

Abstract
Vascular anomalies arise as a consequence of improper development and maintenance of the vasculature. Our knowledge on the pathophysiological bases of vascular anomalies has skyrocketed during the past 5 years. It is becoming clear that common intracellular signaling pathways are often activated by mutations, causing endothelial cell dysfunction. These mutations cause hyperactivation of two major intracellular signaling pathways that may be controlled by inhibitors developed for cancer treatment. Although we do not know yet all the downstream effects, it has become evident that normalization of the abnormal signaling is an interesting target for therapy. This is a major paradigm change, as developmental malformations were considered to be inert to any molecular treatment.
Keywords: vascular malformations, mutations, interventional radiology
Objectives : Upon completion of this article, the reader will be able to discuss the common intracellular signaling pathways that are often activated by mutations, causing endothelial cell dysfunction.
Accreditation : This activity has been planned and implemented in accordance with the Essential Areas and Policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint providership of Tufts University School of Medicine (TUSM) and Thieme Medical Publishers, New York. TUSM is accredited by the ACCME to provide continuing medical education for physicians.
Credit : Tufts University School of Medicine designates this journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit ™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Vascular anomalies arise as a consequence of improper development and maintenance of the vasculature. Effective treatment options are limited and commonly do not provide a cure. Identifying the causes of vascular anomalies and understanding the underlying pathophysiological mechanisms could give new means to develop novel therapies.
Vascular anomalies usually occur sporadically. Yet, rarer familial forms have been instrumental in initiating the discovery of underlying pathophysiology. Inherited loss-of-function (LOF) mutations have been discovered in three genes encoding bone morphogenic protein/transforming growth factor-β (TGFβ) receptor signaling complex linked factors in hereditary hemorrhagic telangiectasia (HHT), 1 2 3 4 5 in glomulin in inherited glomuvenous malformations (GVM), 6 in three genes producing an endothelial cell signaling complex in cerebral cavernous malformations (CCMs), 7 8 9 and in RASA1 in capillary malformation-arteriovenous malformation (CM-AVM). 10 In contrast, weakly activating mutations in TIE2/TEK have been discovered underlying inherited cutaneomucosal venous malformations (VMCMs). 11 As 50% of alleles are affected in these dominantly inherited forms, the mutations were detectable by linkage analyses and Sanger sequencing.
These inherited cases share the following features: multifocality, small size, and an increase in the number of lesions over time. Moreover, some mutation carriers do not have any lesions. The bases of this variability has been shown to be the need of a tissular second-hit, that is, another noninherited mutation that appears on the second allele of the gene. 6 12 13 14 15 Such a tissular event occurs in selected cells of the body, leaving them completely devoid of the encoded protein's normal activity.
The demonstration of this phenomenon to take place led to the hypothesis that the much more frequent, sporadically occurring lesions could be due to somatic changes alone. This was first shown for sporadic venous malformations (VMs), 60% of which have a somatic mutation in TIE2/TEK. 12 These mutations activate the receptor more strongly than the inherited mutations. Since somatic mutations have been identified in all major categories of vascular anomalies, including VMs, capillary malformations (CMs), lymphatic malformations (LMs), as well as certain vascular tumors (pyogenic granuloma [PG], rapidly involuting congenital hemangioma [RICH], and noninvoluting congenital hemangioma [NICH]). However, since the distribution of the mutations is mosaic (not present in all cell types within the lesion), the frequency is sometimes very low (down to 1%), reflecting the inter-cell-type and even intra-cell-type heterogeneity. Therefore, studying a resected tissue fragment needs sensitive techniques. This became possible by the recent development of next-generation sequencing (massively parallel sequencing).
Studies of the majority of genes associated with various familial cases of vascular anomalies indicated that the affected genes/proteins were vascular cell type specific. TIE2/TEK is a tyrosine kinase receptor that is specifically expressed on endothelial cells (EC), and RASA1 and glomulin are enriched in blood vessels. 16 17 18 19 Functional studies also largely viewed pathogenetic mechanisms as singular/separate signaling pathways according to the corresponding disorder. Interestingly, many of the genes identified in sporadic cases are ubiquitously expressed and code for proteins in major pathways with no specificity to the vasculature. Rather, they are also linked to cancers. This surprise can likely be explained by the fact that the somatic mutations that give rise to an isolated vascular anomaly occur in vascular ECs only. This is underscored by the fact that more extensive mosaicism can be seen in syndromic forms, such as Klippel–Trenaunay syndrome (capillary-lymphatic-venous malformation with overgrowth). Most of the activated proteins play a role in two major signaling pathways: the PI3K/AKT and the RAS/MAPK pathways ( Fig. 1 ). This opens the possibility that drugs/inhibitors currently being used in cancer patients may be used to treat vascular anomalies.
Fig. 1.
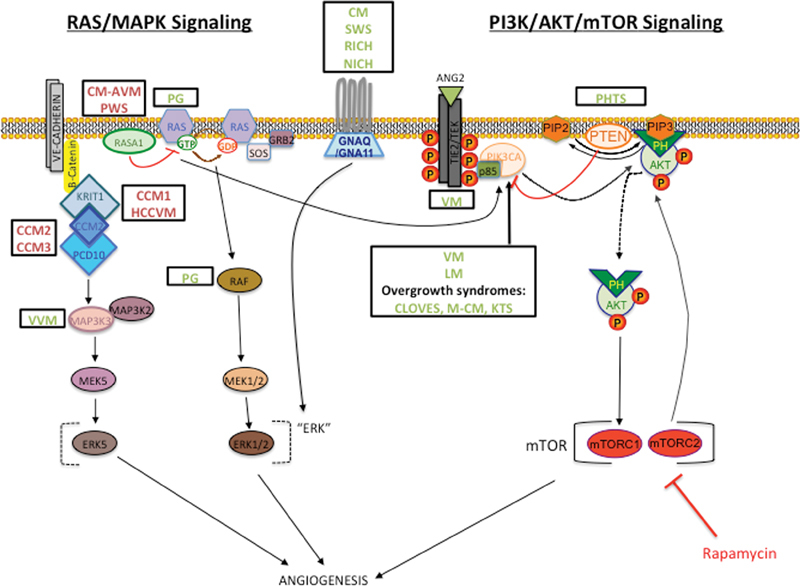
Hyperactivation of the RAS/MAPK or PI3K/AKT/mTOR pathways seen in the majority of vascular anomalies. Schematic of the two pathways displays location of associated protein with names of disorders in boxes. Proteins with loss-of-function mutations indicated in red; those with gain of function in green. Rapamycin (in bright red) is a mammalian target of rapamycin (mTOR) inhibitor effective in treating VMs and LMs.
PI3K/AKT/mTOR Signaling Pathways
The phosphoinositide 3-kinase (PI3K)/AKT pathway plays a role in many cellular processes from cell cycle regulation to proliferation and migration. It is often referred to as the “anti-apoptosis pathway” ( Fig. 1 ). It is triggered when a ligand binds to a receptor tyrosine kinase, which becomes phosphorylated and attracts a PI3K to the plasma membrane. The PI3K interacts either directly to the phosphotyrosine residues on the receptor or adaptors (e.g., p85) within its SH2 domain, turning on the catalytic subunit. The substrate phosphatidylinositol-4,4-bisphosphate (PI-4,5-P2) (PIP2) is converted to phosphatidylinositol-3,4,5-triphosphate (PI-3,4,5-P3) (PIP3), which recruits pleckstrin homology (PH) domain-containing proteins, protein kinase B (PKB), and AKT to the membrane. This complex can regulate cell cycle directly, but in many cases will continue on to initiate the mammalian target of rapamycin (mTOR) signaling. Consequently, several types of proteins involved in a variety of cellular processes are turned on. 20
The discovery that genes mutated in vascular anomalies code for proteins involved in the PI3K/AKT/mTOR signaling pathway underscores its importance in the vasculature, in particular the ECs. The majority of all four classes of VMs (VM, MIM 600221; VMCM, MIM 600195; blue rubber bleb nevus syndrome [BRBN], MIM 112200; and multifocal venous malformation [MVM]) are caused by activating changes in TIE2/TEK (MIM 600221). 11 17 21 In human umbilical vein endothelial cells (HUVECs) that have been transfected with specific TIE2 mutations, AKT activation was induced. 21 22 Thus, the canonical signaling pathway for VMs downstream of TIE2 is the PI3K/AKT signaling pathway. In support of this assertion, it was found that gain-of-function (GOF) mutations in the class I PI3K catalytic subunit p110α (PIK3CA, MIM 171834) were responsible for another 20% of VM patients. 23
Several additional vascular anomalies were found to have somatic mutations in PIK3CA, with five substitutions (c.C420R, p.E542K, p.E545K, p.H1047R, p.H1044L) being the most frequently observed. 24 These changes were also found in isolated LMs and in complex syndromes, such as Klippel–Trenaunay syndrome/capillary-lymphatic-venous malformation with overgrowth (KTS, MIM 149000), congenital lipomatous overgrowth vascular malformation, epidermal nevi, scoliosis/skeletal and spinal (CLOVES, MIM 612918) syndrome, and megalencephaly-capillary malformation (MCM, 602501). 25 26 27 This could be partially explained by differences in the time-point of occurrence of the postzygotic mutation; yet, other instigators, such as environmental cues, or variants in additional genes, should be taken into consideration.
PI3K/AKT/mTOR signaling is negatively regulated by phosphatase and tensin homolog (PTEN, MIM 601728) 28 ( Fig. 1 ). It is a dual specificity phosphatase that suppresses PI3K lipid activity. The loss of PTEN leads to lack of dephosphorylation of the lipid substrate PIP3, causing permanent stimulation of the PI3K/AKT/mTOR pathway. Germline LOF mutations in PTEN cause PTEN hamartoma tumor syndrome (PHTS), which includes Cowden (CS, MIM 158350) and Bannayan–Riley–Ruvalcaba (BRRS, MIM 153480) syndromes. These patients have variable vascular hamartomas, now referred to as PTEN hamartomas of soft tissue (PHOST). 26
The PI3K/AKT/mTOR signaling pathway is overtly activated in many cancers, and several inhibitors have been developed and used in clinical trials, with variable effects, often due to the tumor being a constellation of numerous independent clones, only some of which are responsive. Interestingly, vascular anomalies, even when caused by activation of the same pathway, and by the same driver mutations, do not degenerate into cancer. There is likely less (epi)genetic instability. Therefore, it seems of great interest to test if these inhibitors were relevant for treating vascular anomalies, in which lesional clonality is less pronounced. As a mouse model was developed for MVs, using HUVECs transfected with the most common TIE2/TEK mutation associated with VMs (L914F), preclinical testing could be performed. 29 It was shown that not only was rapamycin capable of normalizing the morphology of the mutant HUVECs in vitro, but it also stopped the growth of the vascular lesions in the xenograft mouse model of VM. Additionally, administrating rapamycin in VM patients successfully ameliorated symptoms and their quality of life. 29 Rapamycin was also effective in improving the quality of life in LM patients. 30 31
Ras/MAPK Signaling Pathways
RAS viral oncogene homolog (Ras) proteins, small guanosine nucleotide bound GTPases, can act within many different intracellular pathways. The class I PI3Ks have a Ras-binding domain in the N-terminal end; thus, RAS can activate the PI3K/AKT signaling pathway. However, the predominant mode is via the RAS/mitogen-activated protein kinase (MAPK) pathway ( Fig. 1 ). Binding of a growth factor to a receptor tyrosine kinase leads to its activation and recruitment of an adaptor protein, growth factor receptor-bound protein-2 (GRB2). GRB2 then attracts a nucleotide exchange factor, son-of-sevenless (SOS) to the plasma membrane. SOS induces activation of GFP-bound Ras form. This leads to the cascade of activation of Raf phosphorylation of mitogen-activated protein kinase (MAPKK/MEK), then MEK of extracellular signaling regulated kinase (ERK). ERK eventually regulates the functions of a multitude of downstream cytosolic and nuclear molecules. 32
The RAS/MAPK pathway is vital in many cellular processes from cell cycle regulation to proliferation and migration. It is often referred to as the “proliferation pathway.” As the Ras/MAPK signaling pathway plays a major role in development, several disorders in which mutations within different genes that code for members of this pathway have been identified. These “rasopathies” essentially result from hyperactivation of the RAS/MAPK pathway and are fairly common, affecting more than 1 in 1,000 people. Rasopathies may share overlapping phenotypes, such as growth defects and vascular malformations. 33
Mutations affecting the RAS/MAPK signaling pathway are also found in isolated vascular anomalies ( Fig. 1 ). LOF mutations in the p120 rasGTPase-activating protein RASA1 are found in capillary malformation-arteriovenous malformations (CM-AVM, MIM 139150) and Parkes Weber syndrome. 10 14 34 p120RASGAP negatively regulates RAS activity by converting GTP-bound RAS to GDP-bound RAS. Thus, lesions with RASA1 mutations have chronic activation of the RAS/MAPK pathway. Mice in which Rasa1 was homozygously knocked out were embryonic lethal at E10.5, while mice mosaic for homozygously absent cells developed abnormal cutaneous vessels. 35
Other vascular anomalies are also linked to RAS/MAPK signaling. Somatic activating mutations in the guanine nucleotide–binding protein G (q/11) (GNAQ, MIM 600998 or GNA11, MIM 139313) are associated with CMs (MIM 16300) and Sturge Weber syndrome (SWS, MIM 185300), as well as RICH and NICH. The most frequent change seen in CM and SWS is an arginine183-to-glycine (R183Q) substitution in GNAQ, 36 37 38 while a glutamine substitution at position 209 (Glu209) in GNAQ and GNA11 is seen in RICH/NICH patients. 39 As the same c.626A > T change in GNAQ and GNA11 was seen in all four anomalies, additional factors (environmental, genetic, cell type, etc.) could still be involved. All these mutations have a weak activating effect and seem to activate ERK more than the PIK3/AKT 40 ( Fig. 1 ).
PGs can have a mutation in different canonical members of the pathway ( Fig. 1 ). A BRAF (MIM 164757) mutation was seen in PG associated with CM, the latter caused by the GNAQ_R183Q change. 41 The same BRAF c.1799T > A mutation is a known “hot-spot” mutation in multiple types of benign nonmelanocytic skin lesions and the specific change in 90% of BRAF-linked human cancers. It leads to chronic ERK1/2 activity. Some PG-associated mutations have been found in N-RAS (neuroblastoma-RAS, MIM 164790) and K-RAS (Kirsten rat sarcoma RAS, MIM 190070). 42 This underscores the implication of overactive RAS/MAPK signaling in these lesions.
The pathophysiological bases of cerebral cavernous malformations (CCM1, MIM 116860; CCM2, MIM 607929; CCM3, MIM 603285) were also recently suggested to be linked to RAS/MAPK signaling. The LOF of three associated proteins (Krev interaction trapped-1 [KRIT, MIM 604214], malcavernin [CCM2, MIM 607929], and programmed cell death 10 [PDCD10, MIM 609118]) results in CCM formation. 43 KRIT1 LOF also causes hyperkeratotic capillary-venous malformation (HCCVM). These molecules likely form a complex near the plasma membrane, with CCM1 interacting with CCM2, and CCM2 with CCM3. CCM2 is a scaffolding protein for MEKK3/MAP3K3. 44 It was found that when Krit1 and CCM2 were deleted in ECs, it resulted in inappropriate activation of MEKK3/MAP3K3. 45 Moreover, MEKK3/MAP3K3 GOF mutations are found in verrucous venous malformations, which mimic HCCVMs. 46 Thus, these malformations may be closely related and specifically inhibiting MAP3K3 may be the best effective treatment option.
TGF-β Signaling Pathway
The TGF-β signaling is ubiquitously present and involved in the regulation of many biological processes. In the vasculature, two main types of ligands, TGF-β and bone morphogenetic proteins (BMPs), can bind to a type II and I serine/threonine kinase receptor complex. In some cases, a type III coreceptor can aid ligand binding. The activated type I receptor then phosphorylates intracellular SMADs (2/3 or 1/5/8) that propagate the signal to the nucleus with the help of the common mediator SMAD4 47 ( Fig. 2 ).
Fig. 2.
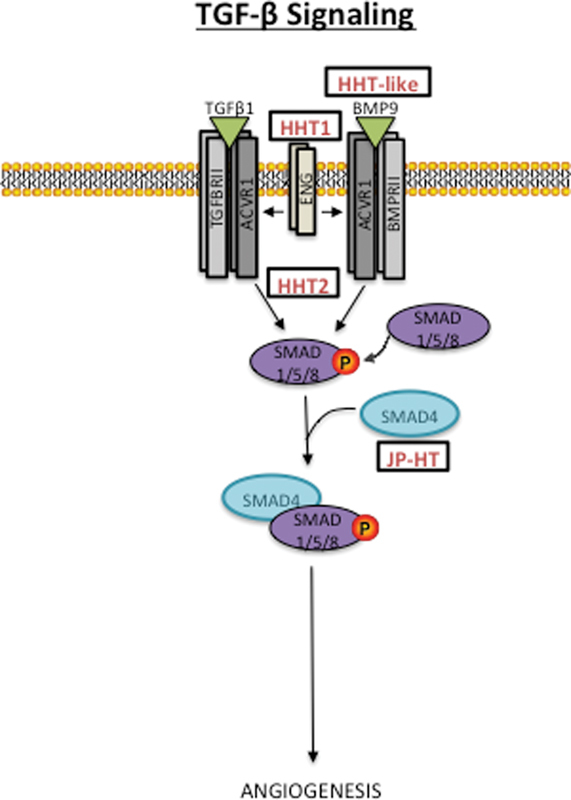
Predicted TGF-β signaling pathway underlying hereditary hemorrhagic telangiectasia (HHT). Loss-of-function mutations within several signaling members. It is uncertain whether pathophysiological pathways underlying HHT overlap with those of other vascular anomalies.
Mutations in several members of the TGF-β signaling superfamily are responsible for HHT. LOF of the type I receptor, activin receptor-like kinase1 (ACVRL1, MIM 601101) and coreceptor endoglin (ENG, 131195), accounts for the majority of HHT cases (HHT2, 600376 and HHT1, 187300). 4 5 48 LOF of SMAD4 (MIM 600993) is linked to a combined syndrome of HHT and juvenile polyposis (JP-HT, MIM 175050). 3 It is BMP signaling that is perturbed in HHT, as BMP9 and BMP10 bind ACVRL1 in vitro, 49 and BMP9/GDF2 (MIM 605120) mutations were identified in three patients with HHT-like phenotypes. 50 Several drugs (e.g., thalidomide and bevacizumab) have been used to alleviate symptoms of HHT, such as bleeding; however, the mechanisms of how these potent antiangiogenic agents help is unknown. 51 52 Overall, how the pathogenic mechanisms of HHT link to those of other vascular anomalies remains unclear.
Conclusion
Our knowledge on the pathophysiological bases of vascular anomalies has skyrocketed during the past 5 years. From deciphering the causes of rare inherited forms, we have moved to unravel those of the most common forms. It is becoming clear that the common intracellular signaling pathways are often activated by mutations, causing EC dysfunction. Although we do not yet know all the downstream effects, it has become evident that normalization of the abnormal signaling is an interesting target for therapy. Rapamycin showed its efficacy for LMs and VMs, and larger studies, such as VASE (Vascular Anomaly—Sirolimus, Europe), have been initiated (L. M. Boon, MD, PhD, unpublished data). It is exciting that the pathways overlap with those related to cancers, as it opens the possibility to study repurposing of cancer drugs for vascular anomalies. This is a major paradigm change, as developmental malformations were considered to be inert to any molecular treatment.
References
- 1.Bayrak-Toydemir P, McDonald J, Akarsu N et al. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006;140(20):2155–2162. doi: 10.1002/ajmg.a.31450. [DOI] [PubMed] [Google Scholar]
- 2.Cole S G, Begbie M E, Wallace G M, Shovlin C L. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42(07):577–582. doi: 10.1136/jmg.2004.028712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gallione C J, Richards J A, Letteboer T G et al. SMAD4 mutations found in unselected HHT patients. J Med Genet. 2006;43(10):793–797. doi: 10.1136/jmg.2006.041517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson D W, Berg J N, Baldwin M A et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13(02):189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 5.McAllister K A, Grogg K M, Johnson D W et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8(04):345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 6.Brouillard P, Boon L M, Mulliken J B et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”) Am J Hum Genet. 2002;70(04):866–874. doi: 10.1086/339492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahoo T, Johnson E W, Thomas J W et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1) Hum Mol Genet. 1999;8(12):2325–2333. doi: 10.1093/hmg/8.12.2325. [DOI] [PubMed] [Google Scholar]
- 8.Liquori C L, Berg M J, Siegel A M et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73(06):1459–1464. doi: 10.1086/380314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergametti F, Denier C, Labauge P et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76(01):42–51. doi: 10.1086/426952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eerola I, Boon L M, Mulliken J B et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73(06):1240–1249. doi: 10.1086/379793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vikkula M, Boon L M, Carraway K L, III et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87(07):1181–1190. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- 12.Limaye N, Wouters V, Uebelhoer M et al. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat Genet. 2009;41(01):118–124. doi: 10.1038/ng.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amyere M, Aerts V, Brouillard P et al. Somatic uniparental isodisomy explains multifocality of glomuvenous malformations. Am J Hum Genet. 2013;92(02):188–196. doi: 10.1016/j.ajhg.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Revencu N, Boon L M, Mendola A et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34(12):1632–1641. doi: 10.1002/humu.22431. [DOI] [PubMed] [Google Scholar]
- 15.Akers A L, Johnson E, Steinberg G K, Zabramski J M, Marchuk D A. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis. Hum Mol Genet. 2009;18(05):919–930. doi: 10.1093/hmg/ddn430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boon L M, Mulliken J B, Vikkula M et al. Assignment of a locus for dominantly inherited venous malformations to chromosome 9p. Hum Mol Genet. 1994;3(09):1583–1587. doi: 10.1093/hmg/3.9.1583. [DOI] [PubMed] [Google Scholar]
- 17.Soblet J, Limaye N, Uebelhoer M, Boon L M, Vikkula M. Variable somatic TIE2 mutations in half of sporadic venous malformations. Mol Syndromol. 2013;4(04):179–183. doi: 10.1159/000348327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wouters V, Limaye N, Uebelhoer M et al. Hereditary cutaneomucosal venous malformations are caused by TIE2 mutations with widely variable hyper-phosphorylating effects. Eur J Hum Genet. 2010;18(04):414–420. doi: 10.1038/ejhg.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McIntyre B A, Brouillard P, Aerts V, Gutierrez-Roelens I, Vikkula M. Glomulin is predominantly expressed in vascular smooth muscle cells in the embryonic and adult mouse. Gene Expr Patterns. 2004;4(03):351–358. doi: 10.1016/j.modgep.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Faes S, Dormond O. PI3K and AKT: unfaithful partners in cancer. Int J Mol Sci. 2015;16(09):21138–21152. doi: 10.3390/ijms160921138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soblet J, Kangas J, Nätynki M et al. Blue rubber bleb nevus (BRBN) syndrome is caused by somatic TEK (TIE2) mutations. J Invest Dermatol. 2017;137(01):207–216. doi: 10.1016/j.jid.2016.07.034. [DOI] [PubMed] [Google Scholar]
- 22.Uebelhoer M, Nätynki M, Kangas J et al. Venous malformation-causative TIE2 mutations mediate an AKT-dependent decrease in PDGFB. Hum Mol Genet. 2013;22(17):3438–3448. doi: 10.1093/hmg/ddt198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Limaye N, Kangas J, Mendola A et al. Somatic activating PIK3CA mutations cause venous malformation. Am J Hum Genet. 2015;97(06):914–921. doi: 10.1016/j.ajhg.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luks V L, Kamitaki N, Vivero M Pet al. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA J Pediatr 2015166041048–540., 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maclellan R A, Luks V L, Vivero M P et al. PIK3CA activating mutations in facial infiltrating lipomatosis. Plast Reconstr Surg. 2014;133(01):12e–19e. doi: 10.1097/01.prs.0000436822.26709.7c. [DOI] [PubMed] [Google Scholar]
- 26.Kurek K C, Luks V L, Ayturk U M et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90(06):1108–1115. doi: 10.1016/j.ajhg.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boscolo E, Coma S, Luks V L et al. AKT hyper-phosphorylation associated with PI3K mutations in lymphatic endothelial cells from a patient with lymphatic malformation. Angiogenesis. 2015;18(02):151–162. doi: 10.1007/s10456-014-9453-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet. 2013;163C(02):114–121. doi: 10.1002/ajmg.c.31364. [DOI] [PubMed] [Google Scholar]
- 29.Boscolo E, Limaye N, Huang L et al. Rapamycin improves TIE2-mutated venous malformation in murine model and human subjects. J Clin Invest. 2015;125(09):3491–3504. doi: 10.1172/JCI76004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lackner H, Karastaneva A, Schwinger W et al. Sirolimus for the treatment of children with various complicated vascular anomalies. Eur J Pediatr. 2015;174(12):1579–1584. doi: 10.1007/s00431-015-2572-y. [DOI] [PubMed] [Google Scholar]
- 31.Adams D M, Trenor C C, III, Hammill A M et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics. 2016;137(02):e20153257. doi: 10.1542/peds.2015-3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sundaram M V.Canonical RTK-Ras-ERK signaling and related alternative pathways WormBook 20131–38.. doi: 10.1895/wormbook.1.80.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tidyman W E, Rauen K A. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19(03):230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boon L M, Mulliken J B, Vikkula M. RASA1: variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev. 2005;15(03):265–269. doi: 10.1016/j.gde.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Henkemeyer M, Rossi D J, Holmyard D Pet al. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein Nature 1995377(6551):695–701. [DOI] [PubMed] [Google Scholar]
- 36.Shirley M D, Tang H, Gallione C J et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368(21):1971–1979. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Couto J A, Huang L, Vivero M P et al. Endothelial cells from capillary malformations are enriched for somatic GNAQ mutations. Plast Reconstr Surg. 2016;137(01):77e–82e. doi: 10.1097/PRS.0000000000001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan W, Nadora D M, Gao L, Wang G, Mihm M C, Jr, Nelson J S. The somatic GNAQ mutation (R183Q) is primarily located within the blood vessels of port wine stains. J Am Acad Dermatol. 2016;74(02):380–383. doi: 10.1016/j.jaad.2015.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ayturk U M, Couto J A, Hann S et al. Somatic activating mutations in GNAQ and GNA11 are associated with congenital hemangioma. Am J Hum Genet. 2016;98(04):789–795. doi: 10.1016/j.ajhg.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas A C, Zeng Z, Rivière J B et al. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Invest Dermatol. 2016;136(04):770–778. doi: 10.1016/j.jid.2015.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groesser L, Peterhof E, Evert M, Landthaler M, Berneburg M, Hafner C. BRAF and RAS mutations in sporadic and secondary pyogenic granuloma. J Invest Dermatol. 2016;136(02):481–486. doi: 10.1038/JID.2015.376. [DOI] [PubMed] [Google Scholar]
- 42.Lim Y H, Douglas S R, Ko C J et al. Somatic activating RAS mutations cause vascular tumors including pyogenic granuloma. J Invest Dermatol. 2015;135(06):1698–1700. doi: 10.1038/jid.2015.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pagenstecher A, Stahl S, Sure U, Felbor U. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet. 2009;18(05):911–918. doi: 10.1093/hmg/ddn420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uhlik M T, Abell A N, Johnson N L et al. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol. 2003;5(12):1104–1110. doi: 10.1038/ncb1071. [DOI] [PubMed] [Google Scholar]
- 45.Zhou Z, Tang A T, Wong W Yet al. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling Nature 2016532(7597):122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Couto J A, Vivero M P, Kozakewich H P et al. A somatic MAP3K3 mutation is associated with verrucous venous malformation. Am J Hum Genet. 2015;96(03):480–486. doi: 10.1016/j.ajhg.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.HHT Mutation Database. Available at:http://www.arup.utah.edu/database/HHT. Accessed: November 2016
- 49.David L, Mallet C, Mazerbourg S, Feige J J, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109(05):1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- 50.Wooderchak-Donahue W L, McDonald J, O'Fallon B et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am J Hum Genet. 2013;93(03):530–537. doi: 10.1016/j.ajhg.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lebrin F, Srun S, Raymond K et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med. 2010;16(04):420–428. doi: 10.1038/nm.2131. [DOI] [PubMed] [Google Scholar]
- 52.Dupuis-Girod S, Ginon I, Saurin J C et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012;307(09):948–955. doi: 10.1001/jama.2012.250. [DOI] [PubMed] [Google Scholar]