

Abstract
A dietary supplement, kelp contains a significant amount of arsenic that is mostly arsenosugars. The determination of arsenosugars is difficult due to a lack of arsenosugar calibration standard, because arsenosugar compounds are not commercially available. This work reports the determination of arsenicals in a kelp extract with traceability to the International System of Units (SI). The hydrophilic fraction of arsenicals was reproducibly extracted from a candidate Standard Reference Material (SRM) 3232 Kelp Powder (Thallus Laminariae) in development at the National Institute of Standards and Technology (NIST). Arsenosugars and dimethylarsinic acid (DMA) were separated into fractions using analytical liquid chromatography (LC) cation and anion columns. The arsenic in the fractions was determined using instrumental neutron activation analysis (INAA). Cation exchange separation was used for INAA determination of arsenosugar 3-[5′-deoxy-5′-(dimethylarsinoyl)-β-ribofuranosyloxy]propylene glycol (As(328)) for the first time, while DMA, arsenosugars 3-[5′-deoxy-5′-(dimethylarsinoyl)-β-ribofuranosyloxy]-2-hydroxypropyl 2,3-dihydroxypropyl hydrogen phosphate (As(482)), and 3-[5′-deoxy-5′-(dimethylarsinoyl)-β-ribofuranosyloxy]-2-hydroxypropanesulfonic acid (As(392)) were determined following anion exchange separation. The contents of DMA, As(328), As(482), and As(392) were 0.41 mg kg−1 ± 0.09 mg kg−1, 1.10 mg kg−1 ± 0.25 mg kg−1, 5.23 mg kg−1 ± 0.46 mg kg−1, and 13.17 mg kg−1 ± 0.67 mg kg−1, respectively. Separately, components of arsenic species in the kelp extract including DMA, As(328), and inorganic arsenic were determined using LC-inductively coupled plasma mass spectrometry. Results of DMA and As(328) were 0.485 mg kg−1 ± 0.024 mg kg−1 and 1.14 mg kg−1 ± 0.03 mg kg−1, respectively, which were in good agreement with those determined by INAA in fractions of LC eluent. The most toxic species, AsIII and AsV were found to be < 0.07 mg kg−1 and 0.231 mg kg−1 ± 0.018 mg kg−1, respectively. Results were traceable to SI.
Keywords: arsenic species, arsenosugar, extraction, LC, INAA, ICP-MS
Graphical Abstract
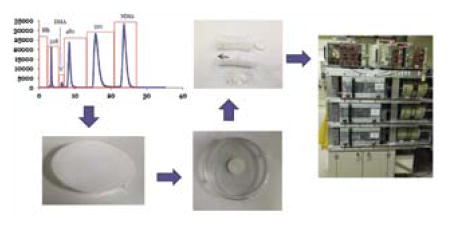
Arsenosugars in kelp extract were collected in fractions and determined by instrumental neutron activation for SI traceability.
Kelp has been in the human diet for centuries. More recently, it is also consumed as a dietary supplement because it is a source of vitamin K, folate, and dietary iodine, which is a vital element for thyroid health [1]. The National Institute of Standards and Technology (NIST) is developing a Standard Reference Material (SRM) 3232 Kelp Powder (Thallus laminariae) in support of US Food and Drug Administration’s (FDA) food safety and nutrition measurements program, and to meet the needs in the food industry for compliance with the current good manufacturing practice (cGMP) and the Food Safety Modernization Act (FSMA) [2]. A toxic element and a known carcinogen, arsenic is routinely measured in food safety assessment. Kelp contains tens of mg kg−1 arsenic, yet most of the arsenic extracted from the kelp is in forms of arsenosugars that are considered practically harmless [3–4]. Therefore, results of total arsenic do not shed light on the dietary safety of kelp. To make the results of arsenic measurement relevant for dietary safety assessment, not only the quantity but also the speciation of the arsenic must be provided [5]. Determination of arsenic species in kelp faces two major difficulties. First, not all arsenic in kelp is extractable [6–9]. Mild solvent yielded up to 70% of the arsenic in the kelp [5]. Harsh chemicals resulted in quantitative extraction; however, changes in arsenic speciation during the extraction process defeated the purpose of assessing arsenic species in kelp [7,8]. Second, measurements of arsenic species in kelp, primarily arsenosugars and arsenolipids [10, 11], are hampered by a lack of calibration standards. Amayo and coworkers reported an ingenious method to correct for the carbon effect that beset the calibration of arsenic species eluted in the gradient of methanol [12]; however, the carbon effect may not be the only culprit besetting the calibration of arsenic species because differences in arsenic species alone resulted in differing instrument response [13]. Despite the challenges, progress was made toward the development of certified reference materials (CRM) that can be used to validate methods for arsenic speciation of edible seaweeds. Madsen and coworkers described the development of an algal (Fucus serratus) reference material intended for use in arsenic speciation studies [14]. Reference material was assigned reference values of arsenosugars 3-[5′-deoxy-5′-(dimethylarsinoyl)-β-ribofuranosyloxy]propylene glycol (As(328)), 3-[5′-deoxy-5′-(dimethylarsinoyl)-β-ribofuranosyloxy]-2-hydroxypropyl 2,3-dihydroxypropyl hydrogen phosphate (As(482)), 3-[5′-deoxy-5′-(dimethylarsinoyl)-β-ribofuranosyloxy]-2-hydroxypropanesulfonic acid (As(392)), and 3-[5′-deoxy-5′-(dimethylarsinoyl)-β-ribofuranosyloxy]-2-hydroxypropyl hydrogen sulfate (As(408)) [14]. Narukawa et al. at the National Metrology Institute of Japan (NMIJ) certified arsenic acid (AsV) in NMIJ CRM 7405-a, trace elements and arsenic compounds in seaweed (Hijiki) [5]. Still, there is no edible seaweed reference material suitable for method validation of the most abundant arsenic species, i.e., arsenosugars.
Although mild solvent such as water-alcohol mixture at neutral pH does not result in a complete extraction of arsenic, it may be the basis for a procedure that reproducibly extracts most of the hydrophilic arsenic species from the kelp [15]. Such extraction procedure may be used for assignment of procedure-defined reference values of arsenic species in kelp [16]. Instrumental neutron activation analysis (INAA) was used to maintain measurement traceability to International System of Units (SI) in the determination of arsenosugars [17]. Because gamma-ray spectrometry is unaffected by the molecular bond of elements, INAA calibrated with a primary inorganic arsenic standard can be used to determine the arsenic in arsenosugars, and the results of the measurement are traceable to the inorganic arsenic primary standard. In conjunction with a reproducible extraction procedure, the species-independent calibration scheme can overcome the obstacles in value assignment of arsenosugars, making possible the development of reference materials much needed for method validation in measurements of edible seaweed commodities [18,19]. Here we describe the first implementation of the approach for the determination of > 98 % of arsenicals in the hydrophilic fraction of SRM 3232 for developing a kelp reference material value-assigned for arsenic species including arsenosugars. We discuss cation exchange and anion exchange for complete separation of each species for INAA determination, and we critically evaluate the offline INAA determination of arsenic in LC fractions ((LC)INAA) by comparing the results to those obtained by online inductively coupled plasma mass spectrometry analysis of the LC eluent (LC-ICP-MS).
Experimental
Instrumentation*
An Agilent (Santa Clara, CA, USA) model 7500 ICP-MS was used for the determination of total arsenic in kelp extracts. A PerkinElmer (Shelton, CT, USA) series 200 quaternary pump equipped with a series 200 autosampler was used for separating arsenic species for neutron activation analysis. A Perkin-Elmer DRCII ICP-MS instrument was used as the LC detector to determine the retention time for fraction collection and to monitor the chromatogram before and after the fraction collection. A Hamilton (Reno, NV, USA) PRP-X 100 250 mm × 4.6 mm (10 μm) column and a Macherey-Nagel (Dueren, Germany) Nucleosil 100-5 SA 250 mm × 4 mm (5 μm) column were used for the separation of arsenic species.
Reagent
ACS HPLC grade methanol, ACS grade pyridine, laboratory grade formic acid, Optima grade nitric acid (HNO3), and 70 mm diameter Whatman 542 filter paper (Cat No. 1542-070) were purchased from Fisher Scientific (Waltham, MA, USA). Puratronic grade ammonium carbonate ((NH4)2CO3, 99.999%) was purchased from Alpha Aesar (Ward Hill, MA, USA). Certified Reference Materials (CRM) SPEC-AS3 containing 1000 mg L−1 arsenous acid (AsIII) and CRM SPEC-AS5 containing 1000 mg L−1 arsenic acid (AsV) were purchased from Spex CertiPrep (Metuchen, NJ, USA). Disodium methylarsonate hexahydrate (MMA) was purchased from Chem Service (West Chester, PA, USA). Cacodylic acid (DMA) was purchased from Sigma-Aldrich (Milwaukee, WI, USA). Arsenobetaine (AB), arsenocholine (AC), and trimethylarsine oxide (TMAO) were purchased from Wako Chemicals (Richmond, VA, USA). Nominal 1.8 mg kg−1 As(328) standard solution was a gift from Jack Creed of the Environmental Protection Agency (EPA) National Exposure Research Laboratory in Cincinnati, OH, USA. SRM 3232 Kelp Powder (Thallus laminariae), SRM 2669 Arsenic Species in Frozen Human Urine, SRM 3103a Arsenic (As) Standard Solution, and SRM 83d Arsenic Trioxide (As2O3) Reductometric Standard were obtained from NIST. Locally prepared sub-boiling distilled water was used for all sample preparations.
Procedure
Arsenic species in the kelp powder was extracted with a solvent containing 50% (v/v) methanol in water. Arsenic species in the extract was separated using cation exchange or anion exchange LC followed by online ICP-MS determination or offline INAA determination of arsenic. For offline determination of arsenic, arsenic species in the LC eluent were collected in fractions. The arsenic in the collected fraction was transferred to a filter paper that was pressed into a pellet for INAA measurement. Each step of the procedure is detailed below.
Extraction optimization – a 0.5 to 1 g portion of kelp was transferred into a 15 mL Falcon tube. The solvent was added to the sample at 10:1 solvent volume to sample mass ratio. The mixture was vortexed at 40 Hz for 1 min, either allowed to stand on the bench (soaking) or sonicated. After soaking or sonication, the contents were vortexed for 30 s and centrifuged at 3600 gn for 30 min. The supernatant was decanted into a fresh 15 mL Falcon tube to yield the extract of the sample. Soaking durations of 0.15 h, 1 h, 2 h, 3.3 h, 3.5 h, and 20 h at ambient temperature of 21 °C were tested. Sonication durations of 0.25 h, 0.5 h, 0.75 h, and 1 h were tested. An ice pack was placed in the ultrasonic bath to maintain the temperature of the water between 20 °C to 24 °C.
Fraction collection – a 0.9 g kelp extract, 0.9 mL H2O, and 0.2 mL internal standard were transferred into a 2-mL vial. The internal standard was A 5 μg g−1 MMA solution or a 5 μg g−1 TMAO solution for separation by anion exchange or cation exchange, respectively. A 50 μL injection of the extract containing the internal standard was eluted isocratically at a flow rate of 1 mL min−1. A mobile phase containing 20 mmol L−1 of (NH4)2CO3 (pH=9) was used as the eluent for PRP X-100 column. The eluent was collected in the following min intervals: 0 – 2.0, 2.0 – 5.3, 5.3 – 6.7, 6.7 – 12.6, 12.6 – 19.6, and 19.6 – 25.0 for blank, As(328), DMA, As(482), As(392), and MMA fractions, respectively. A mobile phase containing 30 mmol L−1 of pyridine (pH=3) was used as the eluent for Nucleosil 100-5 SA column. The eluent was collected in the following min intervals: 0 – 1, 4.5 – 6.5, and 6.5 – 10 for blank, As(328), and TMAO fractions, respectively. Arsenic in extracts and fractions was determined by INAA.
INAA measurement – the solvent of the sample was evaporated in an N-EVAP evaporator (Organomation, Berlin, MA, USA) at 75 °C under a stream of nitrogen. The residue was rinsed with three 0.2 mL aliquots of water successively, and the rinsates were transferred to a Whatman 542 filter paper. The filter paper was dried under a heat lamp and pressed to form a pellet in a 13-mm diameter die. The pellets were sealed in pre-cleaned linear polyethylene for irradiation. Comparator standards were prepared similarly from a solution containing a known amount of SRM 83d. Packages consisting of three to six samples and two standards were irradiated for two 6 h periods in the NIST Center for Neutron Research (NCNR) RT-4 pneumatic irradiation channel at a neutron flux of 3.4×1013 s−1cm−2 with a 180° inversion of the rabbit after the first period for flux homogeneity [20]. After 24 h to 40 h decay the samples and standards were counted on high-rate capable, high resolution gamma spectrometers (35% efficient HPGe detectors with <1.8 keV resolution at 50,000 Hz input count rate) for 4 h to 16 h each [21]. With this approach a limit of detection < 0.2 ng As in each fraction was achieved despite relatively high background from 24Na and 82Br activities in the filter papers. The mass of arsenic in the pellet was determined using NAA software package on a laboratory computer (VMS Genie 2000, Canberra Industries, Meriden, CT, USA).
LC-ICP-MS measurement – extracts containing TMAO internal standard described above were diluted four-fold with water. An aliquot of the diluted extract was spiked with AsV, DMA, and As(328) for quantification by one-point standard addition. Two similarly prepared SRM 2669 Level II samples were used for method validation. Separately, a kelp extract spiked to contain 2.5 ng g−1 each of AsIII, AsV, MMA, DMA, AB, AC, and TMAO was measured for identification and for assessing the detection limit of arsenic species in kelp. Two buffer solutions were used for gradient elution at 1 mL min−1. Buffer A contained 0.10 mmol L−1 pyridine and 2% (v/v) methanol in balance water. Buffer B contained 30 mmol L−1 pyridine and 2 % (v/v) methanol in balance water at pH 3.0. Table 1 lists the instrumental parameter for LC-ICP-MS measurements.
Table 1.
Instrument parameters for the determination of AsV, DMA, and As(328) by LC-ICP-MS
| RF power (W) | 1300 | O2 cell gas flow (mL min−1) | 0.6 |
| Plasma gas flow (L min−1) | 15.0 | RPq | 0.5 |
| Neb gas flow (L min−1) | 0.95 | Measurement mass (u) | 91 |
| Aux gas flow (L min−1) | 1.2 | Injection volume (μL) | 50 |
| Step 0 (Equilibration) | 5 min, 100% buffer A | ||
| Step 1 | 4 min, 100% buffer A | ||
| Step 2 | 1 min, 100% buffer A to 100% buffer B | ||
| Step 3 | 15 min, 100% buffer B | ||
Calibrants mass fraction – the mass fraction of MMA, DMA, TMAO, AB, AC, and As(328) were determined by INAA and LC-ICP-MS using a procedure described in the literature [22]. Briefly, the total arsenic in the candidate calibrant solution was determined by INAA using the procedure described above. Arsenic impurities in the candidate calibrant solution were determined by LC-ICP-MS. The mass fraction of the candidate arsenic species in the solution was calculated by subtracting the arsenic impurities from the total arsenic.
Results and Discussion
Extraction
To estimate the amount of extractable arsenic, six kelp samples of 0.5 g each were extracted successively for five times. Samples were soaked for 2 h for the first extraction, and 1 h for each successive extraction. Figure 1 shows the arsenic found by the successive extractions. The curve approaches an asymptote of approximately 25 mg kg−1, which indicates the maximum amount of extractable arsenic in the kelp. The arsenic remaining in the kelp power was determined by INAA, and the total arsenic in SRM 3232 was calculated to be (mean ± s, n=6) 36.8 mg kg−1 ± 1.1 mg kg−1 by summing the extracted arsenic and the remaining arsenic. The extractable arsenic accounted for approximately 67 % of the total. Without a complete extraction of all arsenic, assignment of a certified value to a species was still possible if the extraction of the species was maximized [5,9]. When arsenosugars and inorganic arsenic were extracted simultaneously, maximizing the extraction efficiency for each species was very difficult if not impossible because of the difference in hydrophobicity of the species [9,15]. For this work, a solvent of 50% (v/v) methanol in water was used for a balanced extraction of inorganic arsenic and arsenosugars [15]. Therefore, the values assigned to the species were defined by the extraction procedure [16].
Figure 1.

The mass fraction of arsenic found through successive extractions of 0.5 g of kelp. Error bars represent 1s (n=6).
The duration and the intensity of solvent-solute interaction were investigated to maximize the extraction efficiency and to minimize the time for the extraction. The effect of soaking time on the amount of arsenic extracted is shown in Figure 2. No significate change in the amount of extracted arsenic was observed when the kelp powder was soaked for 2 h or longer. In a separate trial, samples of kelp powder in the solvent were sonicated for 0 h up to 1 h to assess the effect of sonic energy on the time required of the extraction. The amount of arsenic extracted from the samples increased monotonically with time. The extraction yield after 1 h of sonication was (mean ± s, n=3) 19 mg kg−1 ± 1 mg kg−1, which was approximately the same as that extracted after 1 h of soaking shown in Figure 2. The results suggested that soaking time was the dominant factor over sonication in the extraction. During the 1 h sonication, the temperature of the water bath increased from 20 °C to 24 °C despite the effort of maintaining the temperature with ice. Uncontrolled temperature can result in irreproducible extractions because the solubility and partition coefficient of the solute are functions of the temperature. Moreover, the energy distribution in a sonicator is not homogeneous [23], which can negatively impact the reproducibility of the extraction. Subsequently, extractions were conducted with 2 h of soaking without sonication. The mass of the extracted arsenic species was calculated as the mass concentration of the species in the extract multiplied by the volume of the solvent used in the extraction.
Figure 2.

The mass fraction of the extracted arsenic relative to the time the kelp powder soaked in a solvent of 50% (v/v) methanol in water. Error bars represent 1s (n=3).
(LC)INAA measurements
The stability of retention time is crucial for quantitative collection of arsenic species in fractions at predetermined intervals; therefore, isocratic elution method was exclusively used for (LC)INAA measurements. Figure 3 is a typical chromatogram obtained using the anion exchange separation method. The procedure for identifying the arsenic species in the eluent was described in the literature [17]. The internal standard MMA also served as the calibration standard because there was no detectable MMA in the kelp extract. Per Figure 3, the 2.0 min to 5.3 min time interval used to collect As(328) fraction included unknown U1, which would result in a positive bias for INAA measurements of As(328). A gradient cation exchange LC separation method was modified by eliminating the first gradient so that the isocratic separation would yield a clean fraction of As(328) [22]. Figure 4 shows the cation exchange chromatogram of arsenic species in the kelp extract. The cation exchange method reversed the order of elution relative to the anion exchange method and allowed better separation of the more easily protonated species such as As(328). Facilitated by the knowledge of the identity and the relative peak area of arsenic species shown in Figure 3, the AsV, DMA, and As(328) peaks in the cation exchange chromatogram were identified by spiking. The overlapped As(482) and As(392) peak was identified based on the elution order and the area relative to the As(328) peak. TMAO was below detection in the kelp extract, and the peak in Figure 4 was from the TMAO added as an internal standard. The As(482) and As(392) species were not resolved using the cation exchange column even with the gradient elution procedure discussed below. As(482) and As(392) contain phosphate and sulfonate functional groups with pKa 2 and -3, respectively. Given the working pH range of 2–8, the cation exchange column is unlikely capable of separating the two species based on the cation exchange mechanism.
Figure 3.

Anion exchange separation of arsenic species in the kelp extract. MMA was the internal standard.
Figure 4.

Cation exchange separation of arsenic species in the kelp extract. TMAO was the internal standard.
To determine the arsenic species in the kelp by (LC)INAA, eight packs of candidate SRM 3232 Kelp Powder were sampled in duplicates. Fractions of one LC run were collected for each sample except for the 16th sample. Fractions of five replicate LC runs were collected for the 16th sample to assess the repeatability of the (LC)INAA measurement process. For data reduction, INAA results of the cation exchange and anion exchange fractions were evaluated with Grubb’s test for outliers (Electronic supplemental Table S1). Five values were tested positive, and these values were excluded from the data analysis. The results from the remaining fractions and the total arsenic in the extract were employed to assess the homogeneity of analytes in the kelp using one-way analysis of variance (ANOVA). There was no detectable between-bottle inhomogeneity for arsenic species and total arsenic at 95 % confidence level because all p-values were > 0.05. Therefore, the duplicate samples were treated as independent samples for summary statistics.
Table 2 lists the mass fraction of As(328), DMA, As(482), and As(392) found in the kelp by anion exchange (LC)INAA and As(328) by cation exchange (LC)INAA. The results of As(sum) and As(sample) are statistically identical, suggesting that major components of arsenic species in the kelp extract are accounted for. The replication uncertainty of (LC)INAA measurements varied widely from a minimum of 1.6 % RSD for five replications of As(482) in the 16th extract to 29 % RSD for replications of DMA in sixteen extracts. The sources of uncertainties in (LC)INAA measurement were analyzed to tease out the magnitude of the uncertainty resulting from fraction collection and to understand the reasons behind the disparate replication RSD values. The scounting row lists the counting uncertainty expressed as one standard deviation relative to the mean value of the gamma-ray spectrometric measurements. At the experimental condition of this work, the counting uncertainties for As(328) and DMA were > 10% because of the low mass fraction of the two species in the kelp. In comparison, the mass fraction of As(482) and As(392) was higher, and hence, the < 5% counting uncertainty. The counting uncertainty of As(328) and DMA obscured the other sources of uncertainties; therefore, the results of As(482) and As(392) were investigated for other sources of uncertainties. All replication uncertainties of As(482) and As(392) included the contribution from sample inhomogeneity and (LC)INAA repeatability except the results of the 16th extract that included only (LC)INAA repeatability. Consequently, the repeatability RSD of the (LC)INAA method was estimated to be 1.6 % to 3.2 % based on the result of As(482) and As(392) for the 16th extract. Note that each (LC)INAA result requires two INAA measurements, one for the arsenic in the sample and the other the internal standard, and each measurement contributes 1% to 2% RSD typical for INAA measurement of arsenic. The instrumental measurement repeatability for (LC)INAA is calculated as the square root of sum squared of the two INAA measurement. The resulting range of 1.4% to 2.8% is very close to the 1.6% to 3.2% for As(482) and As(392) in the 16th extract that included the uncertainty of fraction collection in addition to the uncertainty of (LC)INAA repeatability. The implication is that the uncertainty of fraction collection steps including LC separation, fraction collection, and solvent evaporation were negligible relative to the replication uncertainty of (LC)INAA measurements. In addition, the replication RSD % of As(sum) and As(sample) closely matching each other also supported that sample manipulations of the fraction collection procedure contributed negligible uncertainty to (LC)INAA measurements. The time interval for collecting of As(328) fraction from anion exchange separation included the unknown species U1 that was approximately 5 % of the As(328) contents based on the relative peak area shown in Figure 3. However, the mass fraction of As(328) obtained by anion exchange (LC)INAA was statistically identical to that obtained by cation exchange (LC)INAA because expected 5% difference between the two is insignificant relative to >20% replication RSD of As(328) measurements.
Table 2.
Summary results of arsenic species and total arsenic found in the kelp extract by (LC)INAA. Results are in mg kg−1 units.
| As(328)3 | As(328)4 | DMA | As(482) | As(392) | As(sum)5 | As(sample)6 | |
|---|---|---|---|---|---|---|---|
| Sample Mean | 1.12 | 1.10 | 0.41 | 5.23 | 13.17 | 20.0 | 20.5 |
| s (n=14–16) | 0.23 | 0.24 | 0.12 | 0.29 | 0.96 | 1.1 | 1.2 |
| RSD(%) | 21 | 22 | 29 | 5.6 | 7.3 | 5.4 | 5.6 |
| scounting1(%) | 11.4 | 9.9 | 14.9 | 4.3 | 2.0 | ||
| U | 0.24 | 0.25 | 0.086 | 0.46 | 0.67 | 0.69 | 0.64 |
| Rep Mean2 | 1.06 | 1.14 | 0.34 | 5.11 | 13.64 | 20.1 | |
| s (n=5) | 0.22 | 0.29 | 0.063 | 0.084 | 0.43 | 0.47 | |
| RSD(%) | 21 | 25 | 19 | 1.6 | 3.2 | 2.3 |
scounting is the counting uncertainty of INAA measurements expressed as 1 standard deviation relative to the mean.
Rep Mean is the mean of the five repeat measurements of the 16th kelp extract.
Mass fraction of As(328) obtained by anion exchange (LC)INAA.
Mass fraction of As(328) obtained by cation exchange (LC)INAA.
Sum of arsenic species obtained by anon exchange (LC)INAA.
Mass fraction of arsenic in the extract obtained by INAA.
LC-ICP-MS measurements
LC-ICP-MS was used for the determination of AsIII, AsV, and DMA because SI traceable calibration standards for these arsenic species are available. A gradient cation exchange separation procedure was used to achieve baseline separation of DMA, AsIII, and AsV. Figure 5 is a gradient elution chromatogram of a kelp extract spiked with TMAO, which shows an improved resolution for AsV and DMA over the isocratic elution chromatogram shown in Figure 4. The As(482) and As(392) peaks were barely separated with the gradient elution due to technical challenges discussed above. A lack of As(482) and As(392) calibration standards precluded their quantification by LC-ICP-MS, which made the outcome of the separation inconsequential. An unknown peak U2 was observed near the void volume in Figure 5. The short retention time suggested a strong anion consistent with arsenosugar As(408), which was found in a closely related kelp species Laminaria japonica [6]. Two small peaks U3 and U4 were observed between 9 min and 11 min in an expanded view of the chromatogram as shown in Figure 5 inset B. The retention time of U3 was near that of AB, a species found in several seaweeds [24]. The same kelp extract was spiked with 2.5 ng g−1 each of AsIII, AsV, MMA, DMA, AB, AC, and TMAO to assist the identification of the unknowns, and the chromatogram of the spiked kelp was shown as a superimposing red trace in the insets of Figure 5. The superimposed chromatograms of the spiked and the unspiked kelp showed 0.1 min difference in retention time between AB and U3, proving that the species was not AB. The superimposed chromatograms of inset A showed that AsIII in the kelp was below detection, and the detection limit of AsIII in kelp was estimated to be 0.07 mg kg−1.
Figure 5.

A cation exchange chromatogram of gradient elution of kelp extract for LC-ICP-MS measurements. Inset A and B shows the expanded view near the retention time of AsIII and AB, respectively. The red traces in the insets show the chromatogram of the kelp extract spiked with 2.5 ng g−1 AsIII and 2.5 ng g−1 AB.
Table 3 lists the mass fraction of AsV, DMA, and As(328) found in the kelp. The results of As(328) obtained by LC-ICP-MS was in good agreement with those obtained by anion and cation exchange (LC)INAA as indicated by the results within one standard deviation of one another. The expanded uncertainty of the results obtained by LC-ICP-MS was about 1/10 of those by (LC)INAA, because ICP-MS was more sensitive while INAA was counting limited. Because of the difference in sensitivity between (LC)INAA and LC-ICP-MS techniques, columns of different capacity may be considered to improve the measurement. A semi-preparatory column can separate larger mass of analytes into fractions that improves the INAA detection. In contrast, a narrow-bore column can minimize solvent and sample consumption and reduce the analysis time without sacrificing the performance ICP-MS detection [25, 26]. There was 0.231 ±0.018 mg kg−1 of AsV in the kelp. That AsV was the only inorganic arsenic species in the kelp was consistent with inorganic arsenic constituents found in other edible seaweeds [5], and it was consistent with AsV being the primary form of inorganic arsenic species in the sea [9,27]. For SRM 2669 samples used to verify the method accuracy, the measured values (mean ± U, n=2) of AsV, and DMA at (6.53 ± 0.07) μg L−1 and (25.9 ± 0.8) μg L−1 were within the certified range at (6.16 ± 0.95) μg L−1 and (25.3 ± 0.7) μg L−1, respectively, suggesting no detectable bias in the measurement.
Table 3.
Summary results of AsIII, AsV, DMA, and As(328) found in the kelp extract by LC-ICP-MS. Results are in mg kg−1 units.
| AsV | DMA | As(328) | AsIII | |
|---|---|---|---|---|
| Mean | 0.231 | 0.485 | 1.139 | <0.07 |
| s (n=8) | 0.022 | 0.028 | 0.019 | |
| RSD(%) | 9.4 | 5.7 | 1.7 | |
| U | 0.018 | 0.024 | 0.033 |
Conclusion
(LC)INAA and LC-ICP-MS were used for SI traceable determination of arsenic species in the extract of SRM 3232 Kelp Powder. Approximately 95 % of the extractable arsenic in the kelp powder was arsenosugar. Only 1 % of the extractable arsenic was the toxic inorganic species. The unidentified species accounted for about 1.5 % of the extractable arsenic based on the ratio of the peak area.
Supplementary Material
Acknowledgments
The authors thank John T. Creed of EPA in Cincinnati, OH, for the gift of As(328) and the helpful discussion on arsenic species in kelp extract.
Footnotes
Disclaimer: Certain commercial items are identified in this paper to specify adequately the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the equipment identified is necessarily the best for the purpose.
References
- 1.USDA National Nutrient Database for Standard Reference. http://ndb.nal.usda.gov/
- 2.FDA Food Safety Modernization Act (FSMA) [accessed 1/26/2017]; http://www.fda.gov/Food/GuidanceRegulation/FSMA/
- 3.Kaise T, Oya-Ohta Y, Ochi T, Okubo T, Hanaoka K, Irgolic KJ, Sakurai T, Matsubara C. J Food Hyg Soc Jpn. 1996;37:135. [Google Scholar]
- 4.Feldmann J, Krupp EM. Anal Bioanal Chem. 2011;399:1735. doi: 10.1007/s00216-010-4303-6. [DOI] [PubMed] [Google Scholar]
- 5.Narukawa T, Inagaki K, Zhu Y, Kuroiwa T, Narushima I, Chiba K, Hioki A. Anal Bioanal Chem. 2012;402:1713. doi: 10.1007/s00216-011-5584-0. [DOI] [PubMed] [Google Scholar]
- 6.Van Hulle M, Zhang C, Zhang X, Cornelis R. Analyst. 2002;127:634. doi: 10.1039/b110940e. [DOI] [PubMed] [Google Scholar]
- 7.Gamble BM, Gallagher PA, Shoemaker JA, Parks AN, Wei X, Schwegel CA, Creed JT. Analyst. 2002;127:781. doi: 10.1039/b109748b. [DOI] [PubMed] [Google Scholar]
- 8.Gamble BM, Gallagher PA, Shoemaker JA, Parks AN, Freeman DM, Schwegel CA, Creed JT. Analyst. 2003;128:1458. doi: 10.1039/b306931a. [DOI] [PubMed] [Google Scholar]
- 9.Taylor V, Goodale B, Raab A, Schwerdtle T, Reimer K, Conklin S, Karagas MR, Francesconi KA. Sci Total Environ. 2017;580:266. doi: 10.1016/j.scitotenv.2016.12.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raab A, Newcombe C, Pitton D, Ebel R, Feldmann J. Anal Chem. 2013;85:2817. doi: 10.1021/ac303340t. [DOI] [PubMed] [Google Scholar]
- 11.Glabonjat RA, Raber G, Jensen KB, Ehgartner J, Francesconi KA. Anal Chem. 2014;86:10282. doi: 10.1021/ac502488f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amayo KO, Petursdottir A, Newcombe C, Gunnlaugsdottir H, Raab A, Krupp EM, Feldmann J. Anal Chem. 2011;83:3589. doi: 10.1021/ac2005873. [DOI] [PubMed] [Google Scholar]
- 13.Yu LL, Butler TA, Turk GC. Anal Chem. 2006;78:1651. doi: 10.1021/ac051732i. [DOI] [PubMed] [Google Scholar]
- 14.Madsen AD, Goessler W, Pedersen SN, Francesconi KA. J Anal At Spectrom. 2000;15:657. [Google Scholar]
- 15.Yoshinaga J, Shibata Y, Horiguchi T, Morita M. Accredit Qual Assur. 1997;2:154. [Google Scholar]
- 16.van Elteren JT, Šlejkovec Z, Kahn M, Goessler W. Anal Chim Acta. 2007;585:24. doi: 10.1016/j.aca.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 17.Yu LL, Wei C, Zeisler R, Tong J, Oflaz R, Bao H, Wang J. Anal Bioanal Chem. 2015;407:3517. doi: 10.1007/s00216-015-8567-8. [DOI] [PubMed] [Google Scholar]
- 18.Briscoe ML, Ugrai TM, Crewell J, Carter AT. Spectrosc. 2015;30:5. [Google Scholar]
- 19.Raab A, Fecher P, Feldmann J. Microchim Acta. 2005;151:153. [Google Scholar]
- 20.Lindstrom RM, Zeisler R, Greenberg RR. J Radioanal Nucl Chem. 2007;271:311. [Google Scholar]
- 21.Zeisler R. J Radioanal Nucl Chem. 2000;244:507. doi: 10.1007/s10967-017-5342-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis WC, Zeisler R, Sieber JR, Yu LL. Anal Bioanal Chem. 2010;396:3041. doi: 10.1007/s00216-010-3541-y. [DOI] [PubMed] [Google Scholar]
- 23.Santos HM, Capelo JL. Talanta. 2007;73:795. doi: 10.1016/j.talanta.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 24.Nischwitz V, Pergantis SA. Analyst. 2005;130:1348–1350. doi: 10.1039/b509547f. [DOI] [PubMed] [Google Scholar]
- 25.Todolí JL, Grotti M. J Chromatogr A. 2010;1217:7428. doi: 10.1016/j.chroma.2010.09.065. [DOI] [PubMed] [Google Scholar]
- 26.Wangkarn S, Pergantis SA. J Anal At Spectrom. 2000;15:627. [Google Scholar]
- 27.Andreae MO. Deep-Sea Res. 1978;25:391. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.