

ABSTRACT
Protein kinase C-δ (PKCδ) is an allosterically activated enzyme that acts much like other PKC isoforms to transduce growth factor-dependent signaling responses. However, PKCδ is unique in that activation loop (Thr507) phosphorylation is not required for catalytic activity. Since PKCδ can be proteolytically cleaved by caspase-3 during apoptosis, the prevailing assumption has been that the kinase domain fragment (δKD) freed from autoinhibitory constraints imposed by the regulatory domain is catalytically competent and that Thr507 phosphorylation is not required for δKD activity. This study provides a counternarrative showing that δKD activity is regulated through Thr507 phosphorylation. We show that Thr507-phosphorylated δKD is catalytically active and not phosphorylated at Ser359 in its ATP-positioning G-loop. In contrast, a δKD fragment that is not phosphorylated at Thr507 (which accumulates in doxorubicin-treated cardiomyocytes) displays decreased C-terminal tail priming-site phosphorylation, increased G-loop Ser359 phosphorylation, and defective kinase activity. δKD is not a substrate for Src, but Src phosphorylates δKD-T507A at Tyr334 (in the newly exposed δKD N terminus), and this (or an S359A substitution) rescues δKD-T507A catalytic activity. These results expose a unique role for δKD-Thr507 phosphorylation (that does not apply to full-length PKCδ) in structurally organizing diverse elements within the enzyme that critically regulate catalytic activity.
KEYWORDS: Src, apoptosis, protein kinase C, protein phosphorylation
INTRODUCTION
Protein kinase C-δ (PKCδ) sits at the crossroads of signal transduction pathways that play key roles in many cellular responses (1, 2). PKCδ's overall structure consists of an N-terminal regulatory domain (consisting of a C1 domain that binds lipid cofactors and a phosphotyrosine-binding C2 domain [3]) joined by a flexible linker to a C-terminal kinase domain (KD) (2). Like other PKC isoforms, the PKCδ KD contains highly conserved “priming” phosphorylation sites in the activation loop (T507) and at the C terminus (at S645 in the turn motif and S664 in the hydrophobic motif) (Fig. 1). For most PKC isoforms, these priming phosphorylations are stable modifications that are completed during the maturation of the nascent enzyme and required for catalytic activity (4). PKCδ is a notable exception in that constitutive phosphorylation is a feature of the turn and hydrophobic motifs, but endogenous PKCδ is recovered from many resting cell types with no detectable Thr507 phosphorylation. Rather, PKCδ-Thr507 phosphorylation increases dynamically during growth factor receptor activation. Importantly, the growth factor-dependent increase in Thr507 phosphorylation dynamically changes certain aspects of PKCδ's enzymology, but Thr507 phosphorylation is not absolutely required for the catalytic activity of full-length PKCδ (FL-PKCδ) (5–7).
FIG 1.

Domain structure of PKCδ. (Top) Schematic showing the C1 and C2 domains in the regulatory region; the caspase-3 cleavage site in the hinge region; the kinase domain; and the locations of priming phosphorylation sites in the activation loop (Thr507) and C-tail (Ser645 and Ser664), tyrosine phosphorylation sites in the hinge region, and the G-loop phosphorylation site at Ser359. (Bottom) Sequence alignment of the N-terminal αA-helix of PKA and the regulatory domain-kinase linker regions of PKC isoforms, Src, Hck, Csk, Btk, ZAP70, and Raf isoforms. Tyr334, Trp338, and Ser359 in PKCδ are highlighted in red, and residues at the corresponding positions in the other enzymes are emphasized in boldface type.
PKCδ activation is generally attributed to growth factor receptor pathways that promote diacylglycerol accumulation; diacylglycerol interacts with the C1 domain and anchors FL-PKCδ in an active conformation to lipid membranes. This conventional allosteric activation mechanism accounts for PKCδ's membrane-delimited actions, but it does not explain PKCδ's actions in other subcellular compartments. In fact, we and others reported that PKCδ is activated via a distinct lipid-independent mechanism during oxidative stress (6, 8). Oxidative stress leads to the activation of Src and the Src-dependent phosphorylation of PKCδ at Tyr313 and Tyr334 in the V3 hinge region (6). PKCδ-Tyr313 phosphorylation has been implicated in redox-dependent changes in PKCδ activity (5). A functional role for Tyr334 phosphorylation is less obvious, since Tyr334 phosphorylation is not required for the redox-dependent regulation of FL-PKCδ activity (5).
Our recent studies identify the mechanism whereby phosphorylation at Tyr313 (a site outside the catalytic core) alters PKCδ's enzymology (9). We showed that the Tyr313-phosphorylated hinge region functions as a docking site for the phosphotyrosine-binding C2 domain and that the C2-domain–pTyr313 interaction controls PKCδ's enzymology indirectly by inducing a long-range change in the phosphorylation status of Ser359, a site at the tip of the Gly-rich ATP-positioning loop (G-loop) in the KD (Fig. 1). This mechanism contributes to redox-activated PKCδ responses in cardiomyocytes. Specifically, PKCδ is recovered from resting cardiomyocytes as a Ser359-phosphorylated enzyme that shows a strong preference for substrates with a Ser residue at the phosphoacceptor site (P-site). Oxidative stress triggers a redox-induced C2 domain-pTyr313 docking interaction that facilitates PKCδ-Ser359 dephosphorylation and converts PKCδ into a lipid-independent enzyme that displays high levels of both Ser and Thr kinase activities (9).
PKCδ is also proteolytically activated by caspase-3 during apoptosis (10–16). Caspase-3 cleaves PKCδ at a site in the V3 hinge region, liberating a KD fragment (δKD) that is freed from autoinhibitory constraints imposed by the regulatory domain (Fig. 1). The observations that δKD contributes to proapoptotic events and that apoptotic cell death is detected following the overexpression of active, but not kinase-dead, δKD have been interpreted as evidence that δKD is a catalytically competent enzyme (12, 14, 16–18). In fact, detailed studies to determine whether the δKD fragment liberated by caspase-3 invariably retains all of the structural determinants required for catalytic activity have not been reported. This may be important since PKCδ is recovered from many cells with no detectable Thr507 phosphorylation; oxidative stress and proapoptotic stimuli typically do not increase PKCδ-Thr507 phosphorylation (6, 13). With the exception of one study that concluded that Thr507 phosphorylation exerts a rather complex regulatory effect on δKD activity (19), the importance of δKD Thr507 phosphorylation typically has been ignored. This study exposes a unique role for priming phosphorylations at both the activation loop and C terminus as modifications that structure δKD for catalysis. We also show that this interplay between Thr507 and C-terminal tail (C-tail) phosphorylation leads to secondary changes in G-loop Ser359 phosphorylation, and we identify a novel role for Tyr334 as a regulator of δKD catalytic activity.
RESULTS
A T507A substitution leads to a δKD C-tail priming phosphorylation defect.
Given the paucity of information on the role of Thr507 phosphorylation in the context of the δKD fragment, we examined the phosphorylation profiles and enzymology of wild-type δKD (WT-δKD) and δKD-T507A mutant enzymes; the N terminus of the δKD constructs was designed based upon a cleavage event at a consensus D326MQD329 caspase-3 recognition motif (20). (Note that the nomenclature is based upon the sequence of full-length human PKCδ.) Figure 2A shows that WT-δKD expression is detected 24 h following transfection primarily as a single ∼45-kDa fully primed enzyme; WT-δKD is phosphorylated at Thr507 (the activation loop), Ser645 (the C-tail turn motif), and Ser664 (the C-tail hydrophobic motif). WT-δKD protein expression and phosphorylation remain stable for at least 48 h following transfection. In contrast, δKD-T507A is resolved as a doublet. The slower-migrating ∼45-kDa band shows the Thr507 phosphorylation defect but retains phosphorylation at Ser645 and Ser664, whereas the faster-migrating ∼40-kDa band lacks all three priming-site phosphorylations. Both δKD-T507A bands are readily detected 24 to 48 h following transfection. Since C-tail priming-site phosphorylations function to structurally stabilize certain PKC isoforms (21, 22), we examined whether the Thr507 phosphorylation defect (which results in a secondary defect in δKD C-tail phosphorylation) influences δKD stability. Figure 2B shows that the level of the δKD protein decreases progressively in cells treated with the protein synthesis inhibitor cycloheximide and that a T507A substitution does not grossly alter δKD stability; the levels of both WT-δKD and δKD-T507A are reduced by >90% in cells treated with cycloheximide for 24 h. This contrasts with FL-PKCδ, which is a considerably more stable protein; levels of FL-WT-PKCδ and FL-PKCδ-T507A (which is fully phosphorylated at its C-tail priming sites [5]) remain stable during the 24-h cycloheximide treatment. These results indicate that δKD is considerably more labile than FL-PKCδ and that Thr507 phosphorylation does not grossly alter the in vivo stability of truncated or full-length forms of PKCδ.
FIG 2.

A T507A substitution influences δKD phosphorylation at the C-tail, G-loop, and newly exposed N terminus. (A and C) Lysates from HEK293 cells that heterologously overexpress δKD or δKD-T507A for various time intervals (A) or 48 h (C) were subjected to immunoblot analysis to track δKD and δKD-T507A protein expression (with antibodies against a C-terminal epitope on PKCδ or the Flag tag) and phosphorylation at priming sites (Thr507, Ser645, and Ser664) and the G-loop (Ser359). Immunoblots of the 48-h samples are aligned in panel C to emphasize that only the faster-migrating δKD species is phosphorylated at Ser359. (B) HEK293 cells were transfected with plasmids that drive similar expression levels of WT and T507A-substituted forms of FL-PKCδ or δKD. Lysates were prepared for immunoblot analysis of the PKCδ protein following treatment with cycloheximide (Chx) (10 μg/ml) for the indicated intervals. β-Actin served as a loading control. (D) δKD and δKD-T507A were subjected to IVKAs in the presence of Src, and immunoblot analysis was used to track the time course for Src-dependent δKD- or δKD-T507A-Tyr334 phosphorylation. All results are representative of data from 3 or 4 experiments on separate preparations.
We recently identified Ser359 in the G-loop as a phosphorylation site that regulates FL-PKCδ activity (9). Figure 2C shows that Ser359 phosphorylation is detected on the faster-migrating unprimed δKD-T507A construct but not on the slower-migrating C-tail-phosphorylated δKD-T507A construct. Fully primed WT-δKD is not phosphorylated at Ser359.
A C-tail phosphorylation defect facilitates δKD-Tyr334 phosphorylation by Src.
The newly exposed N-terminal tail of δKD retains an Src phosphorylation site at Tyr334 (Fig. 1). Previous studies of FL-PKCδ showed that Tyr334 is a substrate for Src only when the enzyme assumes an active conformation (5–7). Basal/inactive FL-PKCδ is a poor substrate for Src. The notion that Src might directly phosphorylate this site in the isolated δKD fragment has never been considered. Figure 2D shows that WT-δKD is not phosphorylated by Src, but this site is phosphorylated on δKD-T507A. Of note, Tyr334 phosphorylation is detected primarily on the more rapidly migrating δKD-T507A species that is phosphorylated at Ser359 but lacks all three priming-site phosphorylations; the slower-migrating δKD-T507A species with an isolated Thr507 phosphorylation (that retains intact C-tail Ser645 and Ser664 phosphorylations) is a relatively poor substrate for Src. These results indicate that the T507A substitution facilitates Tyr334 phosphorylation indirectly by enhancing Ser359 phosphorylation and/or disrupting C-tail Ser645/Ser664 phosphorylation.
We introduced S645A and S664A substitutions into δKD to directly examine their effects on δKD-Tyr334 phosphorylation. A single S645A substitution produced no discernible phenotype (data not shown), but the combined S645/664A substitution was very destabilizing; δKD-S645/664A was detected only at very low levels as an unprimed, rapidly migrating protein (Fig. 3A). δKD-S645/664A phosphorylation at Thr507 could not be detected even with increased protein loading (data not shown). However, the low levels of δKD-S645/664A protein recovery did not preclude the detection of Src-dependent δKD-S645/664A-Tyr334 phosphorylation. These results emphasize that C-tail priming phosphorylations play a critical role in stabilizing δKD in a conformation that prevents δKD-Tyr334 phosphorylation.
FIG 3.

C-tail priming phosphorylation defects facilitate PKCδ-Tyr334 phosphorylation by Src. The WT or T507A- or S645A/S664A-substituted form of δKD (A) or WT or S645A/S664A-substituted FL-PKCδ (B) was subjected to IVKAs without and with Src; PS-PMA was included in assays in panel B as indicated. Protein expression and phosphorylation were tracked by immunoblot analysis, with each panel depicting results from a single gel exposed for a uniform duration; dashed lines in panel B denote where data from different regions of a single gel were merged for purposes of presentation. Results were replicated in 3 separate experiments.
The effects of S645/664A substitutions on Src-dependent tyrosine phosphorylation of FL-PKCδ were also examined. Figure 3B shows that S645/664A substitutions do not alter FL-PKCδ expression or Thr507-phosphorylated enzymes (i.e., that the S653/664A substitution that severely destabilizes δKD is tolerated in the FL-PKCδ context). The S653/664A substitutions also do not grossly alter FL-PKCδ phosphorylation at Ser359. However, S653/664A substitutions facilitate FL-PKCδ phosphorylation by Src. Specifically, Src phosphorylates FL-WT-PKCδ at Tyr313 and Tyr334 only when it is activated by phosphatidylserine (PS)–phorbol 12-myristate 13-acetate (PMA) (and not when it is in the inactive/closed conformation), whereas Src phosphorylates FL-PKCδ-S645/S664A at Tyr313 and Tyr334 similarly in assays without or with PS-PMA. These results indicate that C-terminal phosphorylations orient tyrosines in the V3 hinge region of FL-PKCδ (or the newly exposed N-tail of δKD) for phosphorylation by Src. Any maneuver that disrupts Ser645/Ser664 phosphorylation results in a structural change that exposes this region to phosphorylation by Src. An additional effect of the G-loop seems unlikely, since an S645/S664A substitution eliminates the lipid requirement for Src-dependent FL-PKCδ-S645/S664A-Tyr313/Tyr334 phosphorylation without grossly altering Ser359 phosphorylation.
A T507A substitution disrupts δKD catalytic activity.
We previously demonstrated that Thr507 phosphorylation is not required for FL-PKCδ Ser kinase activity but that Thr507 phosphorylation confers additional Thr kinase activity. PKCδ's P-site specificity can be discriminated by performing in vitro kinase assays (IVKAs) with the cardiac troponin (cTn) complex (consisting of equimolar amounts of cardiac troponin I [cTnI], cTnT, and cTnC) as the substrate; cTnI and cTnT are physiologically important substrates for PKCδ. Assays that simultaneously track cTnI and cTnT phosphorylations provide a convenient readout of PKCδ's Ser versus Thr kinase activity, since (i) cTnI contains both a Ser phosphorylation cluster at Ser23/Ser24 (recognized by the anti-cTnI-pSer23/Ser24 phosphorylation-site-specific antibody [PSSA]) and a Thr phosphorylation site at Thr144 (which is flanked by an Arg residue at position +2 and therefore is recognized by the anti-TXR motif PSSA) and (ii) cTnT contains three Thr phosphorylation sites flanked by Arg residues at position +2 (that conform to a TXR phosphorylation motif) (Fig. 4).
FIG 4.

A T507A substitution disrupts δKD catalytic activity. (A) IVKAs with FL-PKCδ without and with PS-PMA (left) or WT and T507A-substituted forms of δKD without and with Src (right). Immunoblot analysis was used to track PKCδ protein expression, PKCδ phosphorylation of cTnI at Ser23/Ser24 and Thr144 (detected with an anti-TXR motif antibody), and cTnT phosphorylation at TXR motifs. Schematics are provided to show the locations of threonines in TXR motifs in cTnT (teal) as well as the Ser23/Ser24 and Thr144 phosphorylation motifs in cTnI (pink). Data from a representative experiment are illustrated at the top, and results for PKCδ-dependent 32P incorporation into cTnI are quantified at the bottom, with results being normalized to cTnI phosphorylation by WT-δKD (means ± standard errors of the means; n = 5). (B) CREBtide phosphorylation by δKD or δKD-T507A. Results depict averages from replicates from a single experiment (normalized to maximal 32P incorporation by WT-δKD) and are representative of results from 3 separate experiments.
Figure 4A shows that FL-PKCδ has little-to-no basal catalytic activity (lane 2) and that PMA-treated FL-PKCδ phosphorylates cTnI at Ser23/Ser24, but FL-PKCδ (which is recovered with considerable pSer359 immunoreactivity [9]) has little-to-no Thr kinase activity; it does not phosphorylate cTnI at Thr144, and it does not phosphorylate TXR motifs on cTnT (lane 3). In contrast, δKD displays high levels of cTnI-Ser23/Ser24, cTnI-Thr144, and cTnT-TXR kinase activities (lane 5). A T507A substitution in the δKD backbone dramatically decreases both Ser and Thr kinase activities (lane 8).
Since IVKAs that track the phosphorylation of proteins in the Tn complex are performed at limiting substrate concentrations (practical issues related to cost and protein solubility preclude assays with substrate concentrations that approach the predicted Km for the substrate), we performed additional IVKAs with CREBtide, a peptide substrate based upon the Ser133 phosphorylation site in CREB. Figure 4B shows that the T507A substitution disrupts the δKD phosphorylation of CREBtide even at high substrate concentrations. These results indicate that the T507A substitution disrupts the in vitro catalytic efficiency of δKD.
A δKD fragment that is not phosphorylated at Thr507 accumulates in doxorubicin-treated cardiomyocytes.
As an initial approach to examine the functional significance of δKD-Thr507 phosphorylation in a cellular context, we tracked the molecular species (and phosphorylation status) of δKD species that accumulate in doxorubicin-treated cardiomyocytes. Doxorubicin is a potent chemotherapeutic agent that is widely used for the treatment of hematologic and solid tissue malignancies (23). Doxorubicin binds DNA-associated enzymes, intercalates into nucleic acid side chains, disrupts DNA/RNA synthesis/repair, increases reactive oxygen species (ROS) production, induces cell cycle arrest, and promotes apoptotic cell death. A δKD fragment has been implicated in the proapoptotic response to doxorubicin (and various other DNA-damaging or ROS-producing agents) in noncardiac cell types (10–16). The notion that doxorubicin treatment (which leads to cardiomyocyte apoptosis and clinically important cardiotoxicity [24–26]) leads to the generation of a δKD fragment in cardiomyocytes has never been considered.
Figure 5A shows that native PKCδ is detected as an ∼73-kDa protein that is constitutively phosphorylated at its C terminus (at Ser645) in resting cardiomyocytes. Consistent with our previous results, PKCδ displays little to no Thr507 phosphorylation unless cardiomyocytes are treated with a PKC activator such as PMA (7). Doxorubicin treatment alone does not increase FL-PKCδ phosphorylation at Thr507 or Ser645. Rather, doxorubicin treatment leads to the activation of caspase-3, the cleavage of poly(ADP-ribose) polymerase (PARP), and the generation of a single ∼40-kDa δKD fragment that (like FL-PKCδ) is not phosphorylated at Thr507. This δKD fragment (liberated from Ser645-phosphorylated FL-PKCδ) also displays little-to-no phosphorylation at Ser645; while this result was somewhat surprising, it recapitulates the Ser645 phosphorylation defect displayed by δKD-T507A in HEK293 cells (Fig. 2). The relative abundance of the δKD fragment generated in doxorubicin-treated cardiomyocytes is quite low compared to the expression levels of FL-PKCδ, likely explaining why δKD generation is not associated with a detectable decrease in the abundance of FL-PKCδ.
FIG 5.

δKD fragments accumulate in doxorubicin-treated cardiomyocytes. (A) Cardiomyocyte cultures were treated with the vehicle doxorubicin (Dox) (10 μM for 24 h) or PMA (300 nM); PMA was included during the final 60 min of the stimulation interval or the entire 24-h period, as indicated. Extracts were subjected to immunoblot analysis to track the PKCδ protein, PKCδ-Thr507/Ser645 phosphorylation, as well as caspase-3 and PARP cleavage products as markers of apoptosis. For PKCδ protein and PKCδ-Thr507/Ser645 blots, protein loading and gel exposure times were optimized to visualize either the more abundant FL-PKCδ enzyme (top) or the 40-kDa (◁) and 45-kDa (*) δKD cleavage products (bottom); gels were then aligned in each panel for presentation purposes. (B) Extracts were subjected to immunoblotting with antibodies that track phosphorylation at either the pS-PKC substrate (R/L-X-pS-ϕ-R/L) or pTXR phosphorylation motifs, with β-actin immunoreactivity serving as a loading control. Similar results were obtained with three separate cardiomyocyte culture preparations.
The response to doxorubicin treatment in the presence of an acute (60-min) challenge with PMA (which increases FL-PKCδ-Thr507 phosphorylation) differs. Here, doxorubicin treatment leads to the accumulation of an ∼40-kDa δKD fragment that is not detectably phosphorylated at Thr507 or Ser645 as well as multiple additional δKD fragments with slower electrophoretic migration; the largest ∼40-kDa δKD fragment displays phosphorylation at Thr507 and Ser645. The additional observation that these δKD-immunoreactive species are not detected when doxorubicin challenge is accompanied by chronic (24-h) PMA treatment to downregulate FL-PKCδ validates the conclusion that these immunoreactive species represent bona fide FL-PKCδ cleavage products. These results provide novel evidence that the phosphorylation status of δKD fragments generated in doxorubicin-treated cardiomyocytes is context dependent and influenced by PMA.
We used an immunoblotting approach with PSSAs that screen for PKC substrate phosphorylation (at either Ser or Thr phosphoacceptor motifs) to determine whether the δKD fragment generated in doxorubicin-treated cardiomyocytes is active in a cellular context. Figure 5B shows that acute PMA treatment leads to a marked increase in PKC substrate phosphorylation, but PKC substrate phosphorylation is not increased in doxorubicin-treated cardiomyocytes that contain a δKD fragment that lacks Thr507 phosphorylation.
Thr507 phosphorylation regulates δKD-dependent responses in HEK293 cells.
The observation that doxorubicin treatment leads to the accumulation of δKD fragments that are not Thr507phosphorylated but does not detectably increase PKC substrate phosphorylation suggests that Thr507 phosphorylation is required for in vivo δKD activity. However, a negative result could be inconclusive if the amount of δKD generated from FL-PKCδ is insufficient to induce a detectable increase in PKC substrate phosphorylation in doxorubicin-treated cardiomyocytes. While this alternative interpretation of the data is considered unlikely, since even small amounts of a constitutively active δKD enzyme are expected to produce a functional phenotype, this issue was also addressed by using an overexpression strategy in HEK293 cells. Figure 6 shows that WT-δKD overexpression leads to an increase in overall PKC substrate phosphorylation and the activation/phosphorylation of PKCδ effectors such as extracellular signal-regulated kinase (ERK), stress-activated protein kinase/Jun amino-terminal kinase (SAPK/JNK), and p38–mitogen-activated protein kinase (MAPK). Of note, this is not accompanied by an increase in AKT phosphorylation, indicating that WT-δKD does not nonspecifically stimulate all growth-regulatory pathways. In contrast, δKD-T507A overexpression does not lead to changes in overall PKC substrate phosphorylation or signaling enzyme activation. These studies establish that the T507A substitution that disrupts the in vitro catalytic activity of δKD also disrupts δKD-dependent phosphorylation and signaling responses in vivo in a cellular context.
FIG 6.
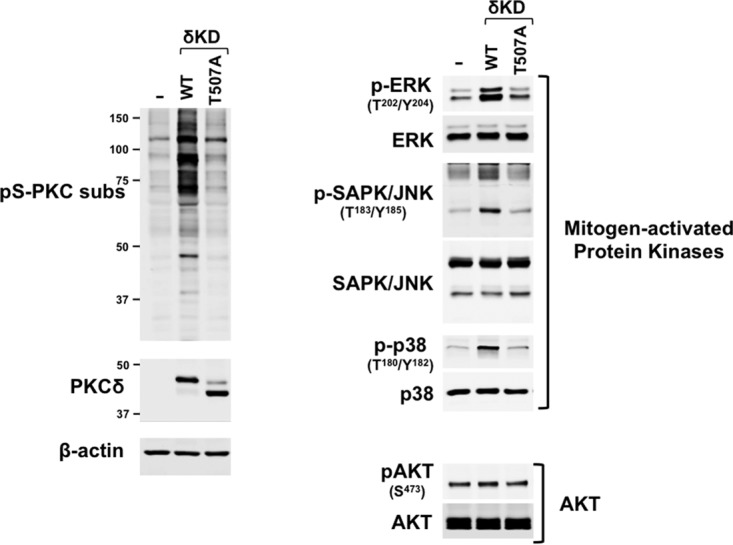
Thr507 phosphorylation regulates δKD-dependent responses in a cellular context. Untransfected HEK293 cell cultures (−) and HEK293 cells that heterologously overexpress similar levels of WT-δKD or δKD-T507A were subjected to immunoblot analysis to screen for overall PKC substrate phosphorylation at the pS-PKC substrate (R/L-X-pS-ϕ-R/L) or TXR motifs (left) or the phosphorylation of various mitogen-activated protein kinases (ERK, SAPK/JNK, and p38-MAPK) or AKT (right). Similar results were obtained in three separate experiments.
δKD-T507A activity is rescued by Src-dependent Tyr334 phosphorylation.
The newly exposed N-terminal region of δKD shares some sequence homology to the N-terminal residues that precede the catalytic core of the protein kinase A (PKA) catalytic subunit as well as the regulatory domain-kinase linkers of several other multidomain cytosolic Ser/Thr or Tyr kinases (Fig. 1). Structural studies of PKA indicate that this region of the N-tail forms an amphipathic helix (the αA-helix) that ends in Trp30; the indole ring of Trp30 protrudes into a hydrophobic cavity between the two lobes of the kinase core, where it makes functionally important contacts with the highly conserved Arg93 residue in the αC-helix in the small lobe of the kinase core and Arg190 at the base of the activation loop in the large lobe (27). These interactions stabilize key sites within the catalytic cleft and are required for PKA activity (28). Of note, Phe26 also fills this hydrophobic pocket and contributes to the allosteric regulation of PKA (27). Since Phe26/Trp30 in PKA corresponds to Tyr334/Trp338 in the newly exposed N terminus of δKD, and the T507A substitution that disrupts δKD catalytic activity is associated with conformational changes that extend to the N terminus (as evidenced by the fact that Tyr334 is a site for Src-dependent phosphorylation in δKD-T507A but not WT-δKD), we examined whether Src-dependent phosphorylation at Tyr334 regulates δKD activity.
Figures 4 (lane 6) and 7 (lane 3) show that Src does not phosphorylate WT-δKD or influence its (already considerable) cTnI-Ser23/Ser24, cTnI-Thr144, and cTnT-TXR kinase activities. Rather, Src phosphorylates δKD-T507A at Tyr334, and this is associated with an increase in its activity toward cTnI-Ser23/Ser24 (Fig. 4A, lane 9, and Fig. 7, lane 6). This is not associated with a detectable increase in cTnI phosphorylation at Thr144 or the phosphorylation of TXR sites on cTnT. While the level of 32P incorporation into cTnI is lower in assays with Src-phosphorylated δKD-T507A than in assays with WT-δKD (Fig. 4A, bottom), this is likely attributable to the absence of any TXR motif phosphorylation (and the fact that only the faster-migrating, completely unprimed pool of δKD-T507A is phosphorylated/activated by Src). Collectively, these results indicate that Src rescues the catalytic activity of δKD-T507A.
FIG 7.

δKD-T507A activity is rescued by Src-dependent Tyr334 phosphorylation or a G-loop S359A substitution. IVKAs were performed with the WT-δKD, δKD-Y334F, δKD-S359A, or δKD-Y334C enzyme in the absence or presence of Src. In each case, the experiment includes constructs harboring either a threonine (WT) or a T507A substitution at the activation loop phosphorylation site. All samples were subjected to immunoblot analysis to track PKCδ protein expression and phosphorylation as well as PKCδ phosphorylation of cTnI at Ser23/Ser24 and cTnT at TXR motifs (as described in the legends to Fig. 2 and 4). Each panel represents results from a single experiment; similar results were obtained in experiments with 3 separate enzyme preparations. Since the large number of samples in panel A could not be included in a single gel, the immunoblots for each antibody in this panel are from two gels run in parallel and exposed for a uniform duration (with the dashed line denoting where data from the different gels were merged for presentation purposes).
Since Tyr334 may not be the only site that Src phosphorylates on δKD-T507A (and Src could in theory regulate δKD-T507A activity through some other mechanism, for example, a direct protein-protein interaction), we generated constructs harboring nonphosphorylatable Y334F or Y334C substitutions on WT and T507A backgrounds. The Y334C substitution was included to mimic a nonsynonymous single-nucleotide polymorphism (SNP) that has been identified in some human populations (see the National Center for Biotechnology Information dbSNP website at http://www.ncbi.nlm.nih.gov/SNP). Control studies show that Y334F and Y334C substitutions alone do not prevent priming phosphorylations; δKD-Y334F and δKD-Y334C are detected as single molecular species that are phosphorylated at Thr507, Ser645, and Ser664 (Fig. 7 and data not shown). Y334F and Y334C substitutions in the WT-δKD background also do not result in gross changes in catalytic activity, tracked as cTnI phosphorylation at Ser23/Ser24 and Thr144 and cTnT phosphorylation at TXR motifs. However, the Y334F and Y334C substitutions in the δKD-T507A background prevent phosphorylation by Src, and they abrogate the Src-dependent increase in cTnI-Ser23/Ser24 activity. These results indicate that Tyr334 in the N-terminal region of δKD-T507A is the only site that is phosphorylated by Src and that Tyr334 phosphorylation mediates the Src-dependent increase in the catalytic activity of δKD-T507A; a Y334C SNP that disrupts this mechanism could have important functional consequences.
An S359A substitution that prevents G-loop phosphorylation rescues δKD-T507A activity.
We previously demonstrated that FL-PKCδ-S359A is a constitutively active enzyme that displays high levels of both Ser and Thr kinase activity (9). Conversely, FL-PKCδ-S359E retains Ser kinase activity, but it does not phosphorylate substrates with a Thr residue at the phosphoacceptor site (9). Since δKD-T507A is recovered as a Ser359-phosphorylated enzyme, we examined whether this increase in G-loop Ser359 phosphorylation restricts δKD-T507A's P-site specificity.
Figure 7A shows that δKD-S359A is detected as a single molecular species that is fully primed (phosphorylated at Thr507, Ser645, and Ser664) and displays high levels of cTnI-Ser23/Ser24 and cTnT-TXR activity. A T507A substitution in the δKD-S359A backbone results in the appearance of a doublet, with the faster-migrating species serving as a substrate for Src. While these properties are similar to those described above for δKD-T507A, the activity profiles of δKD-T507A and δKD-S359A-T507A are very different. δKD-T507A is catalytically inactive, whereas δKD-S359A-T507A retains considerable amounts of cTnI-Ser23/Ser24 and cTnT-TXR activity. The fact that a T507A substitution is tolerated in the δKD-S359A backbone indicates that the T507A substitution disrupts catalytic activity at least in part by increasing G-loop Ser359 phosphorylation. The additional observation that δKD-S359A-T507A retains both Ser and Thr kinase activity indicates that Ser359 phosphorylation plays a similar role in regulating P-site specificity in FL-PKCδ and δKD.
Trp338 regulates the activity of FL-PKCδ but not δKD.
Since a highly conserved Trp residue N terminal to the kinase core functions as an important structural determinant in PKA, various protein tyrosine kinases, and Raf isoforms (Fig. 1) (27, 29, 30), we examined whether Trp338 contributes to the control of δKD activity. Figure 8A shows that δKD-W338A is detected as a single ∼45-kDa fully primed (Thr507/Ser645-phosphorylated) enzyme and that δKD-W338A and δKD display similar high levels of cTnI-Ser23/Ser24 kinase activity. The effect of a T507A substitution is also identical in the δKD or δKD-W338A background. In each case, the enzymes are resolved as doublets (denoting the appearance of a faster-migrating ∼40-kDa unprimed species) with little to no cTnI-Ser23/Ser24 kinase activity; a W336A substitution in the δKD-T507A background does not rescue catalytic activity.
FIG 8.

Trp338 regulates the activity of FL-PKCδ but not δKD. IVKAs were performed with WT-δKD and δKD-W338A (in each case with either a Thr or T507A substitution in the activation loop [A] or with FL-PKCδ or PKCδ-W338A in the absence or presence of PS-PMA and/or Src [B]). Immunoblotting was used to track PKCδ protein expression and phosphorylation and PKCδ activity (measured as cTnI-Ser23/Ser24 or cTnT-TXR motif phosphorylation). Results of a representative experiment are illustrated at the top, and results for PKCδ-dependent 32P incorporation into cTnI are quantified at the bottom, with results being normalized to cTnI phosphorylation by WT-δKD in panel A or FL-PKCδ in the presence of PS-PMA (means ± standard errors of the means; n = 3).
While the W338A substitution has no discernible effect when inserted into δKD, it influences the activity of FL-PKCδ. Figure 8B shows that FL-WT-PKCδ is a lipid-dependent enzyme that displays Tn kinase activity, and becomes a substrate for Src, only when activated by PS-PMA. In contrast, FL-PKCδ-W338A displays considerable lipid-independent Ser and Thr kinase activity, and it is a substrate for Src-dependent Tyr313 and Tyr334 phosphorylation even in assays performed without PS-PMA. These results indicate that Trp338 in the regulatory domain-kinase linker region functions in some way to transmit an inhibitory signal from the regulatory domain that limits FL-PKCδ activity.
DISCUSSION
PKCδ plays pleiotropic roles in the control of cell growth, survival, and proapoptotic responses, depending upon the cellular environment. The growing recognition that traditional models that describe conventional PKCα or PKCβ isoform activation are inadequate to fully account for the diverse cellular actions of PKCδ has provided the rationale to identify regulatory features that are unique to PKCδ and dictate signaling specificity. In this context, we previously implicated phosphorylations at Thr507 and Tyr313 (a tyrosine that is unique to the V3 hinge region of PKCδ and not present in other PKCs) as regulatory modifications that influence the signaling properties of FL-PKCδ (5, 9). While there is considerable evidence that PKCδ can be cleaved by caspase-3 during oxidative or genotoxic stress, the notion that phosphorylation (either at the activation loop or at Tyr334 in the newly exposed δKD N terminus) might also constitute regulatory modifications for δKD has not previously been considered. This study shows that phosphorylation at four distinct sites (the activation loop, C-tail priming sites, Tyr334 in the δKD N terminus, and Ser359 at the tip of the G-loop) plays critical roles in regulating δKD activity.
The activation loop in the catalytic cleft of the enzyme is a highly flexible structure that provides a platform for substrate binding. While activation loops of PKA and most PKCs adopt the proper extended conformation for substrate binding only following phosphorylation at the activation loop phosphorylation motif, FL-PKCδ is a notable exception in that it is catalytically competent even without Thr507 phosphorylation. However, this study shows that a T507A substitution severely disrupts δKD activity. These results suggest that some molecular determinant in the regulatory domain substitutes for Thr507 phosphorylation to stabilize the KD in an active conformation in FL-PKCδ.
Our in vitro studies show that a T507A substitution renders δKD catalytically inactive; cell-based studies indicate that δKD fragments that are not Thr507 phosphorylated accumulate in doxorubicin-treated cardiomyocytes and that the Thr507 phosphorylation defect disrupts δKD's cellular activity. This finding is at odds with results reported previously by Liu et al., which implicated Thr507 phosphorylation as a modification that regulates δKD substrate specificity and δKD-driven cellular responses, but those authors concluded that Thr507 phosphorylation is not absolutely required for δKD catalytic activity (19). While a specific factor that might reconcile these discrepant results is not obvious, it is worth noting that Liu et al. compared δKD and δKD-T507A activities at equal protein concentrations as defined by Western blotting, but immunoblots that would permit the analysis of whether the T507A substitution altered protein mobility (or δKD C-tail priming-site phosphorylation) were not provided. One could speculate that the T507A substitution was tolerated in their experiments because of some cell-specific difference that resulted in the expression of a molecular chaperone or scaffolding protein that stabilized δKD-T507A in a catalytically active conformation (i.e., assumed the role of the regulatory domain of FL-PKCδ). Such protein-protein interactions have been reported to contribute to the control of other PKC enzymes (31, 32).
Cell-based studies link a Thr507 phosphorylation defect to a constellation of changes at the N- and C-terminal tails of δKD. Specifically, we show that δKD-T507A is stabilized in two major conformations in HEK293 cells: a slower-migrating species that retains C-tail Ser645/Ser664 phosphorylation and cannot be phosphorylated by Src at Tyr334 (presumably because the C- and N-terminal tails remain anchored to the kinase core) and a more rapidly migrating species where presumably increased solvent exposure of the C- and N-terminal tails results in decreased Ser645/Ser664 phosphorylation and renders Tyr334 available for phosphorylation by Src. Studies in cardiomyocytes establish that this molecular heterogeneity is physiologically relevant, showing that the phosphorylation status of δKD fragments that accumulate in doxorubicin-treated cardiomyocytes is influenced by PMA. Collectively, these results suggest that the C- and N-terminal tails make functionally important contacts with the δKD kinase core (much like what has previously been described for the catalytic subunit of PKA); Thr507 phosphorylation leads to long-range changes in δKD that influence the conformation of the C- and N-tails. These results also suggest that Src-dependent Tyr334 phosphorylation can be exploited as a convenient biochemical readout to gauge the conformation of the V3 linker region of FL-PKCδ or the N-terminal tail of δKD.
The δKD Thr507 phosphorylation defect also leads to a change in G-loop phosphorylation at Ser359. Specifically, while FL-PKCδ is recovered as a Ser359-phosphorylated enzyme, fully primed (Thr507-phosphorylated) δKD is not phosphorylated at Ser359; Ser359 phosphorylation is confined to δKD fragments that lack priming-site phosphorylation. While this study did not directly examine the mechanism that might contribute to differences in δKD-Ser359 phosphorylation, data from our previous studies suggest that the G-loop Ser359 site is protected from cellular phosphatases by a C2 domain-mediated autoinhibitory interaction (9). In fact, a recent reinterpretation of the crystal lattice packing pattern for PKCβII provides direct evidence that the C2 domain engages in autoinhibitory interactions with the kinase domain that serves to clamp the autoinhibitory pseudosubstrate domain into the substrate-binding cavity (33). Our observation that δKD is not phosphorylated at Ser359 would be consistent with this formulation; in the absence of the regulatory domain, this site would become solvent exposed and susceptible to dephosphorylation by cellular phosphatases.
Since there is as yet no available X-ray crystal structure for δKD, we used a structural model of PKA and a structural representation of δKD (based upon the solved structure of the staurosporine-bound kinase domain PKCθ, PKCδ's closest paralog in the PKC family of enzymes) as a framework to deduce a molecular explanation for the various experimental findings of this study. Figure 9 shows the ribbon structure of PKA, showing the characteristic bilobar kinase fold with an amino-terminal N-lobe and a carboxy-terminal C-lobe. The smaller N-lobe contains the G-loop, which functions to shield ATP from the solvent and position the γ-phosphate of ATP for catalysis. The recognition that the G-loop is a highly flexible portion of the N-lobe (34) and a mutational “hot spot” in certain cancers (35, 36) provides context to appreciate the functional importance of G-loop phosphorylation as a modification that regulates enzyme activation. The αC-helix is another important feature of the N-lobe. It contains a highly conserved Glu residue (Glu91 in PKA and Glu397 in PKCδ) that binds Lys (Lys72 in PKA and Lys378 in PKCδ) in the conserved AxK motif. This ionic interaction is required to correctly orient the αC-helix. The active-site cleft sits at the interface between the N- and C-lobes and contains the ATP-binding site and the adjacent solvent-exposed substrate-binding site. While the N- and C-lobes of PKA and PKCs contain all of the essential catalytic machinery, these enzymes are stabilized in a catalytically active conformation by their N- and C-tails that wrap around both lobes of the kinase core. In the case of PKA, this involves an interaction between the hydrophobic motif at the extreme C terminus of the C-tail and an allosteric pocket at one side of the αC-helix and a second interaction between hydrophobic residues (Tyr26/Trp30) in the N-terminal αA-helix and a hydrophobic pocket at the other side of the αC-helix. While the C-tail hydrophobic motif is conserved in PKCs (although it includes a priming phosphorylation site, FAGFpS664F in PKCδ, that stabilizes these enzymes in a catalytically active conformation), the hydrophobic residues at the N terminus of PKA are conserved in PKCδ but not in other isoforms of PKC (Fig. 1). The observation that all of the regions in δKD affected by the T507A substitution converge on the αC-helix (which has been implicated as a structural integrator of the dynamic behavior of other key regions in PKA [37]) suggests that the relationships between the αC-helix and the activation loop, the C-tail hydrophobic motif, and the G-loop that have been described for PKA are at least partially conserved in δKD.
FIG 9.

Phosphorylation sites in δKD affected by a T507A substitution converge on the highly conserved αC-helix. (Top) PKA catalytic subunit ribbon structure (PDB accession number 1ATP) with the N-lobe oriented on top and the C-lobe at the bottom. The blowup (right) emphasizes key interactions between the αC-helix (red) and conserved motifs in other regions of the enzyme, including (i) the docking site for the hydrophobic motif (FXXF) at the extreme C terminus of the C-tail (slate gray) and an allosteric regulatory pocket at one side of the αC-helix, (ii) a second interaction between hydrophobic residues (Tyr26/Trp30) in the N-terminal αA-helix (tan) and a hydrophobic pocket formed by Arg93/Arg190 (from the αC-helix and activation loop, respectively) at the other side of the αC-helix, and (iii) the position of Ser53 at the tip of the G-loop (green). (Bottom) Two views of a structural model of the corresponding region in δKD based upon the solved structure of PKCθ (PDB accession number 1XJD), with the αA-helix region missing from the PKC structure based upon the αA-helix of PKA. The orientation of the image on the right is similar to that depicted for PKA; the 90° rotation of the image on the left emphasizes the relationship between the αC-helix and phosphorylation sites at the hydrophobic motif (Ser664), G-loop (Ser359), activation loop (Thr507), and N-tail (Tyr334). See the text.
The role of the αC-helix as a docking site for the N-tail αA-helix in PKA is of particular relevance to our studies of δKD, since PKCδ shares considerable sequence homology to PKA at its newly exposed N terminus; this region of other PKC isoforms is highly divergent. Studies of PKA indicate that an interaction between Phe26/Trp30 in the N-tail αA helix and a hydrophobic pocket formed by arginine residues in the αC-helix and the base of the activation loop stabilizes key sites within the catalytic cleft; single-residue substitutions that prevent Trp30 interactions with the kinase core (W30A or W30F) decrease PKA's thermal stability (27, 28, 38). Of note, a Trp residue (similarly positioned N terminal to the catalytic core) is conserved in PKCδ, several Src family kinases (Src and Hck), Csk, Btk, and Raf isoforms (27). In some of these other enzymes, this Trp residue also functions as a structural regulator of catalytic activity, although the mechanism and mode of regulation appear to be highly kinase specific (29, 30, 39–42). Our studies show that a W338A substitution does not grossly alter δKD activity, effectively excluding the similarly placed Trp338 as a direct allosteric regulator of δKD. Rather, the W338A substitution activates FL-PKCδ, indicating that the Trp338 in the regulatory domain-kinase linker region functions in some way to transmit an inhibitory signal from the regulatory domain and limit FL-PKCδ activity.
Finally, this study identifies a novel role for Tyr334 as a regulator of δKD-T507A catalytic activity. Our previous studies identified Tyr334 as an Src phosphorylation site in FL-PKCδ in cardiomyocytes subjected to oxidative stress. However, a functional correlate for this posttranslational modification was not obvious, since biochemical studies failed to link Tyr334 phosphorylation to any detectable changes in FL-PKCδ catalytic activity. Studies reported here show that Tyr334 phosphorylation becomes functionally important in the context of the δKD fragment. We show that Thr507-phosphorylated/catalytically active δKD is resistant to Src-dependent Tyr334 phosphorylation, presumably because Tyr334 in the newly released N-tail is buried in the kinase core. However, unprimed δKD-T507A is phosphorylated at Tyr334 by Src, and this modification rescues kinase activity. These results suggest that Tyr334 in the newly released N-tail acts as an autoinhibitory clamp to limit δKD-T507A activity and that autoinhibition is relieved by Tyr334 phosphorylation. The further observation that a Y334C substitution prevents δKD-T507A phosphorylation by Src suggests that a nonsynonymous SNP that has been identified in certain human populations would function to limit δKD-T507A activity and therefore δKD-T507A-driven proapoptotic cellular responses. These results suggest a heretofore unrecognized biochemical mechanism that could contribute to interindividual differences in cellular injury and/or inflammatory responses and alter clinical outcomes in the setting of myocardial infarction, stroke, neurodegenerative diseases, autoimmune disorders, and certain cancers.
MATERIALS AND METHODS
Materials.
PKCδ and PKCδ-pTyr334 antibodies were obtained from Santa Cruz Biotechnology. Anti-Flag was obtained from Sigma. All other antibodies were obtained from Cell Signaling Technology. The specificity of all anti-PKCδ antibodies, including PSSAs that specifically recognize phosphorylation at Thr507, Tyr313, Tyr334, and Ser359, was validated previously (5, 9). Src was obtained from Oncogene. CREBtide was obtained from Anaspec. PMA and cycloheximide were obtained from Sigma. Other chemicals were of reagent grade.
Plasmids and HEK293 cell culture.
Mutant constructs of a PKCδ-Flag expression plasmid (5) were generated by PCR and validated by sequencing. Expression vectors were introduced into HEK293 cells (maintained in Dulbecco's modified Eagle's medium [DMEM] with 10% fetal bovine serum [FBS]) by using the Effectene transfection reagent (Qiagen) according to the manufacturer's instructions. After 24 h, cells were lysed in homogenization buffer containing 20 mM Tris-Cl (pH 7.5), 0.05 mM EDTA, 0.5 mM dithiothreitol, 0.2% Triton X-100, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 5 μg/ml benzamidine, 1 mM phenylmethylsulfonyl fluoride, and 5 μM pepstatin A.
Cardiomyocyte culture.
Cardiomyocytes were isolated from hearts of 2-day-old Wistar rats by a trypsin dispersion procedure that uses a differential attachment procedure followed by irradiation to enrich for cardiomyocytes (43); the protocol used in this study was approved by the Columbia University Institutional Animal Care and Use Committee. Cells were plated onto protamine sulfate-coated culture dishes at a density of 5 × 106 cells/100-mm dish and grown in minimal essential medium (MEM) (Invitrogen, BRL) supplemented with 10% fetal calf serum for 4 days prior to experiments. Cardiomyocyte extracts were prepared for immunoblot analysis according to methods described previously (44).
IVKAs and Western blotting.
IVKAs were performed with PKCδ immunoprecipitated with anti-Flag from 150 μg of a starting cell extract according to methods described previously (5). Incubations were performed for 30 min at 30°C with 110 μl reaction buffer containing 30 mM Tris-Cl (pH 7.5), 5.45 mM MgCl2, 0.65 mM EDTA, 0.65 mM EGTA, 0.1 mM dithiothreitol (DTT), 1.09 mM sodium orthovanadate, 0.1 μM calyculin, 76 mM NaCl, 3.6% glycerol, 89 μg/ml phosphatidylserine plus 175 nM PMA, and [γ-32P]ATP (10 μCi; 66 μM), with 4 μg of the cTn complex (consisting of equimolar concentrations of cTnI, cTnT, and cTnC; generously provided by John Solaro) as the substrate. Immunoblotting on lysates or immunoprecipitated PKCδ was performed according to methods described previously or the manufacturer's instructions (5). In each figure, each panel represents the results from a single gel (exposed for a uniform duration); detection was performed with enhanced chemiluminescence. CREB kinase assays were performed as described previously (7).
REFERENCES
- 1.Newton AC, Antal CE, Steinberg SF. 2016. Protein kinase C mechanisms that contribute to cardiac remodelling. Clin Sci 130:1499–1510. doi: 10.1042/CS20160036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinberg SF. 2008. Structural basis of protein kinase C isoform function. Physiol Rev 88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benes CH, Wu N, Elia AE, Dharia T, Cantley LC, Soltoff SP. 2005. The C2 domain of PKCδ is a phosphotyrosine binding domain. Cell 121:271–280. doi: 10.1016/j.cell.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 4.Newton AC. 2010. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab 298:E395–E402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sumandea MP, Rybin VO, Hinken AC, Wang C, Kobayashi T, Harleton E, Sievert G, Balke CW, Feinmark SJ, Solaro RJ, Steinberg SF. 2008. Tyrosine phosphorylation modifies PKCδ-dependent phosphorylation of cardiac troponin I. J Biol Chem 283:22680–22689. doi: 10.1074/jbc.M802396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rybin VO, Guo J, Sabri A, Elouardighi H, Schaefer E, Steinberg SF. 2004. Stimulus-specific differences in protein kinase C-δ localization and activation mechanisms in cardiomyocytes. J Biol Chem 279:19350–19361. doi: 10.1074/jbc.M311096200. [DOI] [PubMed] [Google Scholar]
- 7.Rybin VO, Guo J, Gertsberg Z, Elouardighi H, Steinberg SF. 2007. Protein kinase Cε (PKCε) and Src control PKCδ activation loop phosphorylation in cardiomyocytes. J Biol Chem 282:23631–23638. doi: 10.1074/jbc.M701676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, Nishizuka Y. 1997. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci U S A 94:11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gong J, Yao Y, Zhang P, Udayasuryan B, Komissarova EV, Chen J, Sivaramakrishnan S, Van Eyk JE, Steinberg SF. 2015. The C2 domain and altered ATP-binding loop phosphorylation at Ser359 mediate the redox-dependent increase in protein kinase C-δ activity. Mol Cell Biol 35:1727–1740. doi: 10.1128/MCB.01436-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong WW, Kamen R, Weichselbaum R, Kufe D. 1995. Proteolytic activation of protein kinase Cδ by an ICE-like protease in apoptotic cells. EMBO J 14:6148–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denning MF, Wang Y, Nickoloff BJ, Wrone-Smith T. 1998. Protein kinase Cδ is activated by caspase-dependent proteolysis during ultraviolet radiation-induced apoptosis of human keratinocytes. J Biol Chem 273:29995–30002. doi: 10.1074/jbc.273.45.29995. [DOI] [PubMed] [Google Scholar]
- 12.Cross T, Griffiths G, Deacon E, Sallis R, Gough M, Watters D, Lord JM. 2000. PKCδ is an apoptotic lamin kinase. Oncogene 19:2331–2337. doi: 10.1038/sj.onc.1203555. [DOI] [PubMed] [Google Scholar]
- 13.Fukunaga M, Oka M, Ichihashi M, Yamamoto T, Matsuzaki H, Kikkawa U. 2001. UV-induced tyrosine phosphorylation of PKCδ and promotion of apoptosis in the HaCaT cell line. Biochem Biophys Res Commun 289:573–579. doi: 10.1006/bbrc.2001.6025. [DOI] [PubMed] [Google Scholar]
- 14.Vries-Seimon TA, Ohm AM, Humphries MJ, Reyland ME. 2007. Induction of apoptosis is driven by nuclear retention of protein kinase Cδ. J Biol Chem 282:22307–22314. doi: 10.1074/jbc.M703661200. [DOI] [PubMed] [Google Scholar]
- 15.Humphries MJ, Ohm AM, Schaack J, Adwan TS, Reyland ME. 2008. Tyrosine phosphorylation regulates nuclear translocation of PKCδ. Oncogene 27:3045–3053. doi: 10.1038/sj.onc.1210967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeVries TA, Neville MC, Reyland ME. 2002. Nuclear import of PKCδ is required for apoptosis: identification of a novel nuclear import sequence. EMBO J 21:6050–6060. doi: 10.1093/emboj/cdf606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, Pandey P, Datta R, Huang Y, Kharbanda S, Allen H, Kamen R, Wong W, Kufe D. 1996. Proteolytic activation of protein kinase Cδ by an ICE/CED 3-like protease induces characteristics of apoptosis. J Exp Med 184:2399–2404. doi: 10.1084/jem.184.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denning MF, Wang Y, Tibudan S, Alkan S, Nickoloff BJ, Qin JZ. 2002. Caspase activation and disruption of mitochondrial membrane potential during UV radiation-induced apoptosis of human keratinocytes requires activation of protein kinase C. Cell Death Differ 9:40–52. doi: 10.1038/sj.cdd.4400929. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Belkina NV, Graham C, Shaw S. 2006. Independence of protein kinase C-δ activity from activation loop phosphorylation: structural basis and altered functions in cells. J Biol Chem 281:12102–12111. doi: 10.1074/jbc.M600508200. [DOI] [PubMed] [Google Scholar]
- 20.Kato K, Yamanouchi D, Esbona K, Kamiya K, Zhang F, Kent KC, Liu B. 2009. Caspase-mediated protein kinase C-δ cleavage is necessary for apoptosis of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 297:H2253–H2261. doi: 10.1152/ajpheart.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards AS, Faux MC, Scott JD, Newton AC. 1999. Carboxyl-terminal phosphorylation regulates the function and subcellular localization of protein kinase C βII. J Biol Chem 274:6461–6468. doi: 10.1074/jbc.274.10.6461. [DOI] [PubMed] [Google Scholar]
- 22.Bornancin F, Parker PJ. 1997. Phosphorylation of protein kinase C-α on Ser657 controls the accumulation of active enzyme and contributes to its phosphatase-resistant state. J Biol Chem 272:3544–3549. doi: 10.1074/jbc.272.6.3544. [DOI] [PubMed] [Google Scholar]
- 23.Weiss RB. 1992. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol 19:670–686. [PubMed] [Google Scholar]
- 24.Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. 2000. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res 60:1789–1792. [PubMed] [Google Scholar]
- 25.Wang L, Ma W, Markovich R, Chen JW, Wang PH. 1998. Regulation of cardiomyocyte apoptotic signaling by insulin-like growth factor I. Circ Res 83:516–522. doi: 10.1161/01.RES.83.5.516. [DOI] [PubMed] [Google Scholar]
- 26.Chatterjee K, Zhang J, Tao R, Honbo N, Karliner JS. 2008. Vincristine attenuates doxorubicin cardiotoxicity. Biochem Biophys Res Commun 373:555–560. doi: 10.1016/j.bbrc.2008.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veron M, Radzio-Andzelm E, Tsigelny I, Ten Eyck LF, Taylor SS. 1993. A conserved helix motif complements the protein kinase core. Proc Natl Acad Sci U S A 90:10618–10622. doi: 10.1073/pnas.90.22.10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cembran A, Masterson LR, McClendon CL, Taylor SS, Gao J, Veglia G. 2012. Conformational equilibrium of N-myristoylated cAMP-dependent protein kinase A by molecular dynamics simulations. Biochemistry 51:10186–10196. doi: 10.1021/bi301279f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McPherson RA, Taylor MM, Hershey ED, Sturgill TW. 2000. A different function for a critical tryptophan in c-Raf and Hck. Oncogene 19:3616–3622. doi: 10.1038/sj.onc.1203678. [DOI] [PubMed] [Google Scholar]
- 30.Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJ, Kornev AP, Taylor SS, Shaw AS. 2013. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154:1036–1046. doi: 10.1016/j.cell.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoshi N, Langeberg LK, Gould CM, Newton AC, Scott JD. 2010. Interaction with AKAP79 modifies the cellular pharmacology of PKC. Mol Cell 37:541–550. doi: 10.1016/j.molcel.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gould CM, Kannan N, Taylor SS, Newton AC. 2009. The chaperones Hsp90 and Cdc37 mediate the maturation and stabilization of protein kinase C through a conserved PXXP motif in the C-terminal tail. J Biol Chem 284:4921–4935. doi: 10.1074/jbc.M808436200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antal CE, Callender JA, Kornev AP, Taylor SS, Newton AC. 2015. Intramolecular C2 domain-mediated autoinhibition of protein kinase C βII. Cell Rep 12:1252–1260. doi: 10.1016/j.celrep.2015.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Srivastava AK, McDonald LR, Cembran A, Kim J, Masterson LR, McClendon CL, Taylor SS, Veglia G. 2014. Synchronous opening and closing motions are essential for cAMP-dependent protein kinase A signaling. Structure 22:1735–1743. doi: 10.1016/j.str.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torkamani A, Kannan N, Taylor SS, Schork NJ. 2008. Congenital disease SNPs target lineage specific structural elements in protein kinases. Proc Natl Acad Sci U S A 105:9011–9016. doi: 10.1073/pnas.0802403105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dixit A, Yi L, Gowthaman R, Torkamani A, Schork NJ, Verkhivker GM. 2009. Sequence and structure signatures of cancer mutation hotspots in protein kinases. PLoS One 4:e7485. doi: 10.1371/journal.pone.0007485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor SS, Shaw AS, Kannan N, Kornev AP. 2015. Integration of signaling in the kinome: architecture and regulation of the alphaC helix. Biochim Biophys Acta 1854:1567–1574. doi: 10.1016/j.bbapap.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herberg FW, Zimmermann B, McGlone M, Taylor SS. 1997. Importance of the A-helix of the catalytic subunit of cAMP-dependent protein kinase for stability and for orienting subdomains at the cleft interface. Protein Sci 6:569–579. doi: 10.1002/pro.5560060306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin X, Ayrapetov MK, Lee S, Parang K, Sun G. 2005. Probing the communication between the regulatory and catalytic domains of a protein tyrosine kinase, Csk. Biochemistry 44:1561–1567. doi: 10.1021/bi048142j. [DOI] [PubMed] [Google Scholar]
- 40.Guo S, Wahl MI, Witte ON. 2006. Mutational analysis of the SH2-kinase linker region of Bruton's tyrosine kinase defines alternative modes of regulation for cytoplasmic tyrosine kinase families. Int Immunol 18:79–87. doi: 10.1093/intimm/dxh351. [DOI] [PubMed] [Google Scholar]
- 41.LaFevre-Bernt M, Sicheri F, Pico A, Porter M, Kuriyan J, Miller WT. 1998. Intramolecular regulatory interactions in the Src family kinase Hck probed by mutagenesis of a conserved tryptophan residue. J Biol Chem 273:32129–32134. doi: 10.1074/jbc.273.48.32129. [DOI] [PubMed] [Google Scholar]
- 42.Sicheri F, Moarefi I, Kuriyan J. 1997. Crystal structure of the Src family tyrosine kinase Hck. Nature 385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- 43.Rybin VO, Sabri A, Short J, Braz JC, Molkentin JD, Steinberg SF. 2003. Cross regulation of nPKC isoform function in cardiomyocytes: role of PKCε in activation loop phosphorylations and PKCδ in hydrophobic motif phosphorylations. J Biol Chem 278:14555–14564. doi: 10.1074/jbc.M212644200. [DOI] [PubMed] [Google Scholar]
- 44.Rybin VO, Steinberg SF. 1994. Protein kinase C isoform expression and regulation in the developing rat heart. Circ Res 74:299–309. doi: 10.1161/01.RES.74.2.299. [DOI] [PubMed] [Google Scholar]