

Abstract
The ability to harvest single cells from tissues is currently a bottleneck for cell-based diagnostic technologies, and remains crucial in the fields of tissue engineering and regenerative medicine. Tissues are typically broken down using proteolytic digestion and various mechanical treatments, but success has been limited due to long processing times, low yield, and high manual labor burden. Here, we present a novel microfluidic device that utilizes precision fluid flows to improve the speed and efficiency of tissue digestion. The microfluidic channels were designed to apply hydrodynamic shear forces at discrete locations on tissue specimens up to 1 cm in length and 1 mm in diameter, thereby accelerating digestion through hydrodynamic shear forces and improved enzyme-tissue contact. We show using animal organs that our digestion device with hydro-mincing capabilities was superior to conventional scalpel mincing and digestion based on recovery of DNA and viable single cells. Thus, our microfluidic digestion device can eliminate or reduce the need to mince tissue samples with a scalpel, while reducing sample processing time and preserving cell viability. Another advantage is that downstream microfluidic operations could be integrated to enable advanced cell processing and analysis capabilities. We envision our novel device being used in research and clinical settings to promote single cell-based analysis technologies, as well as to isolate primary, progenitor, and stem cells for use in the fields of tissue engineering and regenerative medicine.
Keywords: microfluidics, tissue digestion, single cell
Graphical abstract
Introduction
The past decade has seen a rapid growth in interest to harvest single cells from tissues that has spanned across several biomedical research areas. This has been driven in part by the rise in use of single cell analysis techniques, such as flow cytometry, mass spectroscopy, and single cell sequencing, to identify and profile the diverse cell types typically found within tissues.1–4 For cancer, this has enabled assessment of tumor heterogeneity, metastatic potential, and the presence of rare cell types such as putative cancer stem cells.5–10 These insights obtained at the resolution of single cells are drastically changing our understanding of cancer, and in the future are poised to revolutionize clinical diagnostics and inform personalized patient care.11,12 In the field of tissue engineering, isolation of primary cells from tissues is critical for the creation of new constructs to replace damaged organs, such as skin, liver, heart, pancreas and kidney.13–18 Finally, a major goal of regenerative medicine is to isolate mesenchymal stem cells and progenitor cells from tissues to heal or otherwise replace diseased parts of the body.19–22 A common theme unifying all of these applications is that they require viable single cells that remain as representative of their original phenotypic state as possible. Thus, there is a critical need to develop new technologies that will make it possible to liberate single cells from tissues in a rapid, gentle, and thorough manner.
Microfluidic technologies have emerged as simple yet powerful methods for processing and manipulating cellular samples at the microscale.23,24 However, only a few microfluidic devices have been developed to work with cell aggregates and tissues. The microfluidic cell dissociation chip (μ-CDC) was designed to break down neurospheres under fluid flow using a micro-pillar array.25 However, this device could only be used with aggregates that were less than 300 µm in diameter, and yet still suffered from clogging issues. The Biogrid was designed to mechanically cut neurospheres using sharp silicon knife-edges placed across the device cross-section.26 While more effective, mechanical cutting in this fashion was harsh and only resulted in smaller aggregates, not single cells. In our previous work, we demonstrated a microfluidic device that employed a network of branching channels to achieve highly efficient and rapid dissociation of cancer cell aggregates into viable single cells.27 However, the inlet could not accommodate samples that were greater than 1 mm in size, requiring off-chip mincing and digestion of larger tissue specimens. To date, full scale tissues have only been employed in a single microfluidic application, the culture and enzymatic digestion of rat liver biopsies.28 But this device just provided a means to incubate tissues with enzymes, and suffered from extremely low cell yields, even after prolonged digestion times.
In this work, we present a novel microfluidic device that leverages hydrodynamic fluid flow to rapidly digest tissue specimens into single cells. The device is loaded with tissue specimens up to 1 cm in length and 1 mm in diameter, which can be cut from larger surgical resections or obtained directly from core biopsies. Fluid containing proteolytic enzymes, such as collagenase, is then pumped through the device. The key innovation is the placement of channels directly upstream and downstream of the tissue specimen. The upstream channels drive the fluid into discrete locations of the tissue, effectively mincing it through the application of hydrodynamic shear forces and improved enzyme penetration. The downstream channels act as a sieve that firmly holds the tissue in place while also allowing smaller aggregates and cells to exit the tissue compartment. Using computational fluid dynamics simulations, we evaluate prospective upstream hydro-mincing and downstream sieve channel configurations with respect to number and sizing. We ultimately select three designs with different numbers of hydro-mince channels (3 to 7) that are 200 µm wide and 7 sieve channels that are 500 µm wide. Next, we fabricate the devices by laser-etching acrylic sheets and perform initial tests using beef liver tissue, which indicate that 3 hydro-mince channels is optimal. We then perform validation tests using freshly-harvested mouse liver and kidney samples. We find that our microfluidic digestion device provides superior cell yields compared to conventional scalpel mincing and digestion, while maintaining cell viability. Excellent results can even be obtained after shorter digestion times, including a remarkable 50% cell recovery after only 10 min of device processing. Thus, the digestion device holds exciting potential to eliminate off-chip preparation of tissue samples, automate digestion procedures, and reduce total processing time to preserve cell viability, phenotype, and molecular signatures. In future work, we will integrate downstream operations such as our branching channel dissociation device to better liberate single cells from small aggregates. We will also explore adding cell sorting and analysis components to create point-of-care platforms for cell-based diagnostics and therapies.
Results and Discussion
Device Design
Our device was designed to process 1 cm long × 1 mm diameter tissues, similar to core needle biopsies, directly into cell suspensions without the need for manual processing steps such as scalpel mincing. To achieve this goal, we devised three primary components for the device. First, there is a tissue chamber that holds the sample in place while fluid containing proteolytic enzymes is passed into and over the surface. This was intended to promote sample mixing, enhance enzymatic activity, and apply hydrodynamic shear forces to mechanically dislodge cells and aggregates. Chamber dimensions were 1.5 mm × 1 cm, approximately the size of a Tru-Cut core biopsy needle. Chamber height was also similar to the size of the tissue at ~1 mm. The second feature is a series of fluidic channels located upstream of the chamber, which focuses the fluid into high velocity jets that are directed into the tissue. These jets concentrate hydrodynamic shear forces at discrete locations, breaking the tissue down mechanically and delivering proteolytic enzymes deep inside the tissue. This is analogous to manually mincing the tissue with a scalpel, and therefore we will use the term hydro-mincing. Finally, fluidic channels were also placed downstream of the sample chamber to act as a sieve that selectively retains large pieces of tissue and cellular aggregates for further digestion. Smaller aggregates and single cells would be allowed to pass out of the device for collection, or potentially further microfluidic processing.
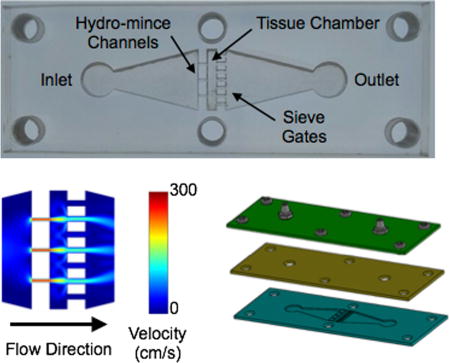
To establish the channel features, we started with the downstream sieves. We reasoned that spacing many sieve channels evenly throughout the device cross-section would firmly hold the tissue in place and minimize backpressure. We chose a channel width of 500 µm, and the device could comfortably accommodate 7 channels across the width of the tissue chamber. Note that 500 µm is comparable to the ~1 mm tissue pieces typically achieved by scalpel mincing. Aggregates of this size would also be ideal for directly inputting into our branching channel array dissociation device.27 For the upstream channels, the goal was to achieve efficient hydro-mincing. Using fewer channels would generate stronger fluidic jets, but would also cover less of the tissue cross-section and lead to higher device backpressures. Since these are competing factors, we chose to use channel number as a test variable and created devices with 3, 5, and 7 hydro-mince channels. As for channel size, smaller widths would generate stronger, more concentrated fluidic jets. Therefore we chose 200 µm, which was the smallest feature resolution that we could reliably achieve with our laser-based fabrication method. A schematic of the tissue digestion device is shown in Figure 1. Computational fluid dynamics simulations were performed using COMSOL Multiphysics software for each 3, 5, and 7 hydro-mince channels using a flow rate of 1 mL/min (Fig. 1B). These simulations were performed with and without a model tissue within the chamber to obstruct flow. As expected, the design with 3 hydro-mince channels generated the highest fluid velocities, or strongest fluidic “cuts.” Increasing channel number provided weaker “cuts” that were better dispersed across the tissue.
Figure 1.

Microfluidic digestion device design and operation. (A) Image of laser-etched acrylic sheet containing the chamber for loading tissue samples and fluidic channels including upstream (left) for hydro-mincing and downstream (right) sieves. (B) Finite-element fluid dynamics simulations showing velocity profiles in devices with different numbers of hydro-mince channels. Simulation results are shown at 1 mL/min flow rate with the chamber empty and partially blocked by a model tissue. Fewer hydro-mince channels will generate stronger fluidic jets to shear the tissue, but with less overall coverage. (C,D) Full digestion device shown in (C) side and (D) exploded views, with a PDMS gasket layer sandwiched between two acrylic sheets. Hose barbs were added to the top layer and nylon screws were used to hold the device together. (E) Experimental set-up for digestion experiments. Flow was driven by a peristaltic pump and tissue digestion was visually monitored with a camera mounted above the device.
Devices were fabricated in hard acrylic sheets using a laser to etch the chamber and channel features (Fig. 1C). Laser power and raster speed were controlled to achieve a depth of approximately 1 mm, establishing channel height. A second layer of acrylic was tapped and fitted with hose barbs to connect inlet and outlet tubing. Finally, a gasket composed of polydimethylsiloxane (PDMS) was sandwiched between the acrylic layers to provide a watertight seal. Note that the deformable nature of PDMS, and likely the tissue itself, should alleviate fluid flow and backpressure issues even while the tissue is initially obstructing the flow path. Finally, the device sandwich was held together using 6 nylon screws. A fully assembled device is shown in Fig. 1D, and the experimental set-up is shown in Fig. 1E. For this initial work, we used a peristaltic pump to recirculate fluid through the device to conserve proteolytic enzyme solution. Alternative operating procedures with continuous flow or removal of cells prior to recirculation could also be implemented, but will be left to future work. A camera was mounted above the device to monitor the progress of tissue digestion.
Initial Device Optimization Using Beef Liver Tissue
Performance of the microfluidic digestion device was first evaluated using beef liver as the sample tissue. Model tissue cores were extracted using a Tru-Cut biopsy needle and loaded into the sample chamber (Fig. 2A). Devices were then primed with PBS buffer containing collagenase, sealed, and flow was initiated at 20 mL/min, the highest flow rate we could achieve with our peristaltic pump. Images of tissue specimens were acquired every 5 min using the camera mounted above the device to monitor digestion, and experiments were performed for a total of 30 min (Figs. 2B). After each image was acquired, flow was briefly reversed to clear tissue that had seeped into the sieve channels. We observed that tissue seeping was most extensive using 3 hydro-mince channels, reflecting the higher hydrodynamic forces being generated. Images were processed using ImageJ and MATLAB to assess the amount of liver tissue remaining in the device at each time point based on tissue area and pixel density (see Supporting Information, Fig. S1). Digestion profiles are plotted in Fig. 2C, after normalizing by initial tissue mass. Results were nominally similar for all three devices, with a dramatic 40% tissue decrease during the first 5 min, followed by a more gradual decrease in tissue by ~10% per 5 min interval. The initial drop primarily correlated to diminished pixel density, which may have reflected tissue debulking or washing out of red blood cells. The subsequent gradual phase primarily reflected a loss of tissue mass. After 30 min, approximately 80% of the tissue had been removed from all three of the device designs. However, the device with 3 hydro-mince channels provided the most consistent results in terms of lower variability between experiments, particularly at later time points, and thus was chosen for further evaluation. Representative micrographs of device effluents collected after 30 min device processing are shown in Fig. 2D. For all cases, sample effluents primarily comprised a mixture of larger tissue aggregates, tissue cells, and red blood cells.
Figure 2.

Digestion device optimization using beef liver cores. (A) Model tissue cores were obtained using a Tru-Cut biopsy needle and placed inside the tissue chamber. (B) Time-lapse images of tissue digestion for devices with 3, 5, and 7 hydro-mince channels. The fluid contained collagenase, and was pumped through the device at 20 mL/min. (C) Tissue loss was quantified from images based on mean gray value and overall tissue area. Trends were similar for each design, but variability was lowest for 3 hydro-mince channels. (D) Micrographs of device effluents after 30 min operation. Scale bar is 100 µm. Error bars represent standard errors from at least three independent experiments.
Evaluation of Cell Suspensions Obtained from Fresh Mouse Organs
Next we tested the 3 hydro-mincing channel design using freshly resected murine liver and kidney samples. These live tissues better represent samples that will be used in future applications, and the resulting cell suspensions can be directly assayed for quality. Liver is generally considered to be among the easiest tissues to dissociate, but hepatocytes are well known to be fragile.13 Kidney is considered to be a difficult tissue to dissociate due to its structure as a dense array of blood vessels and epithelial lined tubules, which function under high physiologic hydrodynamic pressures, have tight intercellular junctions, and have specialized basement membranes.16,17 Immediately after harvesting, tissues were cut into ~1 cm × 1 mm × 1 mm pieces with a scalpel (see Supporting Information, Fig. S2) and weighed. Digestion device experiments were then conducted as described for beef liver, with collagenase recirculated for either 15 or 30 min before sample collection. Images were again taken every 5 min and processed to monitor tissue loss, which was similar to beef liver (see Supporting Information, Fig. S2). Controls were further minced with a scalpel into ~1 mm3 pieces before digesting with collagenase for 15, 30, or 60 min in a conical tube. These samples were constantly agitated, and vortexed every 5 min. A separate control was included in which the tissue was not minced, only digested for 30 min. Following digestion, device-processed and control samples were mechanically treated by vortexing and pipetting, filtered through a 70 µm cell strainer, and treated with DNase to remove extracellular DNA. Cellular content was then assessed based on total genomic DNA (gDNA) extracted using a QIAamp DNA kit. For minced controls, gDNA progressively increased with digestion time (Fig. 3A). Kidney samples yielded approximately 100 ng gDNA per mg of tissue after 60 min digestion, while liver was less than half this value. Slightly less gDNA was obtained from the unminced controls, but differences were not significant. Device treatment yielded dramatically more gDNA than controls when compared at the same digestion time. The difference was approximately 5-fold for both tissue types after 15 min, and 3 to 4-fold after 30 min. Moreover, device treatment produced at least as much gDNA as the minced control at the next longer digestion time point. Thus, the microfluidic digestion device can significantly improve digestion efficiency and shorten digestion time. DNA was also assessed within intact cellular suspensions using the CyQUANT assay, which corroborated gDNA results (see Supporting Information, Fig. S2). Finally, a representative sample of each cellular suspension was treated with red blood cell lysis buffer before quantification of cell number with an automated counter and visualization of cells under phase contrast microscopy. Cell counts, which primarily reflected single cells but may also include some small clusters, were similar to gDNA results (Fig. 3B). The main difference was that liver now provided values that were comparable to kidney. This suggests that a significant portion of kidney cells may have remained in aggregates that could have passed through the cell strainer and be lysed to obtain gDNA. Alternatively, the cell counter may have detected more debris in liver suspensions, which was seen in micrographs for both minced controls and device treated samples (Fig. 3C).
Figure 3.

Cell recovery from fresh mouse kidney and liver tissues. (A) Genomic DNA (gDNA) was extracted and quantified from cell suspensions obtained by digestion only, scalpel mincing and digestion, or device treatment lasting for a total of 15, 30, or 60 min. gDNA increased with treatment time, and overall was higher for kidney samples. Device treatment consistently provided more gDNA than minced controls at the same time point. In most cases, gDNA was also higher than the next digestion time point, although differences were not significant. (B) Cell counter results, showing that single cell numbers largely matched gDNA findings but with higher variability. Also, liver values were now similar comparable to kidney, suggesting that kidney suspensions may have contained more aggregates. (C) Micrographs of minced controls and device effluents after lysing red blood cells. Note the large number of aggregates in the controls, particularly at 60 min. Scale bar is 100 µm. Error bars represent standard errors from at least three independent experiments. * indicates p < 0.05 relative to minced control at the same digestion time.
Analysis of Cell Types, Numbers, and Viability using Flow Cytometry
The final evaluation focused on determining single cell numbers and viability. Fresh mouse kidney and liver samples were prepared and digested as described in the previous section, except the unminced control was removed and a 10 min device treatment was added. Digested cellular suspensions were filtered through a 40 µm cell strainer and labeled with a panel of four fluorescent probes: CellMask Green to stain phospholipid cell membranes, Draq5 to stain DNA within all cells, 7AAD to stain DNA only within dead cells with disrupted plasma membranes, and CD45 to stain leukocytes (Table 1). This panel enabled distinction of tissue cells from non-cellular debris, anucleated red blood cells, and leukocytes, while simultaneously assessing viability. Stained cell suspensions were analyzed with a BD Accuri Flow Cytometer to obtain the number of each cell type using the gating protocol described in the methods section and shown in Supporting Information, Fig. S3. Comparing the relative numbers for each cell type (Fig. 4A and B), red blood cells constituted the majority of all but the minced control that was digested for 15 min. Unexpectedly, red blood cell percentage increased slightly as the tissue was digested more thoroughly, although this effect not significant. Leukocyte percentage remained stable, decreasing slightly with digestion time. Tissue cell counts, which are expected to predominantly be epithelial, were quantified for kidney and liver samples and are presented in Fig. 4C and D; respectively. Tissue cell numbers were 2 to 5 times higher for kidney than liver for the minced controls, which both increased with digestion time. The increases were more than an order of magnitude between 15 to 30 min, and 5-fold between 30 to 60 min. With device treatment, there was little change between 10 and 15 min time points, although 10 min was associated with high variability for kidney samples. Extending processing time to 30 min increased cell number by only ~50% for both tissue types, although differences were not significant. Comparing to the minced controls, device treatment again provided superior results at the same digestion time point. For kidney, cell number differences were 30-fold at 15 min and 4-fold at 30 min. Differences were about half these values for liver. Furthermore, 15 min device treatment yielded similar or better results than the minced control that was digested for 30 min. However, the minced control that was digested for 60 min now provided the highest cell numbers, exceeding the 30 min device treatment by 50% for kidney and 100% for liver. This finding is in contrast to the gDNA results, particularly for kidney, but generally consistent with CyQUANT and cell counter data. Thus, a significant portion of new cells that are liberated by the digestion device likely reside within small aggregates or clusters, which would be reasonable considering the smallest channel feature size is 200 µm. Finally, we assessed viability using a DNA dye that is excluded from healthy cells with intact membranes. Viability was approximately 80% for all kidney samples except the minced control that was digested for 60 min and 30 min device cases, which both dropped to 70% (see Supporting Information, Fig. S4). For liver, viability was approximately 90% for the minced controls, 80% for 10 and 15 min device treatments, and 70% for 30 min device treatment. The number of live tissue cells obtained from each condition is also presented in Fig. 4C and D. For kidney, 30 min device treatment produced approximately the same number of live single tissue cells as the minced control that was digested for 60 min. The 10 and 15 min device treatments produced around half of this value, but in a fraction of the time. For liver, the number of live, single tissue cells did not increase with device treatment beyond 10 min. This was likely due to the fragile nature of liver cells, which may have been damaged or fully destroyed while recirculating through the device. Overall, the microfluidic digestion device performed better for kidney samples despite the fact that this tissue type is generally considered to be more difficult to dissociate. This is likely due to the combination of kidney cells being more robust and the denser kidney tissue requiring higher shear forces to be dissociated.
Table 1.
Flow cytometry probe panel.
| Assay | CellMask Green (Lipid membrane) |
Draq5 (Nucleus) |
CD45-PE (Leukocytes) |
7AAD (Dead cells) |
|---|---|---|---|---|
| Red blood cells | + | − | − | − |
| Leukocytes | + | + | + | +/− |
| Tissue cells | + | + | − | +/− |
Figure 4.

Single cell analysis of mouse kidney and liver cell suspensions. Flow cytometry was used to identify and quantify the number of leukocytes, red blood cells, and single tissue cells in the suspensions obtained from minced controls or device treatment. (A,B) Relative numbers of each cell type are shown for (A) kidney and (B) liver samples. Red blood cells comprised the highest percentage of almost all populations, and there was no statistically significant change in population compositions across all minced control and device conditions. (C,D) Total and live tissue cell numbers per mg of tissue were determined for (C) kidney and (D) liver samples. Tissue cell recovery increased with digestion time for minced controls, but did not change significantly with device processing beyond 10 min. Importantly though, all device conditions yielded more cells than minced controls that were digested for up to 30 min. Viability remained >80% for all but the longest time points, which reached as low as 70%. The x-axis for (A) and (B) are the same as (C) and (D). Error bars represent standard errors from at least three independent experiments. * indicates p < 0.05 relative to minced control at the same digestion time. # indicates p < 0.05 compared to minced control digested for 15 min.
Conclusions
In this work, we have presented a new microfluidic device to isolate single cells from cm × mm-scale tissues using the combination of hydrodynamic shear forces and proteolytic digestion. Upon testing our microfluidic digestion device with kidney and liver tissue samples, we consistently observed improvements in recovery of DNA and single tissue cells relative to standard methods that require mincing with a scalpel. Device performance at short processing times was particularly exciting, as a 10 min treatment yielded results that were within 50% of scalpel mincing and digesting for 1 hour, but with improved viability. Recovery improvement were most striking for DNA, suggesting that the current device design may have left a significant number of cells within small aggregates or clusters. In future work, we will explore ways to improve hydro-mincing such as decreasing channel dimensions, increasing flow rate, and installing valves to direct flow to different regions of the tissue. These approaches could also improve aggregate dissociation, or we could pair the digestion device with another such as our branching channel array with hydrodynamic micro-scalpels.27 We did observe evidence that cells may have been damaged during initial tissue digestion, or more likely while repeatedly recirculating through the device, particularly for liver. Thus, next generation designs will seek to remove single cells as soon as they are liberated via filtration or another means of physical separation. Finally, we will pursue testing of additional tissues such as solid tumors from various cancer types for diagnostic purposes and other healthy tissues such as skin, heart, and fat for use in tissue engineering and regenerative medicine.
Experimental
Fluid Dynamics Simulations
Flow profiles within device channels were simulated using COMSOL Multiphysics software. This involved coupling the Navier-Stokes equations and the continuity equation in finite element fluid dynamics simulations. Fluid flow was assumed to be laminar, and the no-slip boundary condition was enforced at the channel walls. We used a flow rate of 1 mL/min, but flow profiles remain the same at different flow rates up to 20 mL/min as used for experiments. The only difference is a corresponding change in maximum flow velocity.
Device Fabrication
Digestion devices were designed using Onshape software. Fluidic channels and hose barb openings were laser etched using a VLS 4.60 60W CO2 laser (Universal Laser Systems, Scottsdale, AZ). Channel designs were etched in 6” × 6” optically clear cast acrylic sheets (McMaster-Carr, Elmhurst, IL) that served as the bottom layer of the device. Hose barb openings were then tapped to provide threading. A gasket was prepared from PDMS (Ellsworth Adhesives, Germantown, WI) by casting a 5 mm slab and cutting with a scalpel. The device was assembled with the PDMS gasket placed between the top and bottom acrylic layers, and secured with nylon screws. The inlet and outlet of the device were connected to a peristaltic pump that was controlled by a custom-built Arduino Uno R3 microcontroller.
Tissue models
Beef liver was purchased from a local butcher and tissue cores were extracted by using a Tru-Cut biopsy needle (CareFusion, Vernon Hills, IL) in a manner analogous to obtaining a clinical biopsy. Briefly, the obturator was retracted to cover the specimen notch and the cannula handle was held firmly while the needle was inserted into the tissue. The obturator was quickly advanced as far as permitted to position the specimen notch in the tissue and the cannula handle was quickly advanced to cut the tissue. Tissue obtained in the specimen notch was then transferred to device using tweezers. Mouse liver and kidneys were harvested from sacrificed C57B/6 or BALB/c mice (Jackson Laboratory, Bar Harbor, ME) that were deemed waste from a research study approved by the University of California, Irvine, Institutional Animal Care and Use Committee (courtesy of Dr. Angela G. Fleischman). Animal organs were cut with a scalpel into 1 cm long × 1 mm diameter pieces, and the mass of each was recorded. Mouse kidneys were sliced in a symmetrical fashion to obtain histologically similar portions that included both cortex and medulla.
Digestion of tissue samples
The digestion device was first primed with 200 µL collagenase type I (Stemcell Technologies, Vancouver, BC) and heated to 37°C inside an incubator to ensure optimal enzymatic conditions. Tissue was then placed inside the chamber before the device was assembled, secured with nylon screws, and filled with 1 mL collagenase. Experiments were initiated by flowing fluid through the device at 20 mL/min with the peristaltic pump, and every 5 min the flow was reversed to clear tissue from the downstream sieve gates. Device effluents were collected by pumping directly into a conical tube. Controls were digested in a conical tube that contained 1 mL collagenase, either with or without prior mincing with a scalpel into ~1 mm3 pieces. Tubes were placed inside a 37°C incubator and gently agitated on a rotating mixer. Both controls and device treated samples
Every 5 min, the tubes were vortexed to mechanically disrupt tissue and maximize digestion. At the conclusion of digestion procedures, all cell suspensions were repeatedly vortexed and pipetted to mechanically disrupt aggregates and treated with DNase I (10 µL; Roche, Indianapolis, IN) at 37°C for 5 min.
Image analysis to monitor tissue digestion
During device operation, images of the tissue were captured every 5 min using a camera mounted directly above the device. Raw images were processed using ImageJ by first converting to binary to identify the borders of the tissue (see Supporting Information, Fig. S1). Mean gray value was then determined within the tissue border, and multiplied by the area to obtain a single metric accounting for tissue size and density. Results at each time point were normalized by the initial value prior to the experiment, and presented as percent tissue remaining.
Quantification of DNA recovered from cell suspensions
DNA content of digested cell suspensions was assessed by extraction and purification, as well as direct assessment within cells using a fluorescent DNA stain. For both cases, samples were first filtered using a 70 µm cell strainer to remove remaining tissue and large aggregates. Purified genomic DNA (gDNA) was isolated using the QIAamp DNA Mini Kit (Qiagen, Germantown, MD) according to manufacturer’s instructions and quantified using a Nanodrop ND-1000 (Thermo Fisher, Waltham, MA). DNA within cells was labelled using the CyQUANT NF Cell Proliferation Assay Kit (Thermo Fisher, Waltham, MA) according to the manufacturer’s instructions. Briefly, samples were suspended in HBSS supplemented with 35mg/L sodium bicarbonate and 20 mM HEPES and added to an opaque 96-well plate (Corning, Corning, NY) in triplicate. An equal volume of CyQUANT dye was then added to each well, incubated at 37°C for 40 minutes under continuous mixing at 200 RPM, and fluorescence signal was quantified using a Synergy 2 plate reader (BioTek, Winooski, VT). Wells containing only HBSS and CyQUANT dye were used for background subtraction. gDNA and fluorescence intensities were normalized by the initial tissue mass.
Cell counting and imaging of cell suspensions
Digested effluents were collected, filtered using a 70 µm cell strainer, and incubated with red blood cell lysis buffer containing ammonium chloride, potassium carbonate, and EDTA (Biolegend, San Diego, CA) for 5 min at room temperature. Cell concentration was determined using a Moxi Z cell counter with type S cassettes (Orflo, Hailey, ID), and converted to cell number per mass of tissue using the total volume recovered and the initial tissue mass. Imaging was performed by transferring samples to a 12-well plate, waiting for 1 hour for the cells to settle, and capturing images using a Hoffman phase contrast microscope with a 4× objective.
Flow cytometric analysis of single cells
Digested mouse kidney and liver cell suspensions were evenly divided into FACS tubes (Corning, Corning, NY) and resuspended in FACS Buffer (1× PBS, pH 7.4 without Ca and Mg cations) supplemented with 1% BSA and 0.1% NaN3. Samples were first stained with 0.5× CellMask Green (Thermo Fisher, Waltham, MA) and 2.5µg/mL anti-mouse CD45-PE monoclonal antibody (clone 30-F11, (BioLegend, San Diego, CA) for 20 minutes at 37°C and washed twice with FACS Buffer by centrifugation. Cells were then resuspended in FACS buffer supplemented with 12.5 µM Draq5 (BioLegend, San Diego, CA) and 5 µg/mL 7AAD (BD Biosciences, San Jose, CA) and maintained on ice for at least 15 minutes prior to analysis on an Accuri Flow Cytometer (BD Biosciences). An isotype matched, PE-conjugated monoclonal antibody (clone RTK4530, BioLegend, San Diego, CA) was used as a control. Flow cytometry data was compensated and analyzed using FlowJo software (FlowJo, Ashland, OR). Compensation was determined using the kidney and liver tissues that were minced with a scalpel and digested for 60 mi, which were aliquoted into four different preparations to obtain distinct positive and negative subsets for each probe. The four preparations included cell fractions with: 1) negative control CompBeads (3.0–3.4 µm diameter, BD Biosciences, San Jose, CA) and CellMask Green membrane stain, 2) RBCs lysed and CD45-PE antibody, 3) live and dead (heat-killed at 55°C for 30 min) cells with 7AAD stain, and 4) Draq5 stain. Gates encompassing the positive and negative subpopulations within each compensation sample were inputted into FlowJo to automatically calculate the compensation matrix. Finally, a sequential gating scheme was used to identify different cell subpopulations. (see Supporting Information, Fig. S3). A SSC-A vs. FSC-A gate was created to select all cellular events and exclude debris from further analysis. Multicellular aggregates were removed from the analysis population to focus only on single cells using an FSC-H vs. FSC-A gate. Leukocytes were first distinguished from the single cell population based on CD45 expression (FL2-A or PE vs. SSC-H). Anucleate red blood cells were distinguished by their absence of Draq5 nuclear stain (FL4-A or Draq5 vs. SSC-H). The cellularity of the final remaining single cells (CD45 negative, Draq5 positive) was confirmed by detecting cell membranes using CellMask Green stain (FL1-A or CellMask green vs. FSC-H). Finally, live and dead nucleated tissue cell percentages were discriminated based on 7AAD signal (FL3-A or 7AAD vs. SSC-H).
Statistics
Data are represented as the mean ± standard error determined from at least three independent experiments. P-values were calculated using students t-test based on the mean and standard error between different experimental conditions.
Supplementary Material
Figure S1. Image processing algorithm to monitor tissue digestion.
Figure S2. Mouse kidney and liver imaging and CyQUANT data.
Figure S3. Flow cytometry gating scheme.
Figure S4. Mouse kidney and liver cell viability data.
Acknowledgments
The authors wish to thank Dr. Angela G. Fleischman for kindly donating mouse tissue for this project, and to Dr. Thanh (Kim) Nguyen for assistance harvesting organs. We would also like to thank Danny Duong and Griffith Liam Wagner for assisting with experiments and image analyses. The authors would like to acknowledge support from the National Science Foundation and the industrial members of the Center for Advanced Design and Manufacturing of Integrated Microfluidics (NSF I/UCRC award number IIP-1362165), the University of California Cancer Research Coordinating Committee, and the National Cancer Institute of the National Institutes of Health under Award Number P30CA062203. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions
X.Q., E.W., A.K., E.E.H., and J.B.H. devised the concept for the work. E.W. and A.K. fabricated the device. P.P.P. carried out the computational simulations. X.Q. and T.M.W. carried out the experimental work. X.Q., T.M.W., E.L.N., and J.B.H. carried out the experimental analysis. X.Q., T.M.W., and J.B.H. wrote the paper.
References
- 1.Irish JM, Kotecha N, Nolan GP. Nat Rev Cancer. 2006;6:146–55. doi: 10.1038/nrc1804. [DOI] [PubMed] [Google Scholar]
- 2.Bendall SC, Nolan GP. Nat Biotechnol. 2012;30:639–47. doi: 10.1038/nbt.2283. [DOI] [PubMed] [Google Scholar]
- 3.Navin NE. Genome Res. 2015;25:1499–507. doi: 10.1101/gr.191098.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gawad C, Koh W, Quake SR. Nat Rev Genet. 2016;17:175–88. doi: 10.1038/nrg.2015.16. [DOI] [PubMed] [Google Scholar]
- 5.Tirino V, Desiderio V, Paino F, Papaccio G, De Rosa M. Methods Mol Biol. 2012;879:513–29. doi: 10.1007/978-1-61779-815-3_32. [DOI] [PubMed] [Google Scholar]
- 6.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, Regev A, Bernstein BE. Science. 2014;344:1396–401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, Yaswen P, Goga A, Werb Z. Nature. 2015;526:131–5. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M, Dutton-Regester K, Lin JR, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CG, Kazer SW, Gaillard A, Kolb KE, Villani AC, Johannessen CM, Andreev AY, Van Allen EM, Bertagnolli M, Sorger PK, Sullivan RJ, Flaherty KT, Frederick DT, Jané-Valbuena J, Yoon CH, Rozenblatt-Rosen O, Shalek AK, Regev A, Garraway LA. Science. 2016;352:189–96. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, Kong SL, Chua C, Hon LK, Tan WS, Wong M, Choi PJ, Wee LJK, Hillmer AM, Tan IB, Robson P, Prabhakar S. Nat Genet. 2017 doi: 10.1038/ng.3818. [DOI] [PubMed] [Google Scholar]
- 10.Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, Gillespie SM, Dionne D, Luo CC, Ravichandran H, Mylvaganam R, Mount C, Onozato ML, Nahed BV, Wakimoto H, Curry WT, Iafrate AJ, Rivera MN, Frosch MP, Golub TR, Brastianos PK, Getz G, Patel AP, Monje M, Cahill DP, Rozenblatt-Rosen O, Louis DN, Bernstein BE, Regev A, Suvà ML. Science. 2017;355 doi: 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bedard PL, Hansen AR, Ratain MJ, Siu LL. Nature. 2013;501:355–64. doi: 10.1038/nature12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haun JB, Castro CM, Wang R, Peterson VM, Marinelli BS, Lee H, Weissleder R. Sci Transl Med. 2011;3:71ra16. doi: 10.1126/scitranslmed.3002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palakkan AA, Hay DC, Anil Kumar PR, Kumary TV, Ross JA. Liver Int. 2013;33:666–76. doi: 10.1111/liv.12134. [DOI] [PubMed] [Google Scholar]
- 14.Huang HI, Wu CZ. Methods Mol Biol. 2012;879:465–70. doi: 10.1007/978-1-61779-815-3_27. [DOI] [PubMed] [Google Scholar]
- 15.Hawthorne WJ, Williams L, Chew YV. Adv Exp Med Biol. 2016;938:89–122. doi: 10.1007/978-3-319-39824-2_7. [DOI] [PubMed] [Google Scholar]
- 16.Presnell SC, Bruce AT, Wallace SM, Choudhury S, Genheimer CW, Cox B, Guthrie K, Werdin ES, Tatsumi-Ficht P, Ilagan RM, Kelley RW, Rivera EA, Ludlow JW, Wagner BJ, Jayo MJ, Bertram TA. Tissue Eng Part C Methods. 2011;17:261–73. doi: 10.1089/ten.TEC.2010.0399. [DOI] [PubMed] [Google Scholar]
- 17.Van der Hauwaert C, Savary G, Gnemmi V, Glowacki F, Pottier N, Bouillez A, Maboudou P, Zini L, Leroy X, Cauffiez C, Perrais M, Aubert S. PLoS One. 2013;8:e66750. doi: 10.1371/journal.pone.0066750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smits AM, van Oorschot AA, Goumans MJ. Methods Mol Biol. 2012;879:339–49. doi: 10.1007/978-1-61779-815-3_20. [DOI] [PubMed] [Google Scholar]
- 19.Mahla RS. Int J Cell Biol. 2016;2016:6940283. doi: 10.1155/2016/6940283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.West CC, Hardy WR, Murray IR, James AW, Corselli M, Pang S, Black C, Lobo SE, Sukhija K, Liang P, Lagishetty V, Hay DC, March KL, Ting K, Soo C, Péault B. Stem Cell Res Ther. 2016;7:47. doi: 10.1186/s13287-016-0302-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uezumi A, Kasai T, Tsuchida K. Methods Mol Biol. 2016;1460:241–53. doi: 10.1007/978-1-4939-3810-0_17. [DOI] [PubMed] [Google Scholar]
- 22.Hickson LJ, Eirin A, Lerman LO. Kidney Int. 2016;89:767–78. doi: 10.1016/j.kint.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Ali J, Sorger PK, Jensen KF. Nature. 2006;442:403–11. doi: 10.1038/nature05063. [DOI] [PubMed] [Google Scholar]
- 24.Yeo LY, Chang HC, Chan PP, Friend JR. Small. 2011;7:12–48. doi: 10.1002/smll.201000946. [DOI] [PubMed] [Google Scholar]
- 25.Lin CH, Lee DC, Chang HC, Chiu IM, Hsu CH. Anal Chem. 2013;85:11920–8. doi: 10.1021/ac402724b. [DOI] [PubMed] [Google Scholar]
- 26.Wallman L, Akesson E, Ceric D, Andersson PH, Day K, Hovatta O, Falci S, Laurell T, Sundstrom E. Lab Chip. 2011;11:3241–8. doi: 10.1039/c1lc20316a. [DOI] [PubMed] [Google Scholar]
- 27.Qiu X, De Jesus J, Pennell M, Troiani M, Haun JB. Lab Chip. 2015;15:339–50. doi: 10.1039/c4lc01126k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hattersley SM, Dyer CE, Greenman J, Haswell SJ. Lab Chip. 2008;8:1842–6. doi: 10.1039/b809345h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Image processing algorithm to monitor tissue digestion.
Figure S2. Mouse kidney and liver imaging and CyQUANT data.
Figure S3. Flow cytometry gating scheme.
Figure S4. Mouse kidney and liver cell viability data.