

Dear Sir,
Neurodegeneration with brain iron accumulation (NBIA) is a hereditary syndrome characterized by progressive neurodegeneration and iron accumulation in brain tissue with extrapyramidal movement disorders [1]. Currently, ten causative genes for NBIA have been identified. Beta-propeller protein associated neurodegeneration (BPAN, NBIA5), which was previously known as static encephalopathy in childhood with neurodegeneration in adulthood (SENDA), is caused by de novo heterozygous or hemizygous mutation in WDR45 [2]. In patients with NBIA, dystonia is the most commonly recognized involuntary movement. Although parkinsonism was reported in some NBIA patients, pathological substrates for parkinsonism in NBIA cases are not well studied. Here, we report a BPAN patient with a heterozygous mutation in WDR45 showing levodopa-induced dyskinesia (LID) and dopamine loss documented by a dopamine transporter positron emission tomography (PET) study.
A 37-year-old woman was referred to our movement disorders clinic because of generalized dystonia and progressive gait disturbance over the course of 10 years. She had no family history of movement disorders, dementia, or psychiatric illness. She had no history of perinatal injury or seizure. She has had psychomotor retardation from childhood. She could make sound but could not communicate verbally. She partially communicated with others using gestures. Her motor development was also retarded. Although unsteady, she could walk holding an assistant’s hand until she was 27 years old. Thereafter, her gait disturbance and generalized dystonia progressively worsened, and she could not stand at all after age 32. She visited a neurology clinic 3 years ago and was prescribed 0.75 tablets of levodopa/carbidopa 100 mg/ 25 mg BID and Tizanidine 1 mg BID. Her mother reported that her dystonia partially improved; however, she could not tolerate the medication because of generalized dyskinesia, which was newly developed after she was prescribed medication. On examination, she was drooling and showed masked face. She had severe generalized dystonia and hypokinesia with spastic quadriparesis. The Babinski sign was negative. Optokinetic nystagmus was present in all directions. Laboratory tests including complete blood count, blood chemistry, thyroid function test, HIV antibody, ferritin, ceruloplasmin, serum copper level and 24-hour urine copper level were within normal ranges, except for a mildly elevated AST/ALT (54/137). The nerve conduction study and EEG were normal. A brain MRI (Philips Achieva 3.0T MRI System, release 3.2.1.1, Best, The Netherlands) showed generalized cerebral atrophy with ventricular enlargement. T2-weighted and susceptibility-weighed MR images showed marked hypointensity at the substantia nigra region and, to a much lesser extent, at the globus pallidus (Figure 1). T1-weighted 3D MP-RAGE MR images showed a central band of hypointensity at the substantia nigra region but a surrounding halo of hyperintensity was not obvious (Figure 1E). 18F-fluoropropyl-2-beta-carbomethoxy-3-beta-(4-iodophenyl) nortropane (18F-FP-CIT) PET revealed a significant decrease of dopamine transporter in the bilateral striatum. The degree of dopamine transporter loss was relatively symmetrical; however, the posterior part of the putamen was involved more than the anterior part (Figure 1F). PCR and Sanger sequencing of all exons of WDR45 revealed a novel missense mutation (p.N202K, c.606C>G, NM007075) that predicted to be ‘disease-causing’ by Mutation Taster [3]. Her mother was negative for the same mutation, but we could not study her father’s DNA. After she was treated with trihexyphenidryl 1 mg BID, her rigidity mildly improved; however, her mother complained of newly developed intermittent myoclonic jerks. We could not try other medication because follow up was lost.
Figure 1.
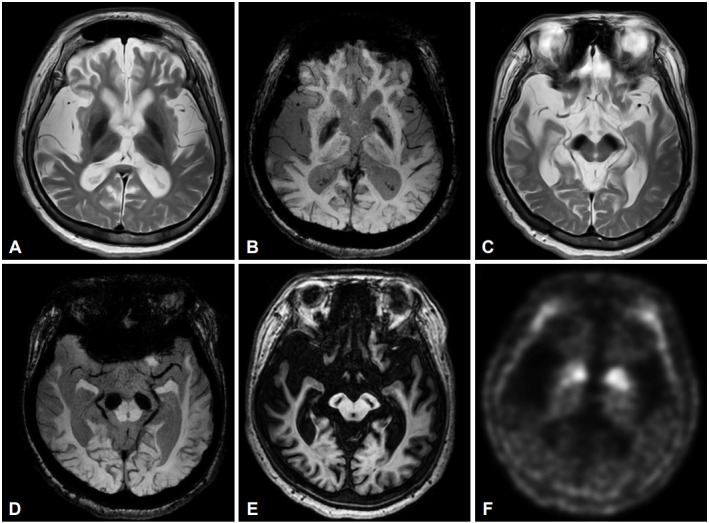
MRI and 18F-FP-CIT PET findings of a patient with beta-propeller protein-associated neurodegeneration. T2-weighted and susceptibility-weighed MR images show marked hypointensity in the globus pallidus (A and B) and the substantia nigra (C and D) in axial slices. T1-weighted 3D MP-RAGE axial images show a central band of hypointensity in the substantia nigra area (E). 18F-FP-CIT PET showed symmetrically reduced dopamine transporter binding activity in the bilateral striatum, where the posterior part of the putamen was more involved than the anterior part (F). 18F-FP-CIT: 18F-fluoropropyl-2-beta-carbomethoxy-3-beta-(4-iodophenyl) nortropane, PET: positron emission tomography, 3D: three-dimential.
Although a functional study for p.N202K mutation in WDR45 in this patient was not performed, we believe that her neurological signs were caused by de novo mutation of WDR45 for the following reasons. Her clinical features such as dystonia, spasticity, mental retardation, parkinsonism and myoclonus, and clinical course showing deterioration in adulthood were characteristic for BPAN. Her MRI features also support BPAN except for T1-weighted images which may be attributable to our parameters for T1-weighted 3D volumetric images. Her de novo mutation in WDR45 was novel and predicted to be disease-causing.
Among NBIAs, parkinsonism was reported in Pantothenate kinase-associated neurodegeneration (PKAN), PLA2G6-assocaited neurodegeneration (PLAN, PARK14), Mitochondrial membrane protein-associated neurodegeneration (MPAN), BPAN, Fatty acid hydroxylase-associated neurodegeneration (FAHN), Coenzyme A synthase protein-associated neurodegeneration (CoPAN), Neuroferritinopathy, Aceruloplasminemia, Woodhouse-Sataki syndrome, and Kufor-Rakeb syndrome (PARK9). In a retrospective review of 23 cases of BPAN, parkinsonism was present in 91% of patients who exhibited neurological deterioration in adolescence or early adulthood [4]. A Japanese study reported that mutations in WDR45 were found in 25% of patients with intellectual disability in childhood and young-onset parkinsonism whose age-at-onset is less than 40 years [5]. Pathological correlates for parkinsonism in BPAN have been poorly studied. Although a unique MRI feature of hyperintense signal of T1-weighted MR images in the substantia nigra in BPAN was argued to reflect degeneration of pigmented neurons, documentation of neuronal loss in the substantia nigra of the autopsied brain was reported only in two cases [4,6]. No functional neuroimaging studies for dopaminergic system such as dopamine transporter PET (or single photon emission computed tomography) or fluorodopa PET have been reported. Although we did not analyze FPCIT PET images quantitatively, images showing severe loss of dopamine transporter with anterior-toposterior gradient are consistent with previous pathological studies [4,6].
Levodopa responsiveness with early development of motor fluctuation and LID was frequently reported in BPAN [4-6]. In some cases, similar to in the patient we presented, LID was disabling which warranted discontinuation of levodopa. Lacking neuronal loss in the putamen and sparing of post-synaptic dopaminergic receptors may be related to these phenotypes [6]; however, the exact mechanism of LID remains unclear.
Footnotes
Conflicts of Interest
The authors have no financial conflicts of interest.
REFERENCES
- 1.Hogarth P. Neurodegeneration with brain iron accumulation: diagnosis and management. J Mov Disord. 2015;8:1–13. doi: 10.14802/jmd.14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013;45:445–449, 449e1. doi: 10.1038/ng.2562. [DOI] [PubMed] [Google Scholar]
- 3.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 4.Hayflick SJ, Kruer MC, Gregory A, Haack TB, Kurian MA, Houlden HH, et al. β-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain. 2013;136(Pt 6):1708–1717. doi: 10.1093/brain/awt095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishioka K, Oyama G, Yoshino H, Li Y, Matsushima T, Takeuchi C, et al. High frequency of beta-propeller proteinassociated neurodegeneration (BPAN) among patients with intellectual disability and young-onset parkinsonism. Neurobiol Aging. 2015;36:e9–2004.e15. doi: 10.1016/j.neurobiolaging.2015.01.020. [DOI] [PubMed] [Google Scholar]
- 6.Paudel R, Li A, Wiethoff S, Bandopadhyay R, Bhatia K, de Silva R, et al. Neuropathology of Beta-propeller protein associated neurodegeneration (BPAN): a new tauopathy. Acta Neuropathol Commun. 2015;3:39. doi: 10.1186/s40478-015-0221-3. [DOI] [PMC free article] [PubMed] [Google Scholar]