

Abstract
Cytochrome P450 aromatase (CYP19A1) plays a key role in the development of estrogen dependent breast cancer, and aromatase inhibitors have been at the front line of treatment for the past three decades. The development of potent, selective and safer inhibitors is ongoing with in silico screening methods playing a more prominent role in the search for promising lead compounds in bioactivity-relevant chemical space. Here we present a set of comprehensive binding affinity prediction models for CYP19A1 using our automated Linear Interaction Energy (LIE) based workflow on a set of 132 putative and structurally diverse aromatase inhibitors obtained from a typical industrial screening study. We extended the workflow with machine learning methods to automatically cluster training and test compounds in order to maximize the number of explained compounds in one or more predictive LIE models. The method uses protein–ligand interaction profiles obtained from Molecular Dynamics (MD) trajectories to help model search and define the applicability domain of the resolved models. Our method was successful in accounting for 86% of the data set in 3 robust models that show high correlation between calculated and observed values for ligand-binding free energies (RMSE < 2.5 kJ mol–1), with good cross-validation statistics.
Introduction
Cytochrome P450 aromatase (CYP19A1; EC 1.14.14.1) is a member of the Cytochrome P450 (CYP) superfamily of mono-oxygenases. This enzyme catalyzes a key step in estrogen biosynthesis, i.e., aromatization of androgens such as androstenedione and testosterone to estrone and 17β-estradiol, respectively.1−7 Common to most CYPs, CYP19A1 can bind a variety of low molecular weight compounds, but it has a high catalytic specificity toward steroid substrates due to the distribution of polar and nonpolar residues within the binding site.8,9
Overexpression of aromatase in tumor tissue was identified to play a key role in the development of estrogen receptor positive breast cancer, endometrial cancer and endometriosis.1,3,5 Around 50–80% of breast cancers have been found to be estrogen-dependent, where estrogen binding to receptor stimulates tumor cell proliferation.10−13 Because the aromatization reaction catalyzed by CYP19A1 is rate limiting, it serves as an ideal target for the development of selective and potent inhibitors that decrease the levels of circulating estrogen.14,15 This has led to the development of four generations of aromatase inhibitors (AIs) in clinical use over the last three decades (Figure 1).14,16,17 Most AIs are nonsteroidal in nature (NSAIs) and derived from aminoglutethimide-like molecules, imidazole/triazole derivatives, or flavonoid analogs.17,18 They act by means of competitive and reversible inhibition (type I) or quasi-irreversible inhibition by coordination with the heme iron (type II).19,20 Steroidal inhibitors are typically based on the adrostenedione scaffold with various chemical substituents at varying positions, which can be functional groups responsible for mechanism based inhibition (e.g., Exemestane, Figure 1f).21
Figure 1.

Members of four generations of clinical steroidal (c,f) and nonsteroidal (a,b,d,e) aromatase (CYP19A1) inhibitors. First generation: aminoglutethimide (a, Cytadren, Novartis).99−101 Second generation: Fadrozole (b, Afema, Novartis),99−101 and Formestane (c, Lentaron, Novartis).14 Third generation: Anastrozol (d, Arimidex, AstraZeneca),102 Letrozole (e, Femara, Novartis),103−105 and Exemestane (f, Aromasine, Pfizer).103
Despite their clinical success, current AIs are associated with various drug related side-effects,22,23 including effects due to inhibition of other members of the CYP family24 that can lead to drug–drug interactions (DDI).25 Therefore, the search for a next generation of AIs with improved potency, higher selectivity and reduced toxicity is still ongoing, and both synthetic as well as natural product derived alternatives such as coumarin, lignin and flavonoids have been explored over the years.17,26−30
The widened search spectrum for AI lead compounds increases the need for effective methods to screen the inhibitory potential and modes of interaction for both the target protein and other CYPs. In particular, the use of in silico methods for the prioritization of compound ideas throughout the lead discovery and optimization stages potentially offers an attractive alternative for extensive use of in vitro methods.31,32 However, especially for CYPs it remains a challenge to train generally applicable predictive models for protein binding due to their substrate promiscuity, catalytic site malleability, different modes of inhibition, and ability to bind the same ligand in multiple orientations.19,25,33,34 The flexibility in both the structural and interaction dimensions limits the applicability of QSAR (Quantitative Structure–Activity Relationship) methods based on molecular descriptors only.33,35,36 The addition of structural information such as in 3D-QSAR, molecular docking or extensive pharmacophore methods has increased predictive capacity, typically yielding local models for structurally similar binders.33,36,37 However, these methods have difficulties dealing with the dynamic nature of the CYP catalytic site and substrate binding modes, providing a limited, static view of protein–substrate interactions.38
On the other hand, molecular dynamics (MD) based free energy calculation methods have the potential to provide an accurate estimate of the binding affinity, even for very flexible systems such as CYP enzymes.39−41 They are valuable methods in the design stage of drug discovery but due to their high CPU costs many of the pathway-based methods (including, e.g., Free Energy Perturbation (FEP) and Thermodynamic Integration (TI)) are unattractive for use in high-throughput settings especially when having to deal with multiple protein and/or ligand conformations that may contribute to binding (as in case of several CYPs).41,42 Alternatively, end-point methods such as Linear Interaction Energy (LIE) theory may provide a trade-off between accuracy and speed by estimating the solvation free energy between the two end points using linear response theory instead of multiple intermediate steps along a pathway.34,43−45 LIE requires empirical calibration of its scaling parameters. An advantage is that the sampling issue can be effectively and efficiently addressed using an iterative version of LIE41 that uses (re)weighted results from multiple short simulations starting from different ligand41 and protein conformations40 as obtained, e.g., by molecular docking.
The LIE method has been applied extensively over the last two decades in the prediction of protein–ligand binding affinities.45,46 Most LIE models have been trained using relatively small and curated data sets ranging from as few as 10–15 up to approximately 50 compounds, yielding predictive affinity models for compounds that are structurally similar or diverse but engage in similar interactions with the target protein.39,40,47−52 However, this situation may differ notably to the much larger data sets obtained in a typical industrial lead development and optimization project. Apart from their size, these compound libraries are often sparse and designed to increase possibilities to discover novel active ligands or scaffolds. It can become a challenge to create predictive LIE models with well-defined applicability domains39 for these data sets and, with high-throughput in mind, to do so in an automated fashion.
In the current study, we present a system of three quantitative binding affinity prediction models for CYP19A1. The models were trained using a data set of 132 putative CYP19A1 inhibitors with known inhibition constants obtained from a lead discovery study by Bayer AG that is subject to the same challenges described above in terms of compound size and chemical diversity. As an additional challenge, we aim here for a comprehensive approach for LIE model inference, parametrization and applicability assessment based on simulation and calibration data only, without the need for data set prefiltering by the user based on other information from experiment such as type of binding, which is not always available a priori. We have dealt with these challenges by developing an automated machine learning workflow that uses a stochastic approach to explore LIE-model parameter landscape. The iterative LIE (iLIE) method is used as a cost function in this search to prioritize the relative contribution of a binding orientation among a series of independent simulations of possible binding orientations. This approach allows to efficiently deal with flexible proteins (such as CYPs) that are able to bind ligands in multiple orientations. The probability of a compound to be part of a particular model is defined by a Bayesian approach that determines the maximum a posteriori estimates for the α and β parameters of the iLIE equation for a set of compounds, in order to maximize the number of explained compounds in one or more LIE models with predefined quality of the correlation between calculated and experimentally observed values for the ligand-binding free energies (in terms of RMSE and r2). We subsequently used protein–ligand interaction profiling to define the applicability domain of the resolved models in terms of protein–ligand interactions.
We show that our machine learning workflow is able to explain 86% of the employed CYP19A1 data set in three fully cross-validated LIE models with distinct combinations of α and β model parameters and an error margin below 3.0 kJ mol–1 of the experimentally observed binding free energy (i.e., within typical experimental accuracy53). Using simulation data only, steroid inhibitors were separated from nonsteroidal inhibitors, and the presented system of binding affinity models together with their protein–ligand interaction profiles provide a comprehensive means of predicting the binding affinity of unknown ligands for CYP19A1.
Methods
Molecular Dynamics (MD) averaged protein–ligand interaction energies for LIE affinity prediction were obtained using our previously published (semi)automated iterative LIE workflow.39,54,64 The methodological details on the protein and ligand structure preparation, molecular docking and MD simulation stages of this workflow are described below, together with the model calibration and validation strategies developed in this study.
Structure Preparation
The crystal structure of CYP19A1 with PDB ID 3EQM(55) was used after removal of the 4-androstene-3-17-dione (ASD) molecule, water oxygen atoms (none in the catalytic cavity) and crystallization buffer additives.
A data set of inhibition constants (Ki) for 132 putative CYP19A1 inhibitors with known stereochemistry (Tables S1 and S4, Supporting Information) was provided by Bayer AG, Berlin. Six of the 132 compounds entered the data set as duplicate (Table S1) but with independently measured Ki values. These compounds were used as internal validation in the study; and indeed most of the duplicates were found within the same (local) model, Table S1. Ki values were determined using the 3H2O release assay with [1β-3H]androstenedione as substrate56 in human placental microsomes according to FDA and UP guidelines, and were used to derive experimental binding free energies ΔGobs (Table S1). Assumptions taken in directly deriving ΔGobs from these inhibition data were supported by the obtained correlations for our final LIE models.
The initially minimized 3D structures for these compounds in their neutral form were generated from their canonical SMILES string using Molecular Operating Environment (MOE) version 2012.10,57 and the MMFF94 force field.58 Subsequent optimization and generation of MD topology and parameter files was performed using the Automated Topology Builder (ATB) server version 1.0.59
Docking and Clustering
Ligands were docked into the active site of CYP19A1 using the PLANTS (Protein–Ligand Ant System) docking software version 1.2.60 The target binding site was defined at the approximate center of the protein active site, at a position 0.7 nm distal to the heme iron center perpendicular to the heme plane, and with a size defined by a sphere with a radius of 1.1 nm. Docking poses were generated with speed 1 settings and evaluated using the ChemPLP61 scoring function. A maximum of 300 docked poses with mutual Root-Mean-Square Deviations (RMSDs) in atomic positions of more than 0.2 nm were retained and clustered using Principle Component Analysis (PCA), using the heavy atom positions as variables. After dimensionality reduction, the scores obtained were used for subsequent k-means clustering.62 During this analysis, an additional component or cluster was taken into account in case it would have led to a further increment of at least 5% of the explained variance in the space of the coordinates or scores, respectively. The medoids of the clusters obtained (4 to 8 per ligand) were chosen as representative binding conformations of the ligand in the CYP19A1 active site.
MD Simulations
The MD simulations for every representative binding pose obtained after docking and clustering were carried out using the GROMACS 4.5.4 package63 and an adapted version64 of the automated MD workflow script obtained from the GROMACS web server.65 The GROMOS 54A7 force field66 was applied to describe the protein and heme group in simulation. Ligand topologies were obtained with ATB59 as described above (see Structure Preparation subsection).
Each complex was energy minimized in a vacuum using steepest descent minimization and solvated in a simulation box with a Near-Densest Lattice Packing (NDLP) optimized volume67,68 (∼6300 solvent molecules) where water was described by the SPC model.69 7 Cl– ions were added to neutralize the system. The system was energy minimized (steepest-descent) and gradually heated up to 300 K in four NvT simulations of 20 ps, in which harmonic potentials were used for heavy-atom positional restrains in the sequence: 100/1000, 200/5000, 300/50 and 300/0 for temperature (K) and positional restraint force constant (kJ mol–1 nm–2), respectively, together with a Berendsen thermostat70 with a separate solute and solvent coupling time of 0.1 ps. Subsequently, three 20 ps equilibration runs were performed at NpT, using heavy-atom positional restraints on the solute with decreasing force constants of 1000, 100 and 10 kJ mol–1 nm–2. To further stabilize the system (as shown to be required before71), a short 500 ps unrestrained NpT simulation preceded the 2 ns production NpT simulation. A leapfrog algorithm72 was employed for integrating the equations of motion. Heavy hydrogens (with a mass of 4.032 amu)73 were used and all bonds were constrained using the LINCS algorithm,74 allowing a time-step of 4 fs. In all NpT simulations, a Berendsen thermostat70 was employed to maintain the temperature of the system close to its reference value of 300 K, using separate temperature baths for the solvent and solute degrees of freedom, with a coupling time of 0.1 ps. A Berendsen barostat70 with a coupling time of 0.5 ps and an isothermal compressibility of 7.5 × 10–4 [kJ mol–1 nm–3]−1 was used to maintain the pressure close to its reference value of 1.013 25 bar during NpT simulations. Van der Waals and short-range electrostatic interactions were explicitly evaluated every time step for pairs of atoms within a 0.9 nm cutoff, and a grid-based neighbor list was used and updated every 2 time steps. Long-range electrostatic interactions were included using a twin-range cutoff (0.9/1.4 nm) with reaction field correction.75 The relative dielectric constant for the medium outside the reaction field was set to 61. Center of mass motion removal was applied to both the solute rotation and translation components every 10 steps. Interaction energies and atomic coordinates were stored every 2 ps.
To evaluate average ligand interaction energies of the unbound ligands in water, each ligand was solvated in a NDLP optimized simulation box filled with approximately 625 SPC water molecules. No counterions were added. The MD protocol was identical to the one described for the simulations of the protein–ligand complex.
Iterative Linear Interaction Energy (iLIE) Method
In LIE, the predicted free energy of binding for a ligand in pose i to a protein (ΔGpredi) is estimated from MD-averaged electrostatic ⟨Vlig–surr⟩ and van der Waals interaction energies ⟨Vlig–surrvdw⟩ between the ligand and its surrounding as obtained in complex with the protein (bound,i) and free in solution (free). Following LIE theory,43,76
| 1 |
Values for the empirical LIE model parameters α and β are obtained by parametrization against a training set of compounds with experimentally determined binding affinities using linear regression analysis. An optional constant γ may be considered and has been used by others to account, e.g., for hydrophobicity of the binding site.76,77
In the iterative version of eq 1,41 the predicted protein–ligand binding free energy ΔGpred is calculated by combining results from N multiple short MD simulations starting from different ligand poses in the protein binding site that represent local minima on the potential energy surface of the protein–ligand complex, using a weighted sum (ΣiNWi). The relative weight (Wi) of each simulation (i) entering the sum is calculated according to its probability using78
| 2 |
with kB Boltzmann’s constant, T the temperature of the simulation system and N the number of independent simulations. Extending eq 1 by including the weighted sum of multiple short simulations yields41
| 3 |
The dependencies of the ΔGpredi on α and β requires eq 3 to be solved iteratively.41
Automated iLIE Binding Affinity Model Inference
LIE models can be trained automatically for an unknown data set of sufficient size by clustering ligands based on similarity in ΔVvdW and ΔVel energy patterns (defined as ⟨Vlig–surrvdw⟩bound (,i) – ⟨Vlig–surr⟩free and ⟨Vlig–surrel⟩bound (,i) – ⟨Vlig–surr⟩free), respectively) as a function of α and β model parameters (and possibly an optional constant γ) using the iLIE equation as a cost function in an iterative regression analysis. This is under the assumptions that (i) ΔVvdw and ΔVel in the data set show multivariate normality and low heteroscedasticity, (ii) there is similarity in the physio-chemical nature of the ligands and their interaction with the protein,76,79 and (iii) the analysis leads to a single model.
Our machine learning workflow expands this automatic training approach with the aim of finding the a posteriori estimates for the α and β parameters of the iLIE equation (eq 3) for a set of compounds that maximizes the number of explained compounds in one or more LIE models within predefined RMSE and r2 margins (RMSE ≤ 5 kJ mol–1 and r2 ≥ 0.6). The workflow (Figure 2) consists of a data curation and filtering stage to deal with assumption (i) (A in Figure 2), the main stochastic sampling routine using a Bayesian inference approach for parameter estimation (B), and a clustering stage where final LIE models are created (C). The latter two stages explore the existence of multiple (independent) models each describing a part of ligand physio-chemical/interaction space (assumptions (ii) and (iii)). The methods used in each of these stages are explained in more detail below.
Figure 2.
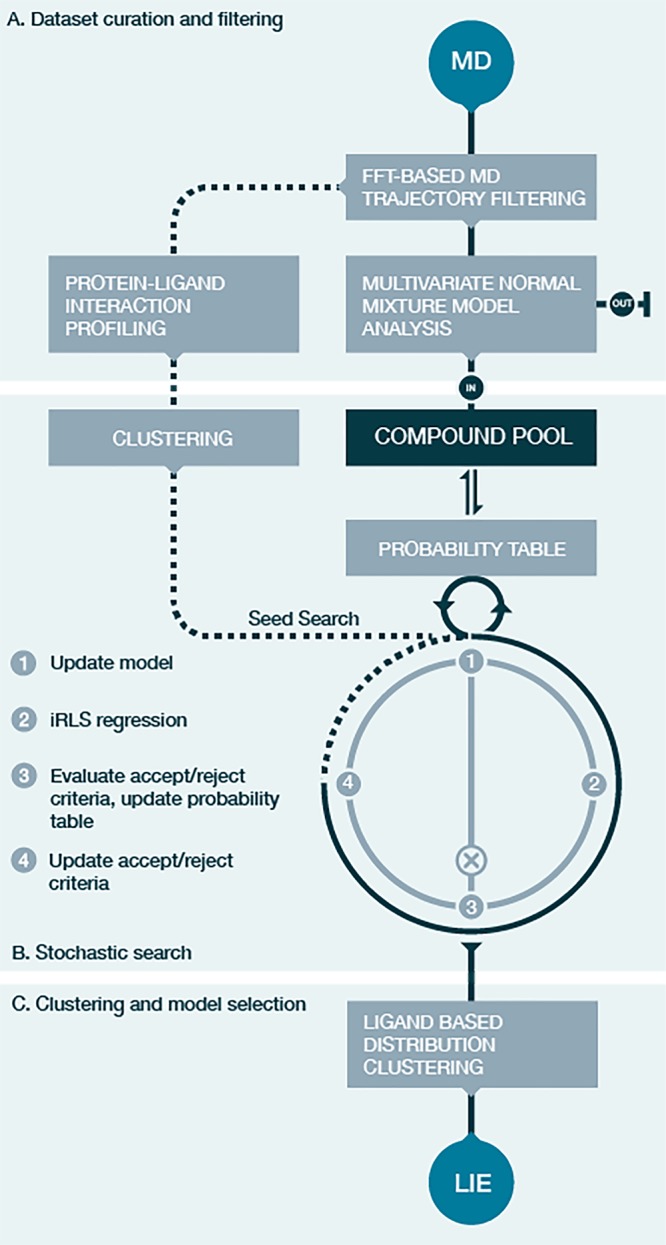
Schematic overview of the automated machine learning workflow aimed at finding the a posteriori estimates for one or multiple combinations of α and β parameters of the iLIE equation for a set of compounds, which maximize the number of explained compounds in one or more LIE models with predefined RMSE and r2 cutoffs. The “data set curation and filtering” stage (A) uses FFT based MD trajectory filtering to obtain stable average values of ΔVvdW and ΔVel. Ligand poses with average ΔVvdW/ΔVel pairs outside a 97.5% confidence interval (Figure S2) identified using multivariate normal mixture model analysis are labeled as outlier (out). Protein–ligand interaction profiling is performed on the FFT based stable energy trajectories (dashed lines). Ligand groups identified by the clustering of the interaction profiles are used as input to the stochastic search (B). The existence of LIE models for these clusters is explored during an iterative four-step stochastic search in which compounds are added to the evolving model from the global compound pool, according to a progressively updated probability using the iRLS weights of the added compounds at every iteration. The charted model landscape is clustered during the last workflow stage (C) and final models are selected.
MD Trajectory Filtering
To enforce the independence of the multiple simulations performed per ligand,78,80 average values ⟨Vlig–surrel⟩bound (,i) and ⟨Vlig–surr⟩bound (,i) are used in eq 3 as obtained from segments of the interaction energy trajectories that are constant in time within a predefined cutoff. For that purpose, Fast Fourier Transform (FFT) filtering was carried out followed by spline fitting.80 FFT smoothens the trajectory using a band-pass filter keeping the first 15 elements around the average in the frequency domain followed by inverse FFT to the original time domain (complex elements are discarded). Double spline fitting on the smoothened energy trajectory followed by gradient analysis identifies transitions when the absolute change in gradient is larger than 0.2 kJ mol–1 ps–1. The gradient was calculated using second-order central differences in the interior and first-order differences at the boundaries. For every protein–ligand MD simulation, the first continuous series of interaction energy data points with a minimum length of 100 ps is used in which both ⟨Vlig–surrvdw⟩bound and ⟨Vlig–surr⟩bound do not show fluctuations (i.e., gradients) larger than the above given cutoff value. Simulations for which no such series could be identified were discarded from further analysis. This event occurred once for a total of 688 CYP19A1 MD simulations performed during this study.
Multivariate normal mixture model analysis deploying the expectation maximization algorithm81,82 was used to identify (possibly multiple) normal distributed subpopulations in the dependent variables (ΔVivdw, ΔVi). We used the algorithm implemented in the sklearn.mixture Python library for this purpose with a “full” covariance type. Individual simulations were labeled as outlier and left out from further analysis if they have variable pairs outside a 97.5% confidence interval of the fitted χ2-distributions for all of the identified populations (out in Figure 2). When applicable, multiple subpopulations are treated as independent data sets in the remainder of the workflow.
Separation of Simulations
The use of multiple poses in iLIE to start short MD simulations allows the ligand to interact with the protein in multiple conformations. To guard the efficiency of the stochastic search when using multiple simulations per compound, a filtering step was performed based on an initial estimate of the α/β model parameter space by sampling a grid of predefined α/β parameters. This allowed to (i) discard results from simulations with low Wi representing poses unlikely to contribute to binding, and (ii) separate the combination of simulations with high Wi values in different regions of sampled α/β model parameter space into unique cases, to increase the chance for related cases to be grouped together in the same regression model. A square grid for the iLIE α and β model parameters was defined between a value of 0 and 1 with a grid spacing of 0.01. To compute ΔGpred and the Wi values, the iLIE equation (eq 3) was evaluated at each grid point using all poses for a given ligand.
Minima in model parameter space were defined as regions in which the deviation of ΔGpred from experiment is within ±5 kJ mol–1 (1.2 kcal mol–1), which equals about one pKi log unit.53 The propagation of the Wi’s for the independent simulations as a function of the model parameters in the defined region were analyzed and categorized as constant across minima or variable (illustrated in Figure 3). If in the latter case there was more than one simulation for which the gradient in Wi propagation is of opposite sign (Figure 3, poses 1 and 2), these are labeled as separate cases, together with a copy of the simulations of the same ligand for which Wi was identified to be constant with significant value (>0.1) across minima (Figure 3, pose 3). Simulations with Wi < 0.1 across minima were discarded for further analysis (Figure 3, pose 4). The separation of poses following this procedure was only used during the stochastic search.
Figure 3.

Propagation of weights Wi for four poses of a compound as a function of α or β model parameter, as obtained by solving the iLIE equation on a fixed grid of α and β parameters and focusing on the grid region where the difference between ΔGpred and experimentally observed ΔGobs is smaller than 5 kJ mol–1.
Stochastic Sampling
A stochastic approach was used to assess a posteriori estimates for combinations of α and β parameters for clusters of compounds in the data set, which together maximize the number of explained compounds in a minimum number of LIE models with predefined RMSE and r2 cutoffs. The stochastic search was seeded with subsets of compounds from every node in the hierarchical cluster tree obtained from protein–ligand interaction profile clustering (see the following). This approach increases the probability to identify predictive LIE models for compounds having similar protein–ligand interaction profiles. The compounds belonging to each node are first filtered for regression outliers according to the same protocol used during the stochastic search (step 3 in the following) to ensure that the start set is of maximum quality from a regression statistics point of view. Each stochastic search is an iterative process (Figure 2, middle pane) that consists of four steps:
-
1.
Increase the current compound set by 20% with a random sample from the main compound pool according to probability p defined as the probability of a compound (c) to be part of a model as p = 1 – (|{c|c ∈ R}|/|R|). R is the set that maintains a counter of the number of times each ligand was labeled as outlier according to the maximum likelihood estimates (weights) of the iterative Reweighted Least Squares regression (iRLS) while iterating (step 3). The probability table is initiated with a probability of 1 for each ligand.
-
2.
α and β model parameters for the new set of compounds are obtained from iRLS using ΔGobs values for the compounds as dependent variable. Andrew’s Wave was used as maximum likelihood estimator.83
-
3.
Use the iRLS weights to determine regression outliers (<0.75) and inliers (≥0.75). Update the global table of outlier counts (R) for each compound and re-evaluate the probability table (p, step 1). Evaluate the model with respect to the ΔGobs/pred RMSE and coefficient of determination r2 and accept the model if statistics are within 10% deviation of the previous model and the α and β parameters are within the range 0 to 1.
-
4.
If accepted, store the new model and start a new iteration until there are no more ligands to be added, statistics constraint limits are reached (5 kJ mol–1 RMSE, 0.6 r2), or no successful ligand addition could be made during 50 consecutive trials.
Model Clustering
The stochastic sampling yields a collection of models describing the probability of ligands and individual poses to occur together with respect to the LIE model parameters and within the predefined statistical model quality. For every ligand in the data set, we collected the iRLS regression weights of the models in which the ligand occurs, normalized by the RMSE of the respective model and summed over individual poses. These values where visualized in a heat map by clustering them with respect to α and β model parameters, illustrating the distribution of ligands over LIE models. The heat map was used to visually select the ligand sets that maximize the number of explained compounds in one or more LIE models. The final models best representing the data set were trained using the iLIE equation (eq 3) with the ligand sets selected from the heat map. Representative simulations for each compound were selected by first training a model using all poses, after which we selected the simulations within a range of 0.2 of the most dominant simulation according to their weights Wi (eq 2). These simulations were used for retraining the final model.
Protein–Ligand Interaction Profiling
Protein–ligand interactions in all MD frames for all ligand poses were analyzed using in-house python software. The software aims to identify protein–ligand contacts as belonging to the following eight interaction types using rule-based protocols that are described in the Supporting Information: hydrogen bonded contacts (hb-ad, hb-da), water mediated hydrogen-bonded contacts (wb-da, wb-ad), halogen-bonded contacts (xb), salt bridging contacts (sb-np, sb-pn), cation-π interactions (pc), aromatic π- (ps) or T-stacking (ts), hydrophobic interactions (hf) and heme-coordination (hm, which is not explicitly accounted for during our classical simulations). Interaction subcategories include donor–acceptor (da), acceptor–donor (ad), negative–positive (np) and positive–negative (pn), for ligand and protein residue, respectively.
Relationships between the interaction profiles within pairs of MD simulations were evaluated by encoding the ligand-protein interaction profile data for each independent simulation as a structured key and by building a pairwise similarity matrix based on them using the following protocol:
-
1.
Discard all protein–ligand interactions that occur for less than 10% of the simulation time, to reduce noise in further analysis due to infrequent interactions.
-
2.
Normalize the frequency of occurrence of the interaction types by their relative abundance in the full data set, to prevent abundant hydrophobic interactions to have an artificially large influence on the final similarity metric.
-
3.
Collect the unique protein residues involved in protein–ligand interactions from all filtered profiles after step 2.
-
4.Collect the resulting (sorted) residue numbers form the primary slots of the structured key. Each slot is divided in 2 × 12 registers subsequently containing:
-
a.The frequency with which any of the 12 interaction types (comprising the 8 main groups and their subcategories mentioned above) occur between the ligand and the given residue over the course of the simulation.
-
b.A list of ligand SYBYL atom types involved in the interaction in the corresponding first 12 registers (4a).
-
a.
-
5.The similarity between two structured keys is evaluated as the sum of similarities between each register, normalized by key length (number of residues). The similarity is calculated differently for the two registry types mentioned in step 4:
-
a.For every residue interaction frequency (4a), the similarity is assigned 1 if they are identical, 0 if any of the registers equals 0, and the minimum frequency of occurrence otherwise.
-
b.Similarity is defined as the percentage of similarity in SYBYL atom types involved in the interaction (4b), multiplied by the frequency of occurrence of the corresponding type in the first 12 registers (4a). This multiplication ensures that the weight of similarity in SYBYL atom types for a given interaction type in the final similarity metric is never larger than that of the interaction type itself.
-
a.
The resulting pairwise similarity matrix is used as input for a hierarchical agglomerative cluster analysis as implemented in the Python scipy.cluster.hierarchy(84) to identify groups of (independent) ligand simulations that show similar interaction profiles, which were used as seeds for the stochastic approach for iLIE model regression described above.
Results and Discussion
The 132 compounds in the data set (Tables S1 and S4 of the Supporting Information) were selected in a lead discovery study as promising AI candidates. The 12 steroid and 121 nonsteroid compounds can be classified as resembling the second to fourth generation of clinical AI’s (Figure 1) including Letrozole (compound 2), Anastrazole (compound 9) and Fadrozole (compound 13). All nonsteroid compounds have one azole ring of which the basic nitrogen atoms (mostly imidazole or triazole) have the potential to apically coordinate the heme iron.85−89 Most of them have an aryl or apolar cyclic moiety mimicking the steroid ring of the substrate, and one or two nitrile groups. Apart from these common features there is considerable structural diversity among the ligands with respect to the topological arrangement and chemical nature of the functional groups.
We initially attempted to train a single iLIE model for our data set of 132 compounds (using all docked poses and a 3/4 test-to-train set ratio), which resulted in models with negative r2 values and RMSE values higher than 7.0 kJ mol–1. Subsequent attempts to group compounds based on their molecular structures into iLIE binding affinity prediction models (i.e., based on SAI/NSAI compound groups or using structurally similar compounds derived from a chemical fingerprinting and clustering analysis) yielded local models with <10 ligands when using all ligand poses, leaving most of the data set unexplained. An initial estimate of optimum α/β parameter ranges was obtained by solving the iLIE equation for every ligand individually, taking into account all poses, on a grid of fixed α/β parameters. The range corresponding to a prediction error in ΔGpred of less than 5 kJ mol–1 for a maximum number of compounds is located in α and β ranges between 0 and 0.5 and 0.55–0.95, respectively, Figure 4. In this broad region, even the most densely populated area of model parameter combinations comprises no more than 50% of the ligands. Furthermore, 101 of the 132 ligands have at least two different dominant binding poses in different parts of this region based on the change in the distribution of weighted propensities Wi for the independent simulations. These observations illustrate the challenges mentioned before associated with LIE model development for diverse and large sets of training compounds with respect to their physicochemical variation, the possibility of ligands to bind in multiple orientations, and the unlikely existence of a single predictive model with sufficient coverage.
Figure 4.
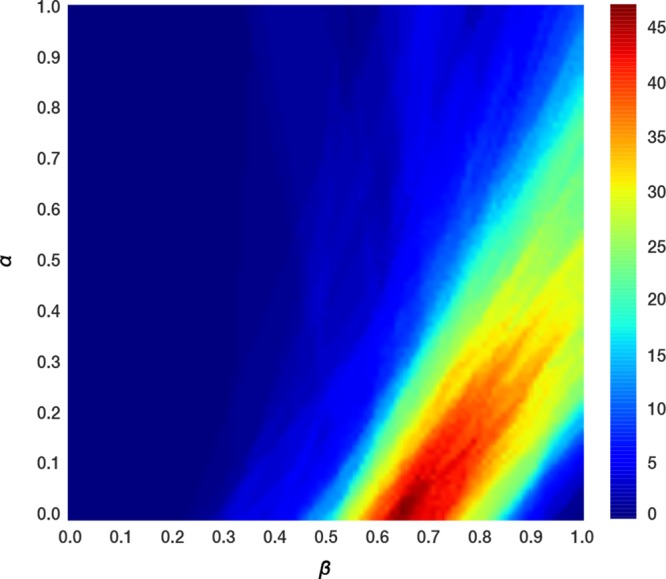
LIE α and β model parameter scan performed on each of the 132 ligands individually by solving the iLIE equation (eq 3), for every point on a square grid of α and β model parameters between a value of 0 and 1 with a grid spacing of 0.01. The color gradient highlights the percentage of ligands having a ΔGpred value within 5 kJ mol–1 of ΔGobs for a given combination of α and β.
To explore the existence of a small subset of iLIE models that cover a substantially larger part of the data set and that have a well-defined protein–ligand interaction profile we developed a machine learning workflow using information extracted from the MD-trajectories only. The philosophy behind the method (see Methods section for details) is described below together with the results obtained after applying it to the data set of aromatase inhibitors.
Data Set Curation and Filtering
⟨Vlig–surrel⟩ and ⟨Vlig–surr⟩ derived from the independent MD simulations used for iLIE are assumed to be averages over well-separated parts of the protein–ligand interaction energy surface.78 Progressive changes in interaction energy values during a simulation may indicate an unstable system or a conformational transition of the ligand away from its initial position. As a remedy, constant interaction energy trajectories were obtained by filtering the MD energy trajectories for large fluctuations using a spline-fitting procedure on the FFT filtered time series.80 Convergence of interaction energy terms is illustrated for a randomly picked subset of simulations in Figure S1 of the Supporting Information.
Subsequently, we performed a multivariate normal distribution analysis to identify possible multiple independent and/or partly overlapping normal distributed subpopulations in the dependent variables (i.e., ΔVvdW and ΔVel derived after FFT filtering, Tables S2 and S3). Using a 97.5% confidence interval, 18 protein–ligand simulations (29 without FFT filtering) were found to be not part of the single identified distribution (Figure S2, Supporting Information), which included all simulations for ligands 156 and 189. These two compounds were therefore not included in further analyses.
From Figure S2, detected outliers (which were left out from further analyses) involve among others the combinations of the ΔV values for compounds 156, and poses 2 of compounds 69 and 300, and pose 3 of compound 148. In these cases, the interaction profiles obtained from simulation indicated ligand interactions with protein residues located near the entrance channel to the catalytic cavity (between β-sheet 4 and helix F) instead of the protein residues and heme group located around the center of the catalytic cavity as defined for docking (see Methods section). Ligands 189 and 233 were found to interact with both regions. From the substantially different average energies and interaction profiles of the simulations involving these ligands, both can be labeled as outliers with respect to the simulations of other ligands in the data set and were left out from further analyses as well.
Stochastic Sampling
The stochastic sampling stage of our machine learning workflow (Methods section and Figure 2C) was initiated with the filtered and curated data set described above and seeded with ligand pose clusters derived from hierarchical clustering of the protein–ligand interaction profiles obtained from the MD trajectories (described in the following).
For the system considered, the full workflow is completed in ∼10 min on one core of a 4 core 2,2 GHz Intel Core i7 desktop machine. The results are visualized as a α and β heat map (Figure 5) showing clusters of ligands with an associated likelihood (color gradient) of constituting a model together for the respective combinations of sampled α and β model parameters. 12 of the 130 ligands used in the search (compounds 159, 166, 182, 194, 210, 220, 223, 232, 233 and 324) could not be placed in any model with a probability larger than 0.5 and are not shown in the heat maps. The ligand sets used to train the final representative models were visually selected based on the clusters in the heat map. This selection is the only manual step within our otherwise automated machine learning workflow.
Figure 5.

Results of the stochastic sampling of α and β model-parameter space for the data set of 132 CYP19A1 inhibitors. The likelihood for a compound (labeled by ID on the x-axis) to be part of a model in a given β (panel A) and α (panel B) range is shown as a heat map. The color gradient is a dimensionless measure of the likelihood calculated as the summed iRLS regression weights of a ligand in each of the sampled models of the stochastic search divided by the Root-Mean-Square Error (RMSE) of the model. The measure of likelihood is plotted as a function of the model α and β parameters with a bin size of 0.02. Regions of highest density used to train the final models are labeled as models 1 to 4 (red dashed rectangular boxes).
Selection of Representative LIE Binding Affinity Models Containing More Than 10 Compounds
The machine learning workflow sampled combinations of α/β model parameters that fall within the broad model-parameter region identified in the grid scan (Figure 4). The workflow identified four posterior distributions (clusters) with centers containing more than 10 components (Figure 5, red dashed rectangular boxes labeled 1 to 4). These four clusters served as training set for representative LIE binding affinity models (Table 1).
Table 1. Final Set of Representative LIE Binding Affinity Models Derived from the Stochastic Approximate Inference of Model Parameters for the Data Set of 132 Putative Aromatase Inhibitorsa.
| Model | n | α | β | γ | RMSE | r2 | q2LOO | SDEPLOO | r2bstr | RMSEbstr |
|---|---|---|---|---|---|---|---|---|---|---|
| 1, center | 16 | 0.203 | 0.663 | 1.30 | 0.95 | 0.94 | 1.47 | 0.960.01 | 1.100.17 | |
| 1, center | 16 | 0.200 | 0.627 | –2.59 | 1.27 | 0.95 | 0.93 | 1.56 | 0.930.10 | 1.410.72 |
| 1, full | 31 | 0.233 | 0.675 | 2.36 | 0.86 | 0.83 | 2.57 | 0.870.03 | 2.230.22 | |
| 1, full | 31 | 0.226 | 0.576 | –7.32 | 2.11 | 0.89 | 0.85 | 2.41 | 0.890.04 | 1.980.23 |
| 2, center | 13 | 0.161 | 0.796 | 1.69 | 0.89 | 0.82 | 2.14 | 0.900.03 | 1.540.28 | |
| 2, center | 13 | 0.162 | 0.830 | 2.45 | 1.69 | 0.89 | 0.78 | 2.38 | 0.900.03 | 1.480.32 |
| 2, full | 52 | 0.107 | 0.748 | 2.59 | 0.76 | 0.76 | 2.59 | 0.730.03 | 2.710.10 | |
| 2, full | 52 | 0.101 | 0.696 | –2.54 | 2.55 | 0.77 | 0.74 | 2.69 | 0.750.02 | 2.650.07 |
| 3, center | 15 | 0.134 | 0.893 | 1.47 | 0.93 | 0.91 | 1.69 | 0.930.02 | 1.460.16 | |
| 3, center | 15 | 0.125 | 0.838 | –3.23 | 1.42 | 0.93 | 0.86 | 2.11 | 0.940.05 | 1.300.12 |
| 3, full | 31 | 0.108 | 0.868 | 1.68 | 0.92 | 0.91 | 1.80 | 0.920.02 | 1.620.17 | |
| 3, full | 31 | 0.110 | 0.877 | 0.495 | 1.68 | 0.92 | 0.90 | 1.89 | 0.920.02 | 1.660.11 |
Model statistics are reported for three representative LIE binding affinity models trained with and without γ parameter (kJ mol–1) for ligands belonging to the full model cluster (full) and the cluster center only (center, within 75% confidence interval). Root-Mean-Square Error (RMSE, kJ mol–1) and coefficient of determination (r2) model statistics are reported for the model, as well as for the bootstrap cross-validated model (bstr) and Leave-One-Out (LOO) cross-validated model (as Standard Error in Prediction (SDEP, kJ mol–1) and cross-validated r2 (q2)). Bootstrap cross validation was performed using a 20-fold random sampling with a training set of 75% of the model data set.
Model 1 with a cluster center of 16 ligands is able to account for a total of 31 ligands and shows little overlap with the other models in terms of α/β model parameter values. Cluster center 4 is able to account for 61% of the ligands not accounted for by model 1, with an r2 of 0.75 and RMSE of 3.3 kJ mol–1. Compared to cluster center 1, the ligands that are part of the area sampled by cluster center 4 show considerably more variation in the values for model parameters that can constitute a predictive model, in particular for α. With α close to 0, the predictive capacity of the model is mostly determined by the electrostatic component of the LIE equation. Furthermore, the dense population of ligands in cluster center 4 is predominantly determined by models with statistics in the less predictive half of the predefined RMSE and r2 margins (with 0.6 ≤ r2 < 0.8 and 3.0 < RMSE ≤ 5 kJ mol–1).
However, the method did resolve two less populated clusters (Figure 5, cluster centers 2 and 3) that show little overlap but are contained within cluster 4. The models derived from these two centers have more favorable statistics than model 4 and together explain 95% of the ligands not explained by model 1. Model 2 and 3 cannot be combined without significantly worsening model statistics (r2= 0.59, RMSE = 3.5 kJ mol–1 for the combined model, versus r2 = 0.76, RMSE = 2.6 kJ mol–1 and r2 = 0.92, RMSE = 1.7 kJ mol–1 for models 2 and 3, respectively; Table 1). Bootstrap analysis confirmed the two models to be derived from partly overlapping but distinct clusters (Figure S3, Supporting Information).
The relatively low values for γ when including an offset parameter (Table 1) indicates that the binding affinity can be modeled in terms of α and β only. Note that β values are higher than the theoretical value43 of 0.5 and than the range of values previously reported for CYP LIE models (0.0–0.5),39−41,54,90 whereas α values are relatively low compared to the corresponding range of values (0.2–0.6),39−41,54,90Table 1. The low α and high β values suggest that the binding represented by the models is mostly determined by electrostatic interactions.
On the basis of the above, models 1, 2 and 3 were selected as representative models. Together, they cover 86% of the original data set with a RMSE between experimental and calculated binding free energy below 2.6 kJ mol–1, which is well within the typical experimental error.53 The LIE models are robust from a statistical point of view shown, e.g., by Leave-One-Out and bootstrap cross-validation, Table 1. Although presented as three separate models, there is overlap in the distributions they were derived from, in particular for models 2 and 3. This becomes evident from the ΔGpred values and errors in prediction (Figure 6, Figure S4 Supporting Information) when making predictions for the same compound using each of the three models. The relationship is also reflected in the protein–ligand interaction profiles of the compounds used to train the models (see below).
Figure 6.

Correlation between the predicted (ΔGpred) and observed (ΔGobs) binding free energies in three models (panels A to C for models 1 to 3, respectively) that were trained using the results from stochastic sampling. The solid diagonal lines indicate ideal correlation and the dashed lines indicate upper and lower error margins of 5 kJ mol–1. Blue filled circles correspond to compounds belonging to the cluster centers as indicated by the red boxes 1–3 in Figure 5, and red filled circles correspond to remaining compounds in that cluster, and gray filled circles correspond to the remaining compounds of the data set as predicted by the model. Blue and red filled crosses in panel C indicate low-affinity Fadrozole-like compounds.
Protein–Ligand Interaction Profiling
The classification of ligand-amino acid interactions occurring for more than 50% of the simulation time for all ligands identified 4 polar interaction sites (“hotspots”, Figure 7) comprising (i) R115 and M374; (ii) Q225; (iii) D309 and T310; and (iv) L477, S478 and the heme prosthetic group (497); and 3 apolar contact sites: (i) I133, F134; (ii) V370; and (iii) F221, W224. This particular combination between apolar and polar interaction sites has also been identified in previous crystallographic, mutagenesis and computational studies on CYP19A1.55,71,91−93 Below, we describe the dominant protein–ligand interactions observed in the simulations used for models 1–3 introduced above, starting with the dominant interactions for models 1 and 3, respectively.
Figure 7.

Cartoon representation of Cytochrome P450 19A1 (PDB code 3EQM(55)) with the natural substrate 4-androstene-3-17-dione (ASD, cyan stick representation) bound. Protein residues (stick representation) involved in polar protein–ligand interactions in more than 50% of the simulation time are grouped in four hotspots (indicated in red, yellow, green and purple), including the heme group (HEME).
Model 1: The steroid based aromatase inhibitors, except ligand 210, form the basis of model 1. The starting poses for the simulations with highest Wi values for these ligands are similar to the natural substrate ASD with respect to binding orientation (i.e., as in the X-ray structure PDB ID 3EQM) and interaction profile,55 cf. Figure S5 of the Supporting Information. Figure 8, panel A shows that hydrogen-bonded contacts dominate the polar interactions, between two carbonyl groups of the steroid (position A3 and D17) and residues M374 and D309 or T310, although not necessarily simultaneously. These findings are in line with previous biochemical studies on steroid binding to CYP19A1.94−96 Steroid 210 has a bulky trifluoride group hindering formation of both hydrogen bonds, resulting in a binding orientation similar to the natural substrate but resulting in a prediction error of 19.0 kJ mol–1 by model 1. Interestingly, 82% of the NSAI ligands that are uniquely described by this model 1 form hydrogen-bonded interactions with M374 and/or D309/T310 as well (forming these interactions in on average 40% of simulation time). They predominantly do so using a carbonyl O atom or N atom acceptor part of a nitrile group and/or azole ring. In particular the presence of a nitrile group allows for simultaneous hydrogen-bond interactions with the aforementioned protein residues. In addition, there are hydrophobic contacts between residues F221 and W224 and aromatic or cyclic apolar ligand groups, and hydrogen bonding interactions with Q225 (Figure 8A). Figure 8 also illustrates that potential heme-coordinating poses of the NSAI ligands are observed less frequently in model 1 in comparison to the other two models.
Figure 8.

Protein–ligand interaction profiles for the compounds in models 1–3 (Table 1) derived from the stochastic approximate inference. The relative interaction frequencies for each protein residue–ligand interaction are represented by vertically stacked bars where the bar colors correspond to a specific interaction type as listed in the graph legend. Hydrophobic contact frequencies are divided by 10 because of their relative abundance with respect to the other classified interactions.
Model 2 and 3 exclusively contain NSAIs. The contact profiles include hydrogen-bonded interactions to the same residues of the protein as in model 1 but the hydrogen bond types and frequencies differ, Figure 8.
Model 3: Hydrogen bonding to residues Q225, M374 as well as S478 predominantly involves the (single) nitrile group of the compounds. The compounds almost exclusively contain imidazole groups of which the N atoms can be involved in hydrogen bonding to T310 and in heme coordination (cf. Figure 8).
22 of the 33 compounds have a Letrozole (Figure 1e) like topology with one imidazole group, one nitrile containing aryl group and a variable apolar moiety. This topology is complementary to the active site and enables contacts with various interaction hotspots simultaneously (Figure 8C). The Letrozole-like compounds can adopt multiple binding orientations due to rotational symmetry, thereby preserving interactions. This is confirmed by the finding that per ligand, several simulations contribute to the calculated binding free energy (Table S1, last column). The importance of interactions for the (predicted) affinity becomes apparent when comparing the compounds in model 3 in the low-affinity region of the correlation plot (Figure 6C, crosses) with those in the high-affinity region. The low-affinity compounds are almost exclusively Fadrozole (Figure 1b) like compounds with either an imidazole or imidazopyridine group that is unable to interact with as many hotspots simultaneously as the Letrozole-like compounds that are in the high-affinity region.
Model 2 shows more variation in the scaffold and functional groups of the compounds included when compared to model 3. Differences are the presence of halogens, carbonyl and hydroxyl groups in model 2 compounds and the replacement of the imidazole by a triazole group, the replacement of the variable apolar moiety by an aryl or thiophene group, and the replacement of a nitrile group by a halogen or keto group. Although the compounds belonging to the cluster center of model 2 are unique to it from a statistical point of view (in terms of α/β combination and other model statistics, Table 1 and Figure S3, Supporting Information), the compounds of the model that are under- and overpredicted are more model 3- and model 1-like, respectively, in terms of topology (Table S1) and interaction profile. As such, model 2 positions itself in between model 1 and 3 as shown by the interaction profile (Figure 8B) and model statistics (Table 1).
In conclusion, the interaction profiles of the MD trajectories provide a basis for understanding and establishing the applicability domain of the resolved models in terms of observed protein–ligand interactions. These differences in interactions can explain why similar compounds fall into different models. For example, compounds 333, 318 and 246 only differ by small variation in their benzylic para-substituent but they are part of models 1, 2 and 3, respectively (Table S1). The p-acetylated compound 318 was found to contribute to model 2 with four different binding poses and accordingly it interacted with a variety of protein residues. In contrast, the slightly larger compound 333 and the nitril containing compound 246 only contributed with a single pose, which mutually differed in terms of binding orientation and interacting residues (310 for compound 246 vs 115 and 309 for compound 333). As another example, a Cl-substituted letrozole analogue (compound 139) contributed to model 2, with its poses differing from the pose that contributed most (to model 1) and that showed the F- and Br-substituents of counterpart compounds 200 and 98 on a distance from methionine 374 suited for halogen-bonding interactions, Table S1.
We note that MD sampling was found to be necessary for the purpose of establishing applicability domains: when performing interaction profiling based on the docked MD starting conformations only, we could, e.g., not classify interaction profiles for models 2 and 3. Despite the diversity in the data set, our machine-learning workflow was successful in grouping together compounds with a steroid scaffold in model 1 and compounds with a Letrozole like topology composed out of an imidazole group, one nitrile containing aryl group, and a variable apolar moiety in model 3. In addition, other compounds present in these models that do not share the same topology show similar interaction profiles in terms of interaction hotspots. These structural characteristics and corresponding interaction profile define the applicability domain for models 1 and 3, which is beneficial for developing predictive LIE models as illustrated by their good model statistics. In contrast, model 2 is more diverse with topologically similar compounds being represented by small subsets (≤5) of compounds. The key protein residues interacting with the compounds comprised in model 2 are the same as for model 3. This could explain e.g. why compounds 78 and 80 fall in models 2 and 3, respectively (Table S1). However, the type of hydrogen-bond acceptors is different (Figure 8), which positions model 2 in between model 1 and 3 in terms of LIE parameters. The diversity in molecular structures covered by the three models is illustrated by a selection of compounds from the models as presented in Figure S6 of the Supporting Information.
Protein–ligand interaction profile results illustrate that the number and type of polar protein–ligand interactions are determinative for the LIE empirical parameters and that the variation in the data set results in a system of three overlapping but distinct models rather than one global model. The importance of polar contacts is shown by the consistent value for α between 0.10 and 0.24 as frequently reported before79,97,98 and a β value as a function of the ligand functional groups97,98 rather than the theoretical value of 0.5.43 With values markedly larger than 0.5, the ligand functional group dependent value for β found in this study are different than those previously proposed for other systems,97,98 which may well be due to choices in the force field and system set up80 and which is in line with spread in LIE parameters previously reported among a variety of other CYPs.39−41,54
All nonsteroidal ligands have one azole ring of which the basic nitrogen atoms have the potential to apically coordinate the heme iron. Despite the inability of commonly used force fields to accurately account for the energetics of heme coordination, a considerable number of compounds in model 2 and 3 adopted a potential coordinating pose during simulation based on interaction profile data (Figure 8, gray bars).
Conclusions
In this work, we present binding affinity prediction models for steroidal and nonsteroidal inhibitors of CYP19A1 aromatase using our automated LIE workflow.39,54,64 The data set of 132 compounds used for training LIE models was acquired from a lead discovery study and poses challenges to the calibration of empirical free energy models because of the sparsity of and large structural diversity within the data set and due to the catalytic site malleability and the substrate and interaction promiscuity of the CYP protein. Nevertheless, it is a data set representative for training of LIE or other models in industrial or other applied settings. In spirit of our automated iLIE approach, we developed an automated machine learning workflow aimed at exploring the model parameter landscape and at maximizing the number of data set compounds explained in one or few LIE models. The method was successful in accounting for 86% of the data set in 3 models showing high correlation between calculated and observed values for binding free energy (r2 of 0.86, 0.76, 0.92; RMSE of 2.36, 2.59, 1.68 kJ mol–1 for the three models, respectively). The use of a maximum likelihood estimator and Bayesian inference maximize the chance of identifying posterior predictive distributions describing the data set and limiting the chance of overfitting of the LIE models. The robustness of the models is illustrated by small variations after bootstrap analysis (segmentation cross validation) and by good leave-one-out cross validation statistics (q2LOO of 0.83, 0.73, 0.92; SDEPLOO of 2.57, 2.59, 1.80 kJ mol–1, respectively). The ligand clusters leading to the final set of models are visually selected from the heat map (Figure 5). In a future iteration of the software we aim to also automate this final step of our workflow, thereby completing its automation.
The resolved clusters, from which the three models were derived separate the data set into a SAI model and two NSAI models that have distinct α and β model parameters. With respect to polar interactions, compounds in model 1 predominantly interact with D309/T310 and/or M374 by hydrogen-bonded interactions with carbonyl oxygens as ligand acceptors, which is common for steroids interacting with CYP19A1.55,71,94−96 On the other hand, compounds in model 3 interact with Q225, M374 and S478 via hydrogen bonding involving the nitrile nitrogen, and with T310 by hydrogen bonding via an imidazole N atom. Rotationally symmetric binding conformations of these compounds that preserve the overall interaction profile are frequently included in the binding affinity prediction. This underlines the flexible nature of the CYP19A1 catalytic cavity and the ability of the iLIE approach to account for this.
Using our machine learning workflow, we have been able to calibrate LIE binding affinity prediction models with a protein–ligand interaction based applicability domain that explains 86% of the data set using experimental affinity data and information extracted from MD only. The detailed structure and interaction based applicability domain provides a frame of reference for the user to determine if a model, and which model, can be used for binding affinity prediction of a novel compound based on the protein–ligand interaction profile and provides an estimate of the reliability of the prediction. The automated nature of the workflow allows it to be used together with our automated iLIE workflow, together creating a pipeline to apply LIE in high-throughput settings.
Acknowledgments
We gratefully acknowledge Thomas Steger-Hartmann and Nikolaus Heinrich for their help in supplying us with experimental data from Bayer AG, and Martin Stroet and Alan Mark for support in ATB topology file generation. This work was supported by the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115002 (eTOX), resources of which are composed of financial contribution from the European Union Seventh Framework Programme (FP7/20072013) and EFPIA companies in kind contribution, and by The Netherlands Organization for Scientific Research (NWO, VIDI grant 723.012.105).
Glossary
Abbreviations:
- AI
Aromatase Inhibitor
- ASD
4-Androstene-3-17-dione
- ATB
Automated Topology Builder
- CYP
Cytochrome P450
- DDI
Drug–Drug Interaction
- FEP
Free Energy Perturbation
- iRLS
Iteratively Reweighted Least Squares
- LIE
Linear Interaction Energy
- MD
Molecular Dynamics
- NSAI
Non-Steroidal Aromatase Inhibitor
- QSAR
Quantitative Structure–Activity Relationship
- RMSD
Root-Mean-Square Deviation (in atomic positions)
- RMSE
Root-Mean-Square Error
- TI
Thermodynamic Integration
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.7b00222.
Description of the rule based protocol for resolving 8 types of protein–ligand interactions used for the interaction profiling analysis described in this work, a selection of time series of Vlig–surrel and Vlig–surr (Figure S1), multivariate normal distribution analysis of ΔVvdW and ΔVel energy values (Figure S2), bootstrap cross-validation results for the combined model 2 and 3 (Figure S3), ΔGpred error values (residuals) for each compound when compared to experiment and when calculated using model 1, 2 or 3 (Figure S4), stick representation of the dominant binding pose of 10 steroidal aromatase inhibitors explained by LIE binding affinity prediction model 1 (Figure S5), molecular structures of compounds in models 1, 2 and 3 (Figure S6), data set of 132 putative aromatase inhibitors with experimentally determined inhibition constants, provided by Bayer AG (Table S1), averaged difference in the Coulomb component of the nonbonded protein–ligand interaction energy for up to 8 selected docking poses for the data set of 132 putative aromatase inhibitors (Table S2), averaged difference in the van der Waals component of the nonbonded protein–ligand interaction energy for up to 8 selected docking poses for the data set of 132 putative aromatase inhibitors (Table S3) (PDF)
Excel spreadsheet containing data from Table S1 together with the canonical SMILES string for all ligands (Table S4) (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Brodie A. M.; Njar V. C. Aromatase inhibitors and their application in breast cancer treatment. Steroids 2000, 65, 171–179. 10.1016/S0039-128X(99)00104-X. [DOI] [PubMed] [Google Scholar]
- Eisen A.; Trudeau M.; Shelley W.; Messersmith H.; Pritchard K. I. Aromatase inhibitors in adjuvant therapy for hormone receptor positive breast cancer: a systematic review. Cancer Treat. Rev. 2008, 34, 157–174. 10.1016/j.ctrv.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Brodie A. Aromatase inhibitors in breast cancer. Trends Endocrinol. Metab. 2002, 13, 61–65. 10.1016/S1043-2760(01)00529-X. [DOI] [PubMed] [Google Scholar]
- Kellis J. T.; Vickery L. E. Purification and characterization of human placental aromatase cytochrome P-450. J. Biol. Chem. 1987, 262, 4413–4420. [PubMed] [Google Scholar]
- Miller W. R.; Mullen P.; Sourdaine P.; Watson C.; Dixon J. M.; Telford J. Regulation of aromatase activity within the breast. J. Steroid Biochem. Mol. Biol. 1997, 61, 193–202. 10.1016/S0960-0760(97)80012-X. [DOI] [PubMed] [Google Scholar]
- Chen S. A.; Besman M. J.; Shively J. E.; Yanagibashi K.; Hall P. F. Human aromatase. Drug Metab. Rev. 1989, 20, 511–517. 10.3109/03602538909103557. [DOI] [PubMed] [Google Scholar]
- Ryan K. J. Conversion of androstenedione to estrone by placental microsomes. Biochim. Biophys. Acta 1958, 27, 658–659. 10.1016/0006-3002(58)90408-6. [DOI] [PubMed] [Google Scholar]
- de Groot M. J.; Ekins S. Pharmacophore modeling of cytochromes P450. Adv. Drug Delivery Rev. 2002, 54, 367–383. 10.1016/S0169-409X(02)00009-1. [DOI] [PubMed] [Google Scholar]
- Simpson E. R.; Clyne C.; Rubin G. Aromatase-a brief overview. Annu. Rev. Physiol. 2002, 64, 93–127. [DOI] [PubMed] [Google Scholar]
- Brueggemeier R. W.; Richards J. A.; Joomprabutra S.; Bhat A. S.; Whetstone J. L. Molecular pharmacology of aromatase and its regulation by endogenous and exogenous agents. J. Steroid Biochem. Mol. Biol. 2001, 79, 75–84. 10.1016/S0960-0760(01)00127-3. [DOI] [PubMed] [Google Scholar]
- Elledge R. M.; Osborne C. K. Oestrogen receptors and breast cancer. BMJ. 1997, 314, 1843–1844. 10.1136/bmj.314.7098.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsey J. L.; Gammon M. D.; John E. M. Reproductive factors and breast cancer. Epidemiol Rev. 1993, 15, 36–47. 10.1093/oxfordjournals.epirev.a036115. [DOI] [PubMed] [Google Scholar]
- Osborne C. K. Steroid hormone receptors in breast cancer management. Breast Cancer Res. Treat. 1998, 51, 227–238. 10.1023/A:1006132427948. [DOI] [PubMed] [Google Scholar]
- Brueggemeier R. W.; Hackett J. C.; Diaz-Cruz E. S. Aromatase inhibitors in the treatment of breast cancer. Endocr. Rev. 2005, 26, 331–345. 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
- Osborne C. K. Aromatase inhibitors in relation to other forms of endocrine therapy for breast cancer. Endocr.-Relat. Cancer 1999, 6, 271–276. 10.1677/erc.0.0060271. [DOI] [PubMed] [Google Scholar]
- Masamura S.; Adlercreutz H.; Harvey H.; Lipton A.; Demers L. M.; Santen R. J.; Santner S. J. Aromatase inhibitor development for treatment of breast cancer. Breast Cancer Res. Treat. 1995, 33, 19–26. 10.1007/BF00666067. [DOI] [PubMed] [Google Scholar]
- Ahmad I.; Shagufta Recent developments in steroidal and nonsteroidal aromatase inhibitors for the chemoprevention of estrogen-dependent breast cancer. Eur. J. Med. Chem. 2015, 102, 375–386. 10.1016/j.ejmech.2015.08.010. [DOI] [PubMed] [Google Scholar]
- Recanatini M.; Cavalli A.; Valenti P. Nonsteroidal aromatase inhibitors: recent advances. Med. Res. Rev. 2002, 22, 282–304. 10.1002/med.10010. [DOI] [PubMed] [Google Scholar]
- de Montellano P. R. O.Cytochrome P450; Kluwer Academic/Plenum Publishers: New York, 2015. [Google Scholar]
- Kao Y. C.; Cam L. L.; Laughton C. A.; Zhou D.; Chen S. Binding characteristics of seven inhibitors of human aromatase: a site-directed mutagenesis study. Cancer Res. 1996, 56, 3451–3460. [PubMed] [Google Scholar]
- Wang X.; Chen S. Aromatase destabilizer: novel action of exemestane, a food and drug administration-approved aromatase inhibitor. Cancer Res. 2006, 66, 10281–10286. 10.1158/0008-5472.CAN-06-2134. [DOI] [PubMed] [Google Scholar]
- Osborne C.; Tripathy D. Aromatase inhibitors: rationale and use in breast cancer. Annu. Rev. Med. 2005, 56, 103–116. 10.1146/annurev.med.56.062804.103324. [DOI] [PubMed] [Google Scholar]
- Jackson J.; Miller W. R.; Dixon J. M. Safety issues surrounding the use of aromatase inhibitors in breast cancer. Expert Opin. Drug Saf. 2003, 2, 73–86. 10.1517/14740338.2.1.73. [DOI] [PubMed] [Google Scholar]
- Jeong S.; Woo M. M.; Flockhart D. A.; Desta Z. Inhibition of drug metabolizing cytochrome P450s by the aromatase inhibitor drug letrozole and its major oxidative metabolite 4,4′-methanol-bisbenzonitrile in vitro. Cancer Chemother. Pharmacol. 2009, 64, 867–875. 10.1007/s00280-009-0935-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich F. P. Cytochrome P450s and other enzymes in drug metabolism and toxicity. AAPS J. 2006, 8, E101–E111. 10.1208/aapsj080112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto T.; Horiguchi H.; Oguma E.; Kayama F. Effects of diverse dietary phytoestrogens on cell growth, cell cycle and apoptosis in estrogen-receptor-positive breast cancer cells. J. Nutr. Biochem. 2010, 21, 856–864. 10.1016/j.jnutbio.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Karkola S.; Wähälä K. The binding of lignans, flavonoids and coumestrol to CYP450 aromatase: a molecular modelling study. Mol. Cell. Endocrinol. 2009, 301, 235–244. 10.1016/j.mce.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Awasthi M.; Singh S.; Pandey V. P.; Dwivedi U. N. Molecular docking and 3D-QSAR-based virtual screening of flavonoids as potential aromatase inhibitors against estrogen-dependent breast cancer. J. Biomol. Struct. Dyn. 2015, 33, 804–819. 10.1080/07391102.2014.912152. [DOI] [PubMed] [Google Scholar]
- Ji J.-Z.; Lao K.-J.; Hu J.; Pang T.; Jiang Z.-Z.; Yuan H.-L.; Miao J.-S.; Chen X.; Ning S.-S.; Xiang H.; et al. Discovery of novel aromatase inhibitors using a homogeneous time-resolved fluorescence assay. Acta Pharmacol. Sin. 2014, 35, 1082–1092. 10.1038/aps.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worachartcheewan A.; Suvannang N.; Prachayasittikul S.; Prachayasittikul V.; Nantasenamat C. Probing the origins of aromatase inhibitory activity of disubstituted coumarins via QSAR and molecular docking. EXCLI J. 2014, 13, 1259–1274. [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Mestres J.; Testa B. In silico pharmacology for drug discovery: applications to targets and beyond. Br. J. Pharmacol. 2007, 152, 21–37. 10.1038/sj.bjp.0707306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti M. D.; Rastelli G. Advances and applications of binding affinity prediction methods in drug discovery. Biotechnol. Adv. 2012, 30, 244–250. 10.1016/j.biotechadv.2011.08.003. [DOI] [PubMed] [Google Scholar]
- Kirchmair J.; Williamson M. J.; Tyzack J. D.; Tan L.; Bond P. J.; Bender A.; Glen R. C. Computational Prediction of Metabolism: Sites, Products, SAR, P450 Enzyme Dynamics, and Mechanisms. J. Chem. Inf. Model. 2012, 52, 617–648. 10.1021/ci200542m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stjernschantz E.; Marelius J.; Medina C.; Jacobsson M.; Vermeulen N. P. E.; Oostenbrink C. Are Automated Molecular Dynamics Simulations and Binding Free Energy Calculations Realistic Tools in Lead Optimization? An Evaluation of the Linear Interaction Energy (LIE) Method. J. Chem. Inf. Model. 2006, 46, 1972–1983. 10.1021/ci0601214. [DOI] [PubMed] [Google Scholar]
- Li H.; Sun J.; Fan X.; Sui X.; Zhang L.; Wang Y.; He Z. Considerations and recent advances in QSAR models for cytochrome P450-mediated drug metabolism prediction. J. Comput.-Aided Mol. Des. 2008, 22, 843–855. 10.1007/s10822-008-9225-4. [DOI] [PubMed] [Google Scholar]
- Stjernschantz E.; Vermeulen N. P. E.; Oostenbrink C. Computational prediction of drug binding and rationalisation of selectivity towards cytochromes P450. Expert Opin. Drug Metab. Toxicol. 2008, 4, 513–527. 10.1517/17425255.4.5.513. [DOI] [PubMed] [Google Scholar]
- Sridhar J.; Liu J.; Foroozesh M.; Stevens C. L. K. Insights on cytochrome p450 enzymes and inhibitors obtained through QSAR studies. Molecules 2012, 17, 9283–9305. 10.3390/molecules17089283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discovery Today 2006, 11, 580–594. 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capoferri L.; Verkade-Vreeker M. C. A.; Buitenhuis D.; Commandeur J. N. M.; Pastor M.; Vermeulen N. P. E.; Geerke D. P. Linear Interaction Energy Based Prediction of Cytochrome P450 1A2 Binding Affinities with Reliability Estimation. PLoS One 2015, 10, e0142232. 10.1371/journal.pone.0142232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perić-Hassler L.; Stjernschantz E.; Oostenbrink C.; Geerke D. P. CYP 2D6 binding affinity predictions using multiple ligand and protein conformations. Int. J. Mol. Sci. 2013, 14, 24514–24530. 10.3390/ijms141224514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stjernschantz E.; Oostenbrink C. Improved ligand-protein binding affinity predictions using multiple binding modes. Biophys. J. 2010, 98, 2682–2691. 10.1016/j.bpj.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodera J. D.; Mobley D. L.; Shirts M. R.; Dixon R. W.; Branson K.; Pande V. S. Alchemical free energy methods for drug discovery: progress and challenges. Curr. Opin. Struct. Biol. 2011, 21, 150–160. 10.1016/j.sbi.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqvist J.; Medina C.; Samuelsson J.-E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng., Des. Sel. 1994, 7, 385–391. 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- Brandsdal B. O.; Osterberg F.; Almlöf M.; Feierberg I.; Luzhkov V. B.; Aqvist J. Free energy calculations and ligand binding. Adv. Protein Chem. 2003, 66, 123–158. 10.1016/S0065-3233(03)66004-3. [DOI] [PubMed] [Google Scholar]
- de Amorim H. L.; Caceres R. A.; Netz P. A. Linear Interaction Energy (LIE) Method in Lead Discovery and Optimization. Curr. Drug Targets 2008, 9, 1100–1105. 10.2174/138945008786949360. [DOI] [PubMed] [Google Scholar]
- Gutiérrez-de-Terán H.; Aqvist J. Linear interaction energy: method and applications in drug design. Methods Mol. Biol. 2012, 819, 305–323. 10.1007/978-1-61779-465-0_20. [DOI] [PubMed] [Google Scholar]
- Kjellgren E. R.; Glue O. E. S.; Reinholdt P.; Meyer J. E.; Kongsted J.; Poongavanam V. A comparative study of binding affinities for 6,7-dimethoxy-4-pyrrolidylquinazolines as phosphodiesterase 10A inhibitors using the linear interaction energy method. J. Mol. Graphics Modell. 2015, 61, 44–52. 10.1016/j.jmgm.2015.06.010. [DOI] [PubMed] [Google Scholar]
- Miranda W. E.; Noskov S. Y.; Valiente P. A. Improving the LIE Method for Binding Free Energy Calculations of Protein-Ligand Complexes. J. Chem. Inf. Model. 2015, 55, 1867–1877. 10.1021/acs.jcim.5b00012. [DOI] [PubMed] [Google Scholar]
- Perdih A.; Wolber G.; Solmajer T. Molecular dynamics simulation and linear interaction energy study of d-Glu-based inhibitors of the MurD ligase. J. Comput.-Aided Mol. Des. 2013, 27, 723–738. 10.1007/s10822-013-9673-3. [DOI] [PubMed] [Google Scholar]
- Su Y.; Gallicchio E.; Das K.; Arnold E.; Levy R. M. Linear Interaction Energy (LIE) Models for Ligand Binding in Implicit Solvent: Theory and Application to the Binding of NNRTIs to HIV-1 Reverse Transcriptase. J. Chem. Theory Comput. 2007, 3, 256–277. 10.1021/ct600258e. [DOI] [PubMed] [Google Scholar]
- van Lipzig M. M. H.; ter Laak A. M.; Jongejan A.; Vermeulen N. P. E.; Wamelink M.; Geerke D.; Meerman J. H. N. Prediction of Ligand Binding Affinity and Orientation of Xenoestrogens to the Estrogen Receptor by Molecular Dynamics Simulations and the Linear Interaction Energy Method. J. Med. Chem. 2004, 47, 1018–1030. 10.1021/jm0309607. [DOI] [PubMed] [Google Scholar]
- Durmaz V.; Schmidt S.; Sabri P.; Piechotta C.; Weber M. Hands-off linear interaction energy approach to binding mode and affinity estimation of estrogens. J. Chem. Inf. Model. 2013, 53, 2681–2688. 10.1021/ci400392p. [DOI] [PubMed] [Google Scholar]
- Shirts M. R.; Pitera J. W.; Swope W. C.; Pande V. S. Extremely precise free energy calculations of amino acid side chain analogs: Comparison of common molecular mechanics force fields for proteins. J. Chem. Phys. 2003, 119, 5740. 10.1063/1.1587119. [DOI] [Google Scholar]
- Vosmeer C. R.; Pool R.; Van Stee M. F.; Perić-Hassler L.; Vermeulen N. P. E.; Geerke D. P. Towards automated binding affinity prediction using an iterative linear interaction energy approach. Int. J. Mol. Sci. 2014, 15, 798–816. 10.3390/ijms15010798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D.; Griswold J.; Erman M.; Pangborn W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 2009, 457, 219–223. 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson E. A.; Siiteri P. K. Utilization of oxygen and reduced nicotinamide adenine dinucleotide phosphate by human placental microsomes during aromatization of androstenedione. J. Biol. Chem. 1974, 249, 5364–5372. [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2012.10; Chemical Computing Group Inc.: Montreal, QC, 2017.
- Halgren T. A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. . [DOI] [Google Scholar]
- Malde A. K.; Zuo L.; Breeze M.; Stroet M.; Poger D.; Nair P. C.; Oostenbrink C.; Mark A. E. J. Chem. Theory Comput. 2011, 7, 4026–4037. 10.1021/ct200196m. [DOI] [PubMed] [Google Scholar]
- Korb O.; Stützle T.; Exner T. E. An ant colony optimization approach to flexible protein–ligand docking. Swarm Intell. 2007, 1, 115. 10.1007/s11721-007-0006-9. [DOI] [Google Scholar]
- Korb O.; Stutzle T.; Exner T. E. Empirical Scoring Functions for Advanced Protein–Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- Lloyd S. Least squares quantization in PCM. IEEE Trans. Inf. Theory 1982, 28, 129–137. 10.1109/TIT.1982.1056489. [DOI] [Google Scholar]
- Hess B.; Kutzner C.; Van Der Spoel D.; Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Capoferri L.; van Dijk M.; Rustenburg A. S.; Wassenaar T.; Kooi D. P.; Rifai E. A.; Vermeulen N. P. E.; Geerke D. P.. eTOX ALLIES: an Automated pipeLine for Linear Interaction Energy-based Simulations. J. Cheminf., submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk M.; Wassenaar T. A.; Bonvin A. M. J. J. A Flexible, Grid-Enabled Web Portal for GROMACS Molecular Dynamics Simulations. J. Chem. Theory Comput. 2012, 8, 3463–3472. 10.1021/ct300102d. [DOI] [PubMed] [Google Scholar]
- Schmid N.; Eichenberger A. P.; Choutko A.; Riniker S.; Winger M.; Mark A. E.; van Gunsteren W. F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Wassenaar T. A.; Mark A. E. The effect of box shape on the dynamic properties of proteins simulated under periodic boundary conditions. J. Comput. Chem. 2006, 27, 316–325. 10.1002/jcc.20341. [DOI] [PubMed] [Google Scholar]
- Bekker H.; van den Berg J. P.; Wassenaar T. A. A method to obtain a near-minimal-volume molecular simulation of a macromolecule, using periodic boundary conditions and rotational constraints. J. Comput. Chem. 2004, 25, 1037–1046. 10.1002/jcc.20050. [DOI] [PubMed] [Google Scholar]
- Berendsen H.; Postma J.; van Gunsteren W. F.. Interaction Models for Water in Relation to Protein Hydration; Pullman B., Ed.; Reidel: Dordrecht, 1981; pp 331–342. [Google Scholar]
- Berendsen H.; Postma J.; van Gunsteren W.; DiNola A.; Haak J. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Suvannang N.; Nantasenamat C.; Isarankura-Na-Ayudhya C.; Prachayasittikul V. Molecular Docking of Aromatase Inhibitors. Molecules 2011, 16, 3597–3617. 10.3390/molecules16053597. [DOI] [Google Scholar]
- Amini M.; Eastwood J. W.; Hockney R. W. Time Integration in Particle Models. Comput. Phys. Commun. 1987, 44, 83–93. 10.1016/0010-4655(87)90019-1. [DOI] [Google Scholar]
- Feenstra K. A.; Hess B.; Berendsen H. Improving efficiency of large timescale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 1999, 20, 786–798. . [DOI] [PubMed] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H.; Fraaije J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. . [DOI] [Google Scholar]
- Tironi I. G.; Sperb R.; Smith P. E.; van Gunsteren W. F. A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys. 1995, 102, 5451–5459. 10.1063/1.469273. [DOI] [Google Scholar]
- Aqvist J.; Luzhkov V. B.; Brandsdal B. O. Ligand Binding Affinities from MD Simulations. Acc. Chem. Res. 2002, 35, 358–365. 10.1021/ar010014p. [DOI] [PubMed] [Google Scholar]
- Carlson H. A.; Jorgensen W. L. An Extended Linear Response Method for Determining Free Energies of Hydration. J. Phys. Chem. 1995, 99, 10667–10673. 10.1021/j100026a034. [DOI] [Google Scholar]
- Hritz J.; Oostenbrink C. Efficient free energy calculations for compounds with multiple stable conformations separated by high energy barriers. J. Phys. Chem. B 2009, 113, 12711–12720. 10.1021/jp902968m. [DOI] [PubMed] [Google Scholar]
- Wang W.; Wang J.; Kollman P. A. What determines the van der Waals coefficient ? in the LIE (linear interaction energy) method to estimate binding free energies using molecular dynamics simulations?. Proteins: Struct., Funct., Genet. 1999, 34, 395–402. . [DOI] [PubMed] [Google Scholar]
- Vosmeer C. R.; Kooi D. P.; Capoferri L.; Terpstra M. M.; Vermeulen N. P. E.; Geerke D. P. Improving the iterative Linear Interaction Energy approach using automated recognition of configurational transitions. J. Mol. Model. 2016, 22, 31. 10.1007/s00894-015-2883-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster A. P.; Laird N. M.; Rubin D. B. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc., Ser. B 1977, 39, 1–38. [Google Scholar]
- McLachlan G.; Krishnan T.. The EM Algorithm and Extensions, Second ed.; John Wiley & Sons Inc.: Hoboken, NJ, 2008. [Google Scholar]
- Huber P. J.; Ronchetti E. M.. Robust Statistics, 2nd ed.; John Wiley & Sons Inc.: Hoboken, NJ, 2009. [Google Scholar]
- Jones E.; Oliphant T.; Peterson P.. SciPy: Open source scientific tools for Python; http://www.scipy.org (accessed 10 Oct. 2015).
- Favia A. D.; Cavalli A.; Masetti M.; Carotti A.; Recanatini M. Three-dimensional model of the human aromatase enzyme and density functional parameterization of the iron-containing protoporphyrin IX for a molecular dynamics study of heme-cysteinato cytochromes. Proteins: Struct., Funct., Genet. 2006, 62, 1074–1087. 10.1002/prot.20829. [DOI] [PubMed] [Google Scholar]
- Guallar V.; Baik M. H.; Lippard S. J.; Friesner R. A. Peripheral heme substituents control the hydrogen-atom abstraction chemistry in cytochromes P450. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 6998–7002. 10.1073/pnas.0732000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole P. A.; Robinson C. H. Mechanism and inhibition of cytochrome P-450 aromatase. J. Med. Chem. 1990, 33, 2933–2942. 10.1021/jm00173a001. [DOI] [PubMed] [Google Scholar]
- Hong Y.; Yu B.; Sherman M.; Yuan Y.-C.; Zhou D.; Chen S. Molecular basis for the aromatization reaction and exemestane-mediated irreversible inhibition of human aromatase. Mol. Endocrinol. 2007, 21, 401–414. 10.1210/me.2006-0281. [DOI] [PubMed] [Google Scholar]
- Galeazzi R.; Massaccesi L. Insight into the binding interactions of CYP450 aromatase inhibitors with their target enzyme: a combined molecular docking and molecular dynamics study. J. Mol. Model. 2012, 18, 1153–1166. 10.1007/s00894-011-1144-y. [DOI] [PubMed] [Google Scholar]
- Vasanthanathan P.; Olsen L.; Jørgensen F. S.; Vermeulen N. P. E.; Oostenbrink C. Computational prediction of binding affinity for CYP1A2-ligand complexes using empirical free energy calculations. Drug Metab. Dispos. 2010, 38, 1347–1354. 10.1124/dmd.110.032946. [DOI] [PubMed] [Google Scholar]
- Recanatini M.; Bisi A.; Cavalli A.; Belluti F.; Gobbi S.; Rampa A.; Valenti P.; Palzer M.; Palusczak A.; Hartmann R. W. A new class of nonsteroidal aromatase inhibitors: design and synthesis of chromone and xanthone derivatives and inhibition of the P450 enzymes aromatase and 17 alpha-hydroxylase/C17,20-lyase. J. Med. Chem. 2001, 44, 672–680. 10.1021/jm000955s. [DOI] [PubMed] [Google Scholar]
- Ghosh D.; Griswold J.; Erman M.; Pangborn W. X-ray structure of human aromatase reveals an androgen-specific active site. J. Steroid Biochem. Mol. Biol. 2010, 118, 197–202. 10.1016/j.jsbmb.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punetha A.; Shanmugam K.; Sundar D. Insight into the enzyme-inhibitor interactions of the first experimentally determined human aromatase. J. Biomol. Struct. Dyn. 2011, 28, 759–771. 10.1080/07391102.2011.10508604. [DOI] [PubMed] [Google Scholar]
- Cepa M. M. D. S.; Tavares da Silva E. J.; Correia da Silva G.; Roleira F. M. F.; Teixeira N. A. A. Structure-activity relationships of new A,D-ring modified steroids as aromatase inhibitors: design, synthesis, and biological activity evaluation. J. Med. Chem. 2005, 48, 6379–6385. 10.1021/jm050129p. [DOI] [PubMed] [Google Scholar]
- Cepa M. M. D. S.; Tavares da Silva E. J.; Correia da Silva G.; Roleira F. M. F.; Teixeira N. A. A. Synthesis and biochemical studies of 17-substituted androst-3-enes and 3,4-epoxyandrostanes as aromatase inhibitors. Steroids 2008, 73, 1409–1415. 10.1016/j.steroids.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Sherwin P. F.; McMullan P. C.; Covey D. F. Effects of steroid D-ring modification on suicide inactivation and competitive inhibition of aromatase by analogs of androsta-1,4-diene-3,17-dione. J. Med. Chem. 1989, 32, 651–658. 10.1021/jm00123a026. [DOI] [PubMed] [Google Scholar]
- Hansson T.; Marelius J.; Aqvist J. Ligand binding affinity prediction by linear interaction energy methods -. J. Comput.-Aided Mol. Des. 1998, 12, 27–35. 10.1023/A:1007930623000. [DOI] [PubMed] [Google Scholar]
- Almlöf M.; Carlsson J.; Aqvist J. Improving the Accuracy of the Linear Interaction Energy Method for Solvation Free Energies. J. Chem. Theory Comput. 2007, 3, 2162–2175. 10.1021/ct700106b. [DOI] [PubMed] [Google Scholar]
- Santen R. J.; Worgul T. J.; Lipton A.; Harvey H.; Boucher A.; Samojlik E.; Wells S. A. Aminoglutethimide as treatment of postmenopausal women with advanced breast carcinoma. Ann. Intern. Med. 1982, 96, 94–101. 10.7326/0003-4819-96-1-94. [DOI] [PubMed] [Google Scholar]
- Santen R. J.; Santner S.; Davis B.; Veldhuis J.; Samojlik E.; Ruby E. Aminoglutethimide inhibits extraglandular estrogen production in postmenopausal women with breast carcinoma. J. Clin. Endocrinol. Metab. 1978, 47, 1257–1265. 10.1210/jcem-47-6-1257. [DOI] [PubMed] [Google Scholar]
- Graves P. E.; Salhanick H. A. Stereoselective inhibition of aromatase by enantiomers of aminoglutethimide. Endocrinology 1979, 105, 52–57. 10.1210/endo-105-1-52. [DOI] [PubMed] [Google Scholar]
- Plourde P. V.; Dyroff M.; Dowsett M.; Demers L.; Yates R.; Webster A. ARIMIDEX: a new oral, once-a-day aromatase inhibitor. J. Steroid Biochem. Mol. Biol. 1995, 53, 175–179. 10.1016/0960-0760(95)00045-2. [DOI] [PubMed] [Google Scholar]
- Dutta U.; Pant K. Aromatase inhibitors: past, present and future in breast cancer therapy. Med. Oncol. 2008, 25, 113–124. 10.1007/s12032-007-9019-x. [DOI] [PubMed] [Google Scholar]
- Trunet P. F.; Bhatnagar A. S.; Chaudri H. A.; Hornberger U. Letrozole (CGS 20267), a new oral aromatase inhibitor for the treatment of advanced breast cancer in postmenopausal patients. Acta Oncol. 1996, 35 (Suppl 5), 15–18. 10.3109/02841869609083962. [DOI] [PubMed] [Google Scholar]
- Lipton A.; Demers L. M.; Harvey H. A.; Kambic K. B.; Grossberg H.; Brady C.; Adlercruetz H.; Trunet P. F.; Santen R. J. Letrozole (CGS 20267). A phase I study of a new potent oral aromatase inhibitor of breast cancer. Cancer 1995, 75, 2132–2138. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.