CEPsh glia which resemble astrocytes in morphology (Fig. 1), in molecular composition (M. Katz and S.S., pers. comm.), and in proximity to synapses16. These similarities between C. elegans CEPsh glia and radial glia/astrocytes are remarkable, as these animals last shared an ancestor over a billion years ago. Therefore, our studies suggest that radial glia may play key roles in pioneering brain assembly by guiding pioneer axons in vertebrates as well.
Methods
C. elegans methods
C. elegans were cultured as previously described52,53. Bristol N2 strain was used as wild type and animals were raised at 20°C (unless otherwise indicated) for at least two generations without starvation. Animals used for phenotypic analysis were hermaphrodites (not males). The age of animals analyzed is indicated in individual sections of the methods presenting of phenotypic scoring and imaging.
Integration of extra-chromosomal arrays was performed using UV with trioxalen (Sigma, T6137). Germ-line transformations to generate unstable extra-chromosomal array (transgenes) were performed using standard protocols of micro-injection54.
Strains and plasmids used in this study
Previously published mutant strains used in this study
Some strains (listed below) were provided from the CGC, funded by the NIH Office of Research Infrastructure Programs (P40 OD010440) and from the National BioResource Project (NBRP, Japan).
The following, previously published mutant alleles were used in this study:
LG.I kpc-1(gk8), bli-4(e937), aex-5 (gk419962), mab-20(ev574)
LG.III: chin(tm1909/+)
LG.IV: ced-3(n717), unc-129(ev557)
LG.V: egl-3(n150), fmi-1(rh308)
LG.X: lin-15(n765), unc-6(ev400) X
New mutant alleles generated in this study
Information is provided in Supplementary Table S6.
Transgenic strains and plasmids used in this study
Information on unstable extra-chromosomal transgenes and stably integrated transgenes respectively is provided in Supplementary Table S7 and S8 respectively.
List of plasmids used in this study
Information is provided in Supplementary Table S9. DNA sequences of pGR plasmids (generated in this study) are available upon request.
Expression patterns of reporters used in this study
Suitable markers were identified by screening transcription-factor, cell-differentiation, and neuropeptide gene reporters. Markers suitable for process imaging in the bean and subsequent embryonic stages were selected based on the pattern reported in previous publications and the criteria of sparse expression, allowing us to easily establish cell identities and track early processes. Pattern was confirmed in this study using embryonic imaging and cell identification was based on cell morphology (axon/dendrite pattern) and cell positioning as previously defined14,68,69, and using the resources of Wormtlas http://www.wormatlas.org/index.html
Information of expression patterns of reporters used in provided in the Supplementary Table S10.
Isolation of Mutants and Genetic Mapping
Animals were mutagenized using 70 mM ethyl methanesulfonate (EMS, Sigma) at 20°C) for 4 hrs.
Primary screen
ntIs1; kyIs136 animals were mutagenized, nonclonal F2 progeny were examined on an fluorescence microscope Zeiss Axioplan 2 (with 63x/1.4 NA objective and FITCH/GFP filter set (Chroma, Set 51019), and animals with aberrant AWC and ASE axon morphologies were recovered.
Secondary enhancer screens
kpc-1(gk8); mgIs18 or chin-1(ns399); mgIs18 animals were mutagenized, nonclonal F2 progeny were examined on a dissecting GFP scope, and animals with defective AIY nerve ring axon were isolated as enhancer candidates. True double mutant enhancers were identified with AIY axon defects of penetrance >60%. DNA samples from mutants were prepared and whole genome sequencing (WGS) was performed at the Genomics Resource Center, at The Rockefeller University. Mutated loci were identified using SNP mapping and rescue experiments with fosmids or plasmid vectors.
Mapping
Mutant animals ntIs1 V; kyIs136 X were crossed into wild-type males of the CB4856 background (Hawaiian isolate) and recombinant progeny were isolated based on AWC and ASE axon guidance defects. In addition to the semi-dominance of the chin-1(ns399) mutation (see Fig. 3 and relevant text), the kpc-1; chin-1 double mutants presented a partial maternal-rescue effect. Specifically, only 6±1.2% of F2 progeny of double heterozygote F1s kpc-1(0/+); chin-1(ns399/+) were defective in AIY axon NR entry. Yet, 11.2±0.8% F2s are expected to be defective, taking into account the Mendelian genetics and the penetrances of the double mutants kpc-1(gk8); chin-1(ns399), equal to 69.3%±6.1%, and the ones of the single mutants kpc-1(gk8) and chin-1(ns399) (17.5%±6.0% and 3.9%±2.3% respectively). The difference of 6±1.2% and 11.25±1.25% (average±SEM, n=4, 200 total animals) is statistically significant (p=0.02). Since this partial maternal rescue effect affected the mutant penetrance of F2 homozygote mutant recombinants, animals of F2 progeny were clonally isolated blindly, and scoring of the relevant defects was performed only on the F3 progeny to isolate homozygous mutant recombinants. Linkage mapping and SNP analysis highlighted two positions linked to the mutant phenotype. One was refined to a ~3.1 cM interval of LG III –25.76 cM [SNP K02F3.2: III:846369] to III-22, 66 cM [SNP Y92C3A:III:1175447 ]. The second one was refined to a ~11.6 cM interval of LG III+2.87 cM [K02B12 I:8,505,227] to III+13, 66 [SNP F15H9: I:12,145,647]. In the relevant LG I region, mutations were detected by WGS in 5 genes. The mutant was rescued with fosmid WRM0635bG07, that included kpc-1, and by a kpc-1 cDNA under the ubiquitous promoter of the gene dpy-30. In the relevant LG III region, 3 mutations were detected by WGS. For 2 of those, fosmids were available but when injected gave no rescue. There was no fosmid available for the mutation present the chin-1 genomic locus. The ns399 mutation was rescued using a chin-1 cDNA under the ubiquitous promoter of the gene dpy-30. fmi-1 mutant alleles recovered in the enhancer screen of the kpc-1(gk8); mgIs18 background were tested for complementation with the reference allele fmi-1(rh308) and failed to complement. Mutants of interest were out-crossed at least 4 times.
CRISPR mutagenesis
CRISPR mutagenesis was performed as previously described79. N2 animals were injected with plasmids dpy-10 sgRNA vector and pGR255B containing the Cas9 and sgRNA target sequences for the genes dpy-10 and mab-20. Repair oligo sequences were designed to reconstitute a dpy-10(cn64) allele, conferring a dominant Roller phenotype and resistant Dpy phenotype. F1s with dpy-10 lesions resulting from active Cas9 were rollers and were cloned and tested for co-CRISPR in the mab-20 locus. CRISPR changes in the targeted region of mab-20 were detected using the Cel-1 enzyme80 and the Surveyor Mutation Detection Kit. CRISPR resulted in generation of the allele mab-20(ns789), that contains a 3 base pair insertion of ATA after 3024C, resulting in change of the motif VHSRVARV into VHSHSVARV abolishing the Arginine247.
Furin motif prediction and deletion
Furins cleave target proteins at RXXR motifs; however, other cleavage sites are known. We identified putative Furin cleavage sites in FMI-1/Flamingo, MAB-20/Sema and UNC-6 proteins by scanning for RXXR motifs and by using predictions of an artificial neural network trained on known Furin cleavage sites (Prop1.0). Disruption of the motif was achieved by deletion of the Arginine predicted to be recognized for cleavage and was verified by accessing the prop1.0 algorithm score of the deleted motif. In cases of adjacent Arginine motifs (RXXRXXR) the intermediate R was deleted to disrupt both of the neighboring motifs.
RNAi Studies
RNAi was performed as previously described, using dsRNA-expressing vectors81 or by feeding on dsRNA expressing bacteria82. For RNAi by feeding, L3 animals were added to plates containing E. coli HT115 with pL4440 expression vectors targeting specific genes (Ahringer library, Vidal Library). Animals were grown at 20°C for 4 days and progeny were scored.
Mosaic analysis
Mosaic analysis was performed as described previously83,84. kpc-1(gk8); chin-1(ns399) mutant animals were injected with unstable extra-chromosomal transgene arrays, stochastically- transmitted to daughter cells during cell divisions. We injected different arrays consisting of a kpc-1 rescuing fosmid WRM0635bG07 or chin-1 under ubiquitous promoter (Pdpy-30::chin-1, since there is no available fosmid bearing the chin-1 locus) and markers of individual lineages as follows: Pelt-2::mCherry for intestine, Pttx-3::mCherry for the AIY neuron respectively, Pgly-18:: myristoylated-GFP for GLR cells, Pmyo-2::GFP for pharyngeal muscle and Pmir-228:: myristoylated-GFP for glia (allows distinguishing CEPsh glia from anterior labial glia and from the Amphid sheath glia). In this strategy informative animals are very rare since they should keep the array overall but lose it only in specific tissues. Due to partial penetrance of the kpc-1(gk8); chin-1(ns399) mutant, only animals with AIY defects were informative. Mutants in which the arrays segregate to intestinal or pharyngeal cells, to GLR mesodermal cells, or to AIY neurons, but not to CEPsh glia and sublateral-commissure neurons, are not rescued for AIY axon NR entry. Similarly, AIY axon defects are not rescued in mutants in which the unstable extra-chromosomal arrays are present in anterior labial or amphid sheath glia but absent from the CEPsh glia (Table S2). In studies of mosaic analysis, presence of the array in the cells directly assayed does not preclude presence of the array in other cells not directly assayed.
Cell ablations
Cell ablation was performed by expressing cDNA of the pro-apoptotic EGL-1/BH3-only protein85 under cell-type-specific promoters active prior and during process outgrowth. Robust expression of the promoters used is detected in bean and later embryonic stages but is not reported in the precursors of the cells of interest. EGL-1/BH3-only protein causes apoptosis when expressed ectopically85. Cell death was confirmed by absence of cell-specific marker expression in newly-hatched larvae. For example, 24/120 EGL-1/BH3-only-expressing animals carrying the CEPsh-glia reporters mir-228 promoter::GFP or ptr-10 promoter::RFP lost fluorescent signal in 1-3 CEPsh glia (animals lacking all 4 were not observed, probably due to lethality). Animals also harboring a ced-3(n717) caspase mutation, preventing apoptosis, did not lose reporter expression (0/125).
Ablations of sublateral commissure neurons
Subsets of sublateral-commissure neurons were ablated by expression of Pceh-17:: egl-1 cDNA, expressed during bean and later stages in the sublateral commissure neurons SIAVL/R, SIADL/R, SIBVL/R and the motoneurons DA5, DA8 and in the non-commissural neurons ALA, RMED starting after the comma stage. Cell ablation was detected by absence of fluorescent reporters Pceh-17::GFP/mCherry labeling post-embryonically the 4 sublateral-commissure neurons SIAVL/R, SIADL/R and the neuron ALA. SIBV neurons express Pceh-17 driven constructs as early and strongly as SIA neurons, and they are sisters to the SIAV neurons. Thus, SIBV most likely retain the provided transgene in cases that SIAV neurons do and are likely killed along with the SIAV neurons. However SIBV ablation was not directly assessed, due to absence of specific SIBV-specific post-embryonic reporters. In addition to SIA and SIBV neurons these ablation experiments most likely result in the killing of the neurons DA5 and DA8 that do not partake in the nerve ring bundle as well as of the NR neurons ALA and RMED. ALA and RMED are located dorsally and further from the AIY and amphid-neuron axon growth and their axon growth is not observed prior to 1.5-fold (G.R. and S.S. unpublished observations; Singhal et al., submitted), thus later than the sublateral commissure axons defining the nerve ring. Moreover, Pceh-17 is expressed in ALA/RMED only during or after but not prior to the 1.5-fold stage, while SIA/SIB axons have already formed the sublateral commissure that defines the nerve ring. Thus, while ALA/RMED were not directly assessed, these are less likely to have direct effects on early axon guidance of AIY and amphid neurons.
Ablations of CEPsh glia
Subsets of glia were ablated by expression of Pmir-228:: egl-1 cDNA. Cell ablation was detected by absence of CEPsh glia expression of fluorescent reporters Pmir-228::GFP (labeling all glia), or Pptr-10::RFP (labeling all but amphid sheath glia (AMsh)), or Phlh-17:: myristoylated-GFP (labeling CEPsh glia). In Supplementary Table S1, ablation of “other glia” refers to the cases where CEPsh reporter expression was not abolished but the Pmir-228::egl-1 was present in the animals (based on co-injection markers), and thus maybe present in other non-CEPsh glia. The large number (46), the close apposition, and the lack of specific reporters for all non-CEPsh glia precluded monitoring closely killing of other glia. Highly penetrant axon guidance defects were correlated with killing of CEPsh glia. We cannot exclude effects of the OLQsh glia on axon guidance, since OLQshD are sisters of the CEPshD glia and thus likely obtain the Pmir-228:: egl-1 array when CEPsh glia do. However, during NR formation (bean-comma-1.5 fold embryos) the OLQshD are positioned in the tip of the nose (5-10 μm anterior to the NR axon growth)14. Moreover none of their processes are reported to be included in the nerve ring structure at any developmental stage during or after NR formation86.
Axon defect scoring
Axon defects were detected by Pceh-24::GFP (labeling sublateral-commissure neurons SIA, SIB and SMD), Pttx-3::mCherry (labeling AIY neurons postembryonically), and Podr-1::RFP (labeling AWC/AWB amphid-commissure axons post-embryonically). Misguided anterior processes were observed for sublateral-commissure neurons, short axons were seen for AIY interneurons, and short axons or unfasciculated axon bundles were observed for AWC and AWB amphid commissure neurons. CEPsh morphology was scored using Phlh-17:: myristoylated-GFP. Animals were scored after the end of embryogenesis. To correlate ablation with defects occurring in embryogenesis, animals at the first larval stage (L1 larvae) containing the ablation constructs were selected by the presence of an embryonically-expressed co-injection marker), and axon defects were scored without scoring cell ablation. Cell ablation was subsequently assessed. CEPsh ablation was specifically assessed in L3 larvae, where the glia reporters allow single resolution of CEPsh glia from other anterior glia. Axon defects significantly correlated with CEPsh absence scored in later stages.
Imaging and defect scoring for post-embryonic axons
Post-developmental live imaging and scoring of the AIY axon defects (by mgIs18 or otIs133) was performed in L3-L4 animals (or L2 larvae when indicated). For scoring of the nerve ring axon defects of neurons SIA, SIB, ASE, AFD, PVQ, or CEPsh glia membrane morphology, animals were anesthetized (20 mM sodium azide in M9 buffer), mounted on pads (2% agarose in H2O) and examined on an Axioscope compound microscope (Zeiss). 20 animals of L3-L4 larval stage (or L2 larvae when indicated) were mounted per slide and examined immediately after anesthetizing.
Quantification of axon defects
For population measurements of nerve ring axon defects, animals were mounted on slides and visualized in an Axioscope compound microscope (Zeiss). Mutant phenotypes were quantified using axon markers to define nerve ring entry and growth based on the meeting point of the dorsal axon part of bilaterally symmetrical neurons. When scoring axons of ASE or AIY neurons, wild type animals show a full axonal ring with a dorsal meeting point of bilaterally symmetrical axons, while mutant animals present an axonal ring with a gap and no dorsal meeting point. Images presented in Fig. 3a–h are of L3–L4 animals for better visualization of the defect. Scoring of mutant phenotypes was performed in L in animals of early larval stages L1–L3; no significant difference of defects was observed in between those early larval stages.
For embryonic axon length measurements, images were acquired on a Deltavision microscope, as described below. Axons were traced and axon length was measured manually using image stacks in ImageJ/Fiji. Axon length was measured from the anterior limit of cell body fluorescence to the anterior limit of axon fluorescent tip.
Deltavision microscopy imaging of embryonic samples
Embryos at late ball stage were mounted on a 5% agar pad without azide and were washed repeatedly to remove bacteria before mounting. Images were collected using a Weatherstation environmental chamber set at 20°C on a DeltaVision Image Restoration Microscope (Applied Precision) with an Inverted Olympus IX-70 microscope, and using a 63x silicone oil-immersion objective and a Photometrics CoolSnap HQ camera (Roper Scientific). Time-lapse images were acquired until the embryo began to twitch at early 1.5-fold stage. A stack of optical sections at 0.5 um spacing was acquired at 12 min intervals, using conditions for non-saturated signal, 10-32% power, and exposure times of 0.2-0.5 sec, depending on the labeling reporter used. Image acquisition did not result in appreciable developmental arrest. Between time points the focal midpoint of the stack was adjusted to compensate for rotation of the embryo and movement of the cell of interest. Deconvolution of DeltaVision Images was performed with Softworx (Applied Precision).
Quantification of FMI-1-GFP localization defect
The same integrated transgene of FMI-1-GFP (under fmi-1 endogenous regulatory sequences) was crossed into genetic backgrounds of chin-1(ns399); kpc-1(gk8) double mutants, or single mutants chin-1(ns399) and kpc-1(gk8). chin-1 single mutants occasionally exhibit embryo-wide FMI-1-GFP mis-expression. This mis-expression is likely irrelevant to the NR defects as it is not observed in kpc-1; chin-1 double mutants and may result from background mutation(s). Embryos presenting this embryo-wide mis-expression were excluded from the imaging analysis to avoid differences in signal localization that are irrelevant to our genes and phenotypes of interest.
Embryo samples expressing integrated FMI-1-GFP (under fmi-1 endogenous regulatory sequences) were collected in M9 buffer and mounted on pads (2% agarose in H2O). Images were acquired in Inverted TCS SP8 laser scanning confocal microscope (Leica), using the same scanning microscope (with same objective, HyD hybrid detector) and the same acquisition parameters (laser wavelength, zoom magnification, exposure time, Z-step size, field size, pinhole=1, etc) between wild-type and mutant samples. Embryos of different genotypes were synchronized at the 1.5-fold stage. Samples where quantitatively analyzed only when comparable based on the following criteria: measurement of the length of the “elongation fold” between the tail and the tail fold-depression should be 16-18 μm. Embryos with smaller or larger length values were excluded, to control sample variability. Sum projections were acquired from whole embryo stacks, region of interest (ROI) were selected and intensities of ROI were quantified, using ImageJ (NIH, USA). ROI sizes (Fig. 5) were of the following sizes: region I (neuropil cell bodies: 7 μm height, 20 μm width), region II (nerve ring bundle: 7 μm height, 20 μm width), region III (sensory dendritic endings: 6 μm height, 15 μm width), region IV (background in the dorsal part of the tail fold: 7 μm height, 20 μm width). In every sum projection, the mean intensity of background (region IV) was subtracted from the mean or maximum intensities of the ROI (I, II, III). Comparison were made between mean or maximum intensities of the regions ROI-I and ROI-II or the ROI-I and the sum of all three ROI I,II, III. A t-test was used for statistical analysis. The background intensity values of (ROI-IV) between samples of different genotypes was not significantly different (t-test).
Image processing
Maximum or sum projections were prepared using ImageJ/Fiji. Top views (top from head) of the nerve ring were rendered by parameter “Stack Reslice” in ImageJ/Fiji. Image projections used for qualitative analysis were adjusted for brightness, contrast and false-color indexing using Photoshop CS4 (Adobe Software). In cases of reporters labeling neurons of different identities (Pttx-3 labeling AIY and SMDD, Phlh-16 labeling both SMDD and AWC), pseudocoloring for purposes of representation was performed as follows: cells were identified based on expression pattern, cell positioning, and process morphology as defined by Sulston et al, 197768 and Sulston et al. 198314, and maximum projections corresponding to each of the two neurons labeled were acquired and colored coded using ImageJ. Merged color images were assembled using the layer mode in Photoshop. Movies were assembled using QuickTime Pro and Final cut Pro.
Embryo/L1 sample preparation for stage synchronization
Adults were cut-open and embryos were selected and synchronized at 2-cell or 4-cell. These embryos were grown at 20°C and selected after 310-320 min (for 2-cell stage embryos) or 290-310 min (for 4-cell stage embryos) for “late ball” stage embryos (Fig. 4g) and 20 min or 40 min or 60 min later for bean stage, comma stage and 1.5-fold stage embryos respectively. For L1 animals, laid embryos were selected and synchronized by morphology at the 1.5-fold stage. L1 animals were picked after growing at 20°C for 400–450 min.
Electron microscopy sample preparation and imaging
Embryos were synchronized as described above. Samples were prepared for electron microscopy using standard methods87. Ultra-thin serial sections (70 nm) were collected by using a Leica Ultracut UCT Ultramicrotome. Sections at head regions were examined. Electron microscopy images were acquired using a FEI Tecnai G2 Spirit BioTwin transmission electron microscope operating at 120 kV with a Gatan 4K × 4K digital camera.
FIB-SEM microscopy
Focused ion beam Scanning Electron Microscope (FIB-SEM) was performed at the NY Structural Biology Center using a Dual beam FEI Helios NanoLab 650 instrument. A modified high-contrast en bloc staining OTO method88 was applied to the FIB-SEM specimen preparation. Sodium thiocarbohydrazide (TCH) was used to bind the primary osmium stain. Then, the en-block stain was enhanced by a second round of osmium fixation. Glutaraldehyde (1%) was added to provide effective protein crosslinking in tissues. The fixed samples were embedded in Eponate 12™ Resin (TedPella). The serial sections prior to the region-of-interest were searched and confirmed by TEM imaging before the critical portion of the specimen had been reached. Sample blocks were mounted on scanning electron microscope (SEM) stubs using double sided carbon sticky tape, then painted with colloidal silver (EMS). The sample blocks were made conductive by coating with Palladium in a Denton sputter coater (25 mA current for 5 min at 50-60 milliTorr) then inserted into the FEI Helios NanoLab 650 (FEI) vacuum chamber. Areas were identified by viewing with the e-beam at 20 keV then coated with 1μm thick platinum for specimen surface protection. The FEI AutoSlice and View™ software was used to collect serial images using a slice width of 30 nm and a horizontal field width of 18.7 micron. Sections were imaged at an e-beam voltage of 2 keV and 100 picoamperes (pA) current. The focus and stigmation of the electron beam is continuously checked and optimized by the AutoSlice and View™ as the FIB continues to mill into a sample and the imaging face recedes. Following collection, accurate image registration was achieved by applying a scale-invariant features algorithm (Lowe, 1999) to the raw image stacks. The image x-y resolution of the acquired FIB-SEM images is 4.56nm/pixel. L1 animals of N2 (wild-type) or chin-1(ns399); kpc-1(gk8) strains were used for the FIB-SEM, synchronized as described above.
Sample preparation for heat-shock studies
Synchronization was performed as described above. Transgenic embryos expressing rescuing constructs were distinguished from the non-transgenics prior to morphogenesis, by the extrachromosomal expression of RFP under a ubiquitous promoter driving early embryonic expression (Pubq-1). These embryos were grown at 20°C and relevant embryonic stages were heat-shocked at 30°C incubator for 5 min, then allowed to recover for 34 h at 20°C. 2 days later, hatched animals were scored as L3s for nerve ring defects of the AIY axon.
Dye filling assays
Animals were washed off NGM plates with M9 medium and spun down briefly in a micro-centrifuge. The supernatant was removed, and the lipophilic dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI, prepared in N,N-dimethylformamide) was added at 10 ug/ml in M9. Animals were then soaked in dye for 60 minutes in the dark, washed twice with M9 and mounted on slides to be scored for amphid commissure axon defects in Axioscope compound microscope (Zeiss). Amphid commissure presented variable defects: thinner bundle, indicating at least some amphid axons prematurely stop; or aberrant processes, primarily guided to the anterior of the nerve ring bundle, likely following aberrant sublateral neurons.
Yeast 2-Hybrid screen
Yeast 2-hybrid screening was performed using the Dual Y2H kit (DUALSYSTEMS Biotech). N-terminal fusions of LexA and Gal4 domains were used. Candidate interactions were tested between dominant GAD-CHIN-1(R270A) and DBD-GTPase fusions of dominant Q61L mutant variants that stabilize GAP-GTPase binding89. The following RhoGTPases were assayed: CED-10, RAB-2, RAB-6.1, RAB-6.2, RAB-7, RAB-11, CDC-42. Plates with or without Histidine (His + or His −) were used, respectively, for growth or to detect interactions.
Statistics
The sample size and statistical tests were chosen based on previous studies with similar methodologies and the data met the assumptions for each statistical test performed. No statistical method was used in deciding sample sizes. All experiments were performed 3-4 times, as indicated, yielding similar results and comprised of biological replicates. Independent transgenic lines or individual days of scoring for mutant strains were treated as independent experiments for standard error of the mean. Fisher’s test (GraphPad) was used for statistical significance of AIY or SubL axon nerve-ring defects. For comparison of normally distributed values of intensities of FMI-1-GFP, student’s t-test (GraphPad) was used. For all figures, the mean +/− standard deviation (s.d.) is represented unless otherwise noted. P values are calculated using GraphPad software. Prism calculates the difference between each set of pairs, keeping track of sign. The t ratio for a paired t test is the mean of these differences divided by the standard error of the differences. The number of degrees of freedom equals the number of pairs minus 1.
Blinding and randomization during data analysis
Blinding during data analysis was not performed except of the following:
In cell-ablation experiments (Fig. 2e–h), axon defects were quantified prior to determining presence or absence of CEPsh glia. During experiments of electron microscopy in L1 animals (FIB-SEM, Fig. 2i–l), the genotype of L1 animals analyzed was unknown to the researcher performing EM preparation and imaging. During yeast-two-hybrid screening experiments (Fig. 6h), analysis of Y2H interactions of CHIN-1 with candidate GTPases was performed blindly; the genetic identity of the clones analyzed was unknown to the researcher quantifying strength of interactions. Samples were allocated to groups of the genetic background (genotype), detected by standard genetic/genomic approaches. Otherwise samples were randomly selected within these groups, based on previous studies with similar methodologies.
Life Sciences Reporting Summary
A summary of Experimental design and Software is also provided in the Life Sciences Reporting Summary by Nature Research Publishing Group.
Supplementary Material
- Supplementary Figures S1–S8 (Integrated Suppl. Figures template)
- Supplementary Tables S1–S5
- Supplementary Movies S1, S2
- Supplementary Tables S6–S10 (XLS files)
- Supplementary References (51–89)
Summary sentence.
Radial glia-like cells and pioneer neurons drive hierarchical assembly of the C. elegans brain through Chimaerin/Furin-dependent guidance cue trafficking.
Acknowledgments
We thank C. Bargmann, V. Bertrand, L. Cochella, L. Chen, J. Culotti, O. Hobert, H. Hutter, L. Kutscher, J. Malin, G. Oikonomou, N. Pujol, P. Sengupta, B. Tursun, WG. Wadsworth, S. Wallace, and M. Zhen for reagents as well as Menachem Katz for sharing unpublished information. Some strains were provided by the CGC, funded by NIH (P40 OD010440). We thank the Rockefeller University Bio-Imaging and Electron Microscopy Resource Centers for technical help, William J. Rice at the Simons Electron Microscopy Center (NYSBC) for help with FIB/SEM imaging and C. Bargmann and the Shaham lab for insights. G.R. was a Shelby White and Leon Levy Foundation postdoctoral fellow. This work was supported in part by NIH grants NS064273 and NS073121 to S.S.
Footnotes
AVAILABILITY OF DATA
The datasets generated and/or analyzed during the current study are either included in this published article (and its supplementary information files) or available from the corresponding author on reasonable request.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Easter SS, Ross LS, Frankfurter A. Initial tract formation in the mouse brain. J Neurosci. 1993;13:285–299. doi: 10.1523/JNEUROSCI.13-01-00285.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chedotal A, Richards LJ. Wiring the Brain: The Biology of Neuronal Guidance. Cold Spring Harb Perspect Biol. 2010;2:a001917–a001917. doi: 10.1101/cshperspect.a001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacobs JR, Goodman CS, Ii CNS. Embryonic Development of Axon Pathways Behavior of Pioneer Growth Cones in the Drosophila. 1989:2412–2422. doi: 10.1523/JNEUROSCI.09-07-02412.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hidalgo A, Urban J, Brand AH. Targeted ablation of glia disrupts axon tract formation in the Drosophila CNS. 1995;3712:3703–3712. doi: 10.1242/dev.121.11.3703. [DOI] [PubMed] [Google Scholar]
- 5.Hidalgo A, Booth GE. Glia dictate pioneer axon trajectories in the Drosophila embryonic CNS. Development. 2000;127:393–402. doi: 10.1242/dev.127.2.393. [DOI] [PubMed] [Google Scholar]
- 6.Whitington PM, Quilkey C, Sink H. Necessity and redundancy of guidepost cells in the embryonic Drosophila CNS. Int J Dev Neurosci. 2004;22:157–163. doi: 10.1016/j.ijdevneu.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Takizawa K, Hotta Y. Pathfinding analysis in a glia-less gcm mutant in Drosophila. 2001:30–36. doi: 10.1007/s004270000117. [DOI] [PubMed] [Google Scholar]
- 8.Placzek M, Briscoe J. The floor plate: multiple cells, multiple signals. Nat Rev Neurosci. 2005;6:230–240. doi: 10.1038/nrn1628. [DOI] [PubMed] [Google Scholar]
- 9.Minocha S, et al. Nkx2.1-derived astrocytes and neurons together with Slit2 are indispensable for anterior commissure formation. Nat Commun. 2015;6:6887. doi: 10.1038/ncomms7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolodkin AL, Tessier-Lavigne M. Mechanisms and Molecules of Neuronal Wiring: A Primer. Cold Spring Harb Perspect Biol. 2011;3:a001727–a001727. doi: 10.1101/cshperspect.a001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwasato T, et al. Rac-GAP α-Chimerin Regulates Motor-Circuit Formation as a Key Mediator of EphrinB3/EphA4 Forward Signaling. Cell. 2007;130:742–753. doi: 10.1016/j.cell.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 12.Jaworski A, et al. Operational redundancy in axon guidance through the multifunctional receptor Robo3 and its ligand NELL2. Science (80-) 2015;350:961–965. doi: 10.1126/science.aad2615. [DOI] [PubMed] [Google Scholar]
- 13.Jorgensen EM, Mango SE. The art and design of genetic screens: caenorhabditis elegans. Nat Rev Genet. 2002;3:356–369. doi: 10.1038/nrg794. [DOI] [PubMed] [Google Scholar]
- 14.Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 15.Oikonomou G, Shaham S. The Glia of Caenorhabditis elegans. Glia. 2010;59:1253–1263. doi: 10.1002/glia.21084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White JG, Southgate E, Thomson JN, Brenner S. The Structure of the Nervous System of the Nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1986;314:340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 17.MacNeil LT, Hardy WR, Pawson T, Wrana JL, Culotti JG. UNC-129 regulates the balance between UNC-40 dependent and independent UNC-5 signaling pathways. Nat Neurosci. 2009;12:150–155. doi: 10.1038/nn.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zallen JA, Kirch SA, Bargmann CI. Genes required for axon pathfinding and extension in the C. elegans nerve ring. Development. 1999;126:3679–3692. doi: 10.1242/dev.126.16.3679. [DOI] [PubMed] [Google Scholar]
- 19.Hedgecock EM, Culotti JG, Hall DH. The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron. 1990;4:61–85. doi: 10.1016/0896-6273(90)90444-k. [DOI] [PubMed] [Google Scholar]
- 20.Kennerdell JR, Fetter RD, Bargmann CI. Wnt-Ror signaling to SIA and SIB neurons directs anterior axon guidance and nerve ring placement in C. elegans. Development. 2009;136:3801–3810. doi: 10.1242/dev.038109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wadsworth WG, Bhatt H, Hedgecock EM. Neuroglia and pioneer neurons express UNC-6 to provide global and local netrin cues for guiding migrations in C. elegans. Neuron. 1996;16:35–46. doi: 10.1016/s0896-6273(00)80021-5. [DOI] [PubMed] [Google Scholar]
- 22.Durbin RM. Studies on the development and organisation of the nervous system of Caenorhabditis elegans. Cambridge: 1987. [Google Scholar]
- 23.Yoshimura S, Murray JI, Lu Y, Waterston RH, Shaham S. mls-2 and vab-3 Control glia development, hlh-17/Olig expression and glia-dependent neurite extension in C. elegans. Development. 2008;135:2263–2275. doi: 10.1242/dev.019547. [DOI] [PubMed] [Google Scholar]
- 24.Troemel ER, Sagasti A, Bargmann CI. Lateral signaling mediated by axon contact and calcium entry regulates asymmetric odorant receptor expression in C. elegans. Cell. 1999;99:387–398. doi: 10.1016/s0092-8674(00)81525-1. [DOI] [PubMed] [Google Scholar]
- 25.Schroeder NE, et al. Dauer-Specific Dendrite Arborization in C. elegans Is Regulated by KPC-1/Furin. Curr Biol. 2013;23:1527–1535. doi: 10.1016/j.cub.2013.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thacker C, Rose AM. A look at the Caenorhabditis elegans Kex2/Subtilisin-like proprotein convertase family. BioEssays. 2000;22:545–553. doi: 10.1002/(SICI)1521-1878(200006)22:6<545::AID-BIES7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 27.Mason C, Erskine L. Growth cone form behavior, and interactions in Vivo: Retinal axon pathfinding as a model. J Neurobiol. 2000;44:260–270. doi: 10.1002/1097-4695(200008)44:2<260::aid-neu14>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 28.Kumfer KT, et al. CGEF-1 and CHIN-1 regulate CDC-42 activity during asymmetric division in the Caenorhabditis elegans embryo. Mol Biol Cell. 2010;21:266–277. doi: 10.1091/mbc.E09-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roy PJ, Zheng H, Warren CE, Culotti JG. mab-20 encodes Semaphorin-2a and is required to prevent ectopic cell contacts during epidermal morphogenesis in Caenorhabditis elegans. Development. 2000;127:755–767. doi: 10.1242/dev.127.4.755. [DOI] [PubMed] [Google Scholar]
- 30.Steimel A, et al. The Flamingo ortholog FMI-1 controls pioneer-dependent navigation of follower axons in C. elegans. Development. 2010;137:3663–3673. doi: 10.1242/dev.054320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Organisti C, Hein I, Kadow ICG, Suzuki T. cooperates with Netrin/Frazzled in Drosophila midline guidance. 2014:50–67. doi: 10.1111/gtc.12202. [DOI] [PubMed] [Google Scholar]
- 32.Feng J, et al. Celsr3 and Fzd3 Organize a Pioneer Neuron Scaffold to Steer Growing Thalamocortical Axons. 2016:3323–3334. doi: 10.1093/cercor/bhw132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferrario JE, et al. Axon guidance in the developing ocular motor system and Duane retraction syndrome depends on Semaphorin signaling via alpha2-chimaerin. Proc Natl Acad Sci. 2012;109:14669–14674. doi: 10.1073/pnas.1116481109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan X, Eathiraj S, Munson M, Lambright DG. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature. 2006;442:303–306. doi: 10.1038/nature04847. [DOI] [PubMed] [Google Scholar]
- 35.Chen W, Lim HH, Lim L. The CDC42 homologue from Caenorhabditis elegans. Complementation of yeast mutation. J Biol Chem. 1993;268:13280–13285. [PubMed] [Google Scholar]
- 36.Lipschutz JH, Mostov KE. Exocytosis: the many masters of the exocyst. Curr Biol. 2002;12:R212–4. doi: 10.1016/s0960-9822(02)00753-4. [DOI] [PubMed] [Google Scholar]
- 37.Hosaka M, et al. Arg-X-Lys/Arg-Arg motif as a signal for precursor cleavage catalyzed by furin within the constitutive secretory pathway. J Biol Chem. 1991;266:12127–12130. [PubMed] [Google Scholar]
- 38.Adams RH, Lohrum M, Klostermann A, Betz H, Püschel AW. The chemorepulsive activity of secreted semaphorins is regulated by furin-dependent proteolytic processing. EMBO J. 1997;16:6077–6086. doi: 10.1093/emboj/16.20.6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sadeqzadeh E, et al. Furin processing dictates ectodomain shedding of human FAT1 cadherin. Exp Cell Res. 2014;323:41–55. doi: 10.1016/j.yexcr.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 40.Hung WL, Wang Y, Chitturi J, Zhen M. A Caenorhabditis elegans developmental decision requires insulin signaling-mediated neuron-intestine communication. Development. 2014;141:1767–1779. doi: 10.1242/dev.103846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tassew NG, Charish J, Seidah NG, Monnier PP. SKI-1 and Furin Generate Multiple RGMa Fragments that Regulate Axonal Growth. Dev Cell. 2012;22:391–402. doi: 10.1016/j.devcel.2011.11.022. [DOI] [PubMed] [Google Scholar]
- 42.Riccomagno MM, et al. The RacGAP &β2-Chimaerin Selectively Mediates Axonal Pruning in the Hippocampus. Cell. 2012;149:1594–1606. doi: 10.1016/j.cell.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyake N, et al. Human CHN1 Mutations Hyperactivate 2-Chimaerin and Cause Duane’s Retraction Syndrome. Science (80-) 2008;321:839–843. doi: 10.1126/science.1156121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barry DS, Pakan JMP, McDermott KW. Radial glial cells: Key organisers in CNS development. Int J Biochem Cell Biol. 2014;46:76–79. doi: 10.1016/j.biocel.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 45.Rakic P. Neuron-glia relationship during granule cell migration in developing cerebellar cortex. A Golgi and electronmicroscopic study in Macacus Rhesus. J Comp Neurol. 1971;141:283–312. doi: 10.1002/cne.901410303. [DOI] [PubMed] [Google Scholar]
- 46.Kuwajima T, et al. Optic Chiasm Presentation of Semaphorin6D in the Context of Plexin-A1 and Nr-CAM Promotes Retinal Axon Midline Crossing. Neuron. 2012;74:676–690. doi: 10.1016/j.neuron.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Métin C, Deléglise D, Serafini T, Kennedy TE, Tessier-Lavigne M. A role for netrin-1 in the guidance of cortical efferents. Development. 1997;124:5063–5074. doi: 10.1242/dev.124.24.5063. [DOI] [PubMed] [Google Scholar]
- 48.Dominici C, et al. Floor-plate-derived netrin-1 is dispensable for commissural axon guidance. Nature. 2017;545:350–354. doi: 10.1038/nature22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varadarajan SG, et al. Netrin1 Produced by Neural Progenitors, Not Floor Plate Cells, Is Required for Axon Guidance in the Spinal Cord. Neuron. 2017;94:790–799.e3. doi: 10.1016/j.neuron.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Voigt T. Development of glial cells in the cerebral wall of ferrets: direct tracing of their transformation from radial glia into astrocytes. J Comp Neurol. 1989;289:74–88. doi: 10.1002/cne.902890106. [DOI] [PubMed] [Google Scholar]
- 51.Duckert P, Brunak S, Blom N. Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel. 2004;17:107–112. doi: 10.1093/protein/gzh013. [DOI] [PubMed] [Google Scholar]
- 52.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stiernagle T. Maintenance of C. elegans. C elegans. 1999;2:51–67. doi: 10.1895/wormbook.1.101.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Altun-Gultekin Z, et al. A regulatory cascade of three homeobox genes, ceh-10, ttx-3 and ceh-23, controls cell fate specification of a defined interneuron class in C. elegans. Development. 2001;128:1951–1969. doi: 10.1242/dev.128.11.1951. [DOI] [PubMed] [Google Scholar]
- 56.Pujol N, Torregrossa P, Ewbank JJ, Brunet JF. The homeodomain protein CePHOX2/CEH-17 controls antero-posterior axonal growth in C. elegans. Development. 2000;127:3361–3371. doi: 10.1242/dev.127.15.3361. [DOI] [PubMed] [Google Scholar]
- 57.Wenick AS, Hobert O, York N, York N. Genomic cis- Regulatory Architecture and trans- Acting Regulators of a Single Interneuron-Specific Gene Battery in C elegans. 2004;6:757–770. doi: 10.1016/j.devcel.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 58.Yu S, Avery L, Baude E, Garbers DL. Guanylyl cyclase expression in specific sensory neurons: a new family of chemosensory receptors. Proc Natl Acad Sci U S A. 1997;94:3384–3387. doi: 10.1073/pnas.94.7.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Viveiros R, Hutter H, Moerman DG. Membrane extensions are associated with proper anterior migration of muscle cells during Caenorhabditis elegans embryogenesis. Dev Biol. 2011;358:189–200. doi: 10.1016/j.ydbio.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 60.Cochella L, Hobert O. Embryonic Priming of a miRNA Locus Predetermines Postmitotic Neuronal Left/Right Asymmetry in C. elegans. Cell. 2012;151:1229–1242. doi: 10.1016/j.cell.2012.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chalasani SH, et al. Neuropeptide feedback modifies odor-evoked dynamics in Caenorhabditis elegans olfactory neurons. Nat Neurosci. 2010;13:615–621. doi: 10.1038/nn.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim K, Li C. Expression and regulation of an FMRFamide-related neuropeptide gene family inCaenorhabditis elegans. J Comp Neurol. 2004;475:540–550. doi: 10.1002/cne.20189. [DOI] [PubMed] [Google Scholar]
- 63.Tsalik EL, Hobert O. Functional mapping of neurons that control locomotory behavior inCaenorhabditis elegans. J Neurobiol. 2003;56:178–197. doi: 10.1002/neu.10245. [DOI] [PubMed] [Google Scholar]
- 64.Lanjuin A, VanHoven MK, Bargmann CI, Thompson JK, Sengupta P. The C. elegans Otx-related genes specify the identities of distinct sensory neuron types. Int Worm Meet. 2003;5:621–633. doi: 10.1016/s1534-5807(03)00293-4. [DOI] [PubMed] [Google Scholar]
- 65.Bertrand V, Hobert O. Linking Asymmetric Cell Division to the Terminal Differentiation Program of Postmitotic Neurons in C. elegans. Dev Cell. 2009;16:563–575. doi: 10.1016/j.devcel.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fukushige T, Hawkins MG, McGhee JD. The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev Biol. 1998;198:286–302. [PubMed] [Google Scholar]
- 67.Loria PM, Hodgkin J, Hobert O. A Conserved Postsynaptic Transmembrane Protein Affecting Neuromuscular Signaling in Caenorhabditis elegans. 2004;24:2191–2201. doi: 10.1523/JNEUROSCI.5462-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 69.Hedgecock EM, Culotti JG, Thomson JN, Perkins LA. Axonal guidance mutants of Caenorhabditis elegans identified by filling sensory neurons with fluorescein dyes. Dev Biol. 1985;111:158–170. doi: 10.1016/0012-1606(85)90443-9. [DOI] [PubMed] [Google Scholar]
- 70.Abdus-Saboor I, et al. Notch and Ras promote sequential steps of excretory tube development in C. elegans. Development. 2011;138:3545–3555. doi: 10.1242/dev.068148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pierce ML, et al. MicroRNA-183 family conservation and ciliated neurosensory organ expression. MicroRNA-183 Fam Conserv ciliated neurosensory organ Expr. 2008;10:106–113. doi: 10.1111/j.1525-142X.2007.00217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nonet ML, et al. Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J Neurosci. 1997;17:8061–8073. doi: 10.1523/JNEUROSCI.17-21-08061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stefanakis N, Carrera I, Hobert O. Regulatory Logic of Pan-Neuronal Gene Expression in C. elegans. Neuron. 2015;87:733–750. doi: 10.1016/j.neuron.2015.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hsu DR, Chuang PT, Meyer BJ. DPY-30, a nuclear protein essential early in embryogenesis for Caenorhabditis elegans dosage compensation. Development. 1995;121:3323–3334. doi: 10.1242/dev.121.10.3323. [DOI] [PubMed] [Google Scholar]
- 75.Bertrand V, Bisso P, Poole RJ, Hobert O. Notch-Dependent Induction of Left/Right Asymmetry in C. elegans Interneurons and Motoneurons. Curr Biol. 2011;21:1225–1231. doi: 10.1016/j.cub.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Warren CE, Krizus A, Dennis JW. Complementary expression patterns of six nonessential Caenorhabditis elegans core 2/I N-acetylglucosaminyltransferase homologues. Glycobiology. 2001;11:979–988. doi: 10.1093/glycob/11.11.979. [DOI] [PubMed] [Google Scholar]
- 77.Murray JI, et al. Multidimensional regulation of gene expression in the C elegans embryo Multidimensional regulation of gene expression in the C elegans embryo. 2012:1282–1294. doi: 10.1101/gr.131920.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mckay SJ, et al. Gene Expression Profiling of Cells, Tissues, and Developmental Stages of the Nematode C. elegans. Cold Spring Harb Symp Quant Biol. 2003;68:159–170. doi: 10.1101/sqb.2003.68.159. [DOI] [PubMed] [Google Scholar]
- 79.Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods. 2013;10:1028–1034. doi: 10.1038/nmeth.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ward JD. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 2015;199:363–377. doi: 10.1534/genetics.114.172361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tavernarakis N, Wang SL, Dorovkov M, Ryazanov A, Driscoll M. Heritable and inducible genetic interference by double-stranded RNA encoded by transgenes. Nat Genet. 2000;24:180–183. doi: 10.1038/72850. [DOI] [PubMed] [Google Scholar]
- 82.Kamath RS, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 83.Heiman MG, Shaham S. DEX-1 and DYF-7 Establish Sensory Dendrite Length by Anchoring Dendritic Tips during Cell Migration. Cell. 2009;137:344–355. doi: 10.1016/j.cell.2009.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yochem J, Herman RK. Investigating C. elegans development through mosaic analysis. Development. 2003;130:4761–4768. doi: 10.1242/dev.00701. [DOI] [PubMed] [Google Scholar]
- 85.Conradt B, Horvitz HR. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell. 1998;93:519–529. doi: 10.1016/s0092-8674(00)81182-4. [DOI] [PubMed] [Google Scholar]
- 86.Ward S, Thomson N, White JG, Brenner S. Electron microscopical reconstruction of the anterior sensory anatomy of the nematode Caenorhabditis elegans. ?2UU. J Comp Neurol. 1975;160:313–337. doi: 10.1002/cne.901600305. [DOI] [PubMed] [Google Scholar]
- 87.Bargmann CI, Hartwieg E, Horvitz HR. Odorant-selective genes and neurons mediate olfaction in C. elegans. Cell. 1993;74:515–527. doi: 10.1016/0092-8674(93)80053-h. [DOI] [PubMed] [Google Scholar]
- 88.Seligman AM, Wasserkrug HL, Hanker JS. A new staining method (OTO) for enhancing contrast of lipid–containing membranes and droplets in osmium tetroxide–fixed tissue with osmiophilic thiocarbohydrazide(TCH) J Cell Biol. 1966;30:424–432. doi: 10.1083/jcb.30.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Scheffzek K, et al. The Ras-RasGAP Complex: Structural Basis for GTPase Activation and Its Loss in Oncogenic Ras Mutants. Science (80-) 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
- Supplementary Figures S1–S8 (Integrated Suppl. Figures template)
- Supplementary Tables S1–S5
- Supplementary Movies S1, S2
- Supplementary Tables S6–S10 (XLS files)
- Supplementary References (51–89)