

Abstract
As legacy toxicogenomics databases have become available, improved data mining approaches are now key to extracting and visualizing subtle relationships between toxicants and gene expression. In the present study, a novel “aggregating bundles of clusters” (ABC) procedure was applied to separate cholestatic from non-cholestatic drugs and model toxicants in the Johnson & Johnson (Janssen) rat liver toxicogenomics database [3]. Drug-induced cholestasis is an important issue, particularly when a new compound enters the market with this liability, with standard preclinical models often mispredicting this toxicity. Three well-characterized cholestasis-responsive genes (Cyp7a1, Mrp3 and Bsep) were chosen from a previous in-house Janssen gene expression signature; these three genes show differing, non-redundant responses across the 90+ paradigm compounds in our database. Using the ABC procedure, extraneous contributions were minimized in comparisons of compound gene responses. All genes were assigned weights proportional to their correlations with Cyp7a1, Mrp3 and Bsep, and a resampling technique was used to derive a stable measure of compound similarity. The compounds that were known to be associated with rat cholestasis generally had small values of this measure relative to each other but also had large values of this measure relative to non-cholestatic compounds. Visualization of the data with the ABC-derived signature showed a very tight, essentially identically behaving cluster of robust human cholestatic drugs and experimental cholestatic toxicants (ethinyl estradiol, LPS, ANIT and methylene dianiline, disulfiram, naltrexone, methapyrilene, phenacetin, alpha-methyl dopa, flutamide, the NSAIDs–—indomethacin, flurbiprofen, diclofenac, flufenamic acid, sulindac, and nimesulide, butylated hydroxytoluene, piperonyl butoxide, and bromobenzene), some slightly less active compounds (3′-acetamidofluorene, amsacrine, hydralazine, tannic acid), some drugs that behaved very differently, and were distinct from both non-cholestatic and cholestatic drugs (ketoconazole, dipyridamole, cyproheptadine and aniline), and many postulated human cholestatic drugs that in rat showed no evidence of cholestasis (chlorpromazine, erythromycin, niacin, captopril, dapsone, rifampicin, glibenclamide, simvastatin, furosemide, tamoxifen, and sulfamethoxazole). Most of these latter drugs were noted previously by other groups as showing cholestasis only in humans. The results of this work suggest that the ABC procedure and similar statistical approaches can be instrumental in combining data to compare toxicants across toxicogenomics databases, extract similarities among responses and reduce unexplained data varation.
Keywords: Cluster analysis, Cholestasis, Gene signature, Microarray, Prediction, Toxicogenomics
1. Introduction
Cholestasis, or the reduction of bile flow, can progress to serious hepatotoxicity in patients and is a particular concern in evaluation of novel drug candidates. Extrahepatic cholestasis occurs within the bile duct, forming blockages via concentrated, precipitated drugs, or by damage of the biliary epithelial cells, and loss of bile transport and flow. Although a variety of drugs are concentrated in the bile, drug-induced obstructive cholestasis is often readily observed preclinically, and can be avoided. Intrahepatic cholestasis associated with hepatotoxicity generally results from bile salt export pump (BSEP) inhibition, which if not adequately handled by accessory pathways such as MRP3, results in accumulation of detergent-like bile salts, that are toxic to hepatocytes. At high dose levels, novel drug candidates can bind to multiple hepatic transporters and trigger cholestatic signals. Therefore, screening for transporter inhibition in membrane vesicles is a typical step in the drug discovery and development process [21], [22], [25], [33]. Identification and translational understanding of cholestatic responses in preclinical species is key step in the nonclinical safety assessment before initiating pharmaceutical clinical development.
The rat is a well-characterized model in which to study cholestasis, and is a principal species for preclinical toxicology studies. There are some differences from human: bile salts are conjugated by different enzymes (cholic acid and chenodeoxycholic acid are predominantly conjugated with glycine in man versus with taurine in the rat), the molecular weight “filter” for drug biliary secretion is generally lower than in humans (∼350 molecular weight in rat, versus ∼500 in humans), and the rat lacks a gall bladder [1], [17]. This latter difference is an experimental advantage in that there is more consistent bile flow regulation in rats. Additionally, laboratory rats eat a well-defined, carbohydrate-rich rodent chow and feed most heavily during the dark cycle, more continually through the day than most humans. These feeding habits contribute to less variation in rat biliary transport and a continual and higher volume of bile flow [10]. Despite numerous species differences when comparing drug effects in rat to human, most transporter inhibition studies show remarkable consistency; for example, no rat versus human differences were observed in a large study of drug inhibition of BSEP transport [21].
A number of models of cholestasis in the rat are well studied and established: biliary ligation, which models bile duct obstruction; ANIT, which damages/destroys biliary epithelium; LPS, which down-regulates much of the hepatic metabolism including most transporters; and glucuronidated ethinyl estradiol-induced cholestasis by inhibition of BSEP [2], [7], [11], [20], [23], [34], [36], [39]. All of these models have been characterized by gene expression on microarrays [7], [11], [34], [39]. There is a misleading tendency to generalize the specifics of each of these different models to all forms of cholestasis. For characterization of novel drug candidates, there is a need to capture as much of the invariant cholestasis gene response as possible across a wide range of pharmaceutical structural classes.
At Janssen we have developed a predictive, 24 h treatment rat toxicogenomic database using approximately one hundred non-proprietary treatments and four times as many proprietary compound treatments, the latter having supporting exposure, clinical chemistry and histopathology data, often out to a month of dosing. We have published predictive gene signatures for non-genotoxic carcinogens [24], [27] macrophage and PPARα receptor activation [18] and particularly oxidative stress/reactive metabolite responses for detecting idiosyncratic hepatotoxicants [14], [15]. We previously developed a cholestasis gene expression signature using in-house proprietary compounds that induced cholestasis in rat studies (unpublished data). In the present study, we have used three robust non-redundant genes (Cyp7a1, Mrp3 and Bsep) from this previous rat signature which have been implicated in cholestasis [13], [19], [29], [35]. Non-redundant genes differentially respond with large variances to compounds across our database, therefore a single one of these genes suffices for relevant transcriptomics information. Protein expression of MRP3 is up-regulated during cholestasis and genetic defects in MRP3 have been linked with pregnancy or estrogen hormone-induced cholestasis [28]. Mutations of the gene encoding the human bile salt export pump are implicated in progressive familial intrahepatic cholestasis type 2 (PFIC2), and inhibition of human and/or rat BSEP transporter constructs have been demonstrated to be correlated to cholestatic potential [4]. CYP7A1 is an important participant in transcriptional regulation, due to bile acid synthesis via nuclear hormone receptors and modulates cholestasis response via the classical pathway of bile enzyme synthesis [12], [26], [31].
From this starting point of three genes, we then applied the “aggregating bundles of clusters” (ABC) statistical approach [3] to develop a rat cholestasis gene expression signature that better discriminates cholestatic from non-cholestatic compounds. Both our unpublished proprietary signature and the present 100-gene rat cholestasis signature of this work yield good separation of compounds, and can discriminate many drugs known to cause cholestasis in man.
2. Materials and methods
2.1. Animals
Male Sprague-Dawley rats, seven to eight weeks old, and approximately 275 g body weight (Charles River Laboratories, Inc.) were used for experiments. Animals were individually housed in wire-bottom cages, on a 12 h/light/dark cycle, and fed Certified Rodent Diet 5002 (LabDiet) ad libitum, with free access to water. On the day prior to dosing, animals were randomized by weight and allocated to groups (n = 3 rats/group). The route of administration for each test article is denoted in Table 1. Oral gavage was the most commonly utilized mode of administration as it is the typical route for the majority of compounds (predominantly pharmaceuticals) included in the training set. Animals were dosed in the morning hours, with health checks performed at 1 h and 4 h after dosing, and the end of the workday. Any animals deemed to be in poor health status were evaluated by a veterinarian: in these experiments no animals were prematurely terminated due to poor health.
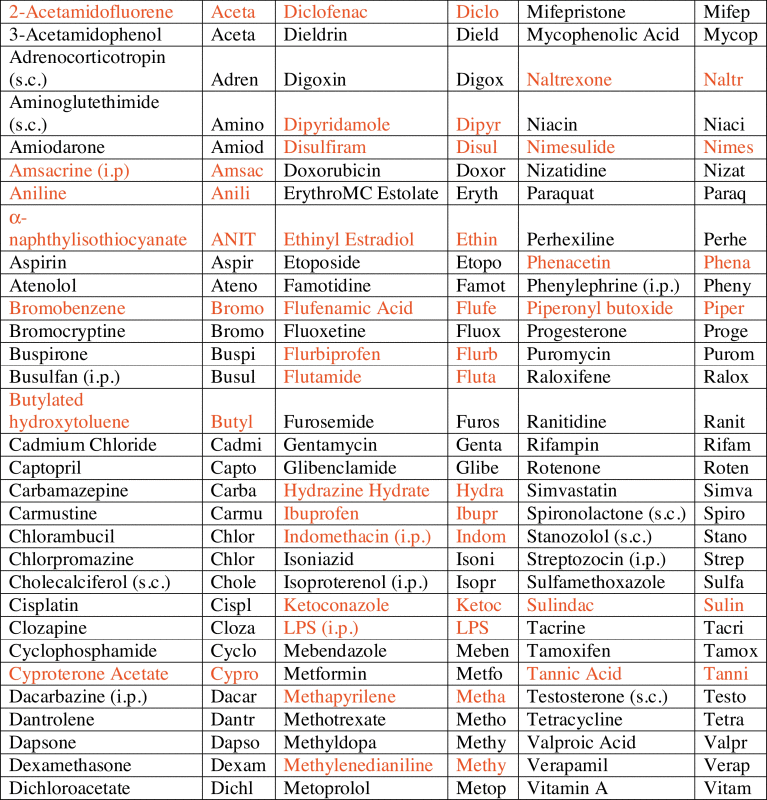
Table 1.
Figure abbreviations for cholestatic (red) and non-cholestatic (black) pharmaceuticals and model toxicants used in this study. Compounds were dosed via oral gavage (p.o.) unless otherwised noted (subcutaneous—s.c.; intraperitoneal—i.p.).
 |
Necropsy was performed on fasted rats, 24 h following dosing. Rats were killed by exsanguination, severing the vena cava under CO2 analgesia, and liver sections (approximately 200 mg of the right medial lobe) were transfered to labeled cryo tubes and snap frozen in liquid nitrogen.
The selected dose level was the maximum tolerated dose, as elucidated from the literature [18], [24]. Compound selection rationale, dose levels, route of exposure, group size, and experiment number are documented within Johnson and Johnson publically-accessible NIEHS-hosted database http://cebs.niehs.nih.gov.
In all instances, the animals were humanely handled and according to institutional guidelines, in accordance with the IACUC and NRC Guide for the Care and Use of Laboratory Animals.
2.2. Generation of microarray data
Total RNA was extracted from liver samples using Qiagen RNEasy Midi kits (Qiagen, Inc., Valencia, CA) as per kit instructions. The amount of RNA in the samples was determined spectrophotometrically by absorbance ratio at 260 and 280 nm. Quality of RNA in the samples was assessed using rRNA peaks determined by an Agilent 2100 Bioanalyzer.
Reverse transcription of RNA with a T7 promoter oligo (dT) primer, followed by in vitro transcription using the RiboBeast 1-Round Aminoallyl-aRNA Amplification Kit was used for one round of RNA amplification (Epicentre, Madison, WI). Superscript II (Invitrogen, Carslbad, CA) was used as the reverse transcriptase, and the Rneasy96 block was used for purification (Qiagen, Valencia, CA) as previously described [24]. Purified biotin-labeled cRNA was fragmented at 94 °C for 20 min and added to Codelink hybridization buffer A and B (GE Healthcare, Chandler, AZ). Each cRNA sample was applied to two Codelink Rat Whole Genome microarrays (GE Healthcare) using the manufacturer’s protocol, and hybridized at 37 °C overnight. Microarrays were washed, stained with an Alexa555-streptavidin conjugate (Invitrogen), and then scanned at 532 nm with an Agilent G2565BA Microarray Scanner (Agilent Technologies, Palo Alto, CA). Fluorescence intensities for the microarray features were determined using Codelink EXPv4.1 software (GE Healthcare). Additional details regarding experimental procedures may be found in Ref. [18].
2.3. Preprocessing of microarray data
The data for this study was part of a larger set of toxicogenomic experiments which spanned several hybridization batches. There were several vehicle controls run concurrently in each hybridization batch to establish baseline gene expression levels. The normalization procedure consisted of the following steps within each hybridization batch: (i) spot winsorization on the log-intensities to remove outliers; (b) quantile normalization to the median of the vehicle controls in that batch; (c) mean summarization of the log-intensities for each gene of the technical replicates for each RNA sample; (d) log ratios of the mean in (c) with the median of the vehicle control within the hybridization set. Details of the winsorization and quantile normalization can be found in Ref. [3]. The dataset consists of log-ratios of the expression levels to the median vehicle control of the corresponding hybridization batch as described above, for each sample, for the 35129 probesets on the microarray chip. All analyses described below were conducted on the probesets. Compounds that were known PPAR agonists and macrophage activators (many of the acutely hepatototoxic compounds) were omitted from analysis despite cholestatic properties, since their robust effects on gene expression interfered with subtle signatures, as noted previously [24], [27]. LPS was added to the analysis since it is an important cholestatic model compound in the rat.
2.4. Public availability of microarray data
The data used in this study is from the non-proprietary Johnson & Johnson (Janssen) toxicogenomics Codelink database, available in normalized ratio format in collaboration with Strand Life Sciences, Pvt., Ltd., at the weblink: http://pubdata.strandls.com/ in the folder “jnj codelink data/” [30].
Raw data related to this project are also publically available in the CEBS at the NIEHS website.
To access the CEBS website:
-
•
Go to http://cebs.niehs.nih.gov and select the “Open CEBS” option. Select the “CEBS accession number” option under the “Search Study” area.
-
•
Enter ‘004-00009-0000-000-3’ and select “Search.”
-
•
The folder “J&J Compounds of Known Toxicity” will appear. Open folder by clicking on the arrow to view the studies in the database. Details on each study may be obtained by double clicking on that file.
-
•
To access specific data by compound, organ, and Unigene ID, select “J&J Codelink Data” in the “Workflows” area.
-
•Data can be directly downloaded via the CEBS FTP site in two ways. Go to http://cebs.niehs.nih.gov.
-
•Select “Download Data” option. All user submitted microarray data and quantile normalized files are available at: ftp://157.98.192.110/ntp-cebs/datatype/microarray/J&J/004-00009-0000-000-3/ or can be found by selecting the options: Download data > microarray > J&J > 004-00009-0000-000-3.
-
•
-
•
Alternatively, microarray data organized by study are available at the link: ftp://157.98.192.110/ntp-cebs/FileList/004-00009-0000-000-3/.
2.5. Biostatistical analysis of the microarray data
A novel ABC-like procedure was applied [3]. The following three steps were performed R times:
-
•
N compounds were selected at random with replacement. Any repeats and/or replicates were discarded. Assume that this left N* compounds (N* ≤ N). This is the “bag” of compounds for this run.
-
•
An association score Ak was assigned to each gene k. Ak was taken to be the maximum of the correlations between the kth gene and each of the three focal genes across the N* samples; any gene that was highly correlated to at least one of the three focal genes would have a high association score. Under the premise that genes with relatively large values of Ak were more likely to carry cholestasis related information than genes with relatively small values of Ak, a weight wk was calculated for each gene k: wk = 1/(Rk + √G), where Rk was the rank of Ak.
-
•
√G genes were selected using weighted random sampling without replacement with weights (wk). This small set of genes included an overabundance of genes correlated with the three focal genes. For only these N* compounds and G* genes, a cluster analysis was run using a standard clustering algorithm such as Ward’s hierarchical clustering procedure to get a set of √N base clusters.
Once these steps were run R times, the results were collated. Of the number of times that the ith and jth compounds were both included in the same bag (in step (1) of the procedure), the proportion Cij of times that they also fell into the same cluster (in step (3) of the procedure) was determined.
Let Dij = 1 − Cij. A value of Dij close to zero indicated that compounds i and j often clustered together in the base clusters, thereby implying that they were relatively similar to each other. Alternatively, a value of Dij close to 1 indicated that compounds i and j rarely clustered together in the base clusters, thereby implying that they were relatively dissimilar to one another. Thus Dij could serve as a dissimilarity measure. Multidimensional scaling of (Dij) was then used to generate a two-dimensional representation of the similarities and dissimilarities of the compounds.
Following the ABC-like procedure, a boosting procedure incorporating an elastic net approach [16], [41] was applied and performed 1000 times, with frequencies calculated for all signature sets.
3. Results
Although an in-house proprietary cholestasis signature was available our goal was to generate a robust gene expression signature using only non-proprietary drugs and compounds. Table 1 provides a list of cholestatic (red) and non-cholestatic (black) compounds. Initially expression profiles of only three genes (Cyp7a1, Mrp3 and Bsep), well characterized as cholestasis-responsive genes, were examined. Their expression profiles, plotted in a three-dimensional scatterplot, with known cholestatic compounds highlighted in red, did not reveal a useful separation of the compounds in any readily interpretable fashion (Fig. 1).
Fig. 1.

mRNA expression profiles of 3 genes (Cyp7a1, Mrp3 and Bsep) to cholestatic (red) and non-cholestatic (black) training set pharmaceuticals and model toxicants in male rat liver. Note that no clear separation was observed between classes.
We next examined the simultaneous expression profiles of all genes using several methods. One approach was to reduce the dimensionality of the data using principal component analysis and to then plot the first two components (Fig. 2). Another was to calculate the dissimilarity between every pair of compounds using either Euclidean distance (Fig. 3) or 1-correlation (Fig. 4) and to then use multidimensional scaling to display the compounds on a two-dimensional scatterplot [37]. This indicated fair separation between groups, typical of many gene expression signatures, however many compounds in the two groups overlapped, with no obvious basis for the way cholestatic and non-cholestatic compounds distributed in space. Clearly while it was insufficient to consider three key genes (Fig. 1), consideration of all genes was also unproductive (Fig. 2, Fig. 3, Fig. 4). Although such mild separations were considered acceptable in past studies [24], a better statistical analysis method was sought.
Fig. 2.

Principal component analysis of training set (plot of first two components). The applied methods generate a composite representative point with arbitrary units within a two-dimensional space reflecting the similarity of one compound to another.
Fig. 3.

Two-dimensional scatterplot of training set using multidimensional scaling to illustrate dissimilarity between every pair of pharmaceuticals and model toxicants based on Euclidean distance. The applied methods generate a composite representative point with arbitrary units within a two-dimensional space reflecting the similarity of one compound to another.
Fig. 4.

Two-dimensional scatterplot of training set using multidimensional scaling to illustrate dissimilarity between every pair of pharmaceuticals and model toxicants based on 1-correlation. The applied methods generate a composite representative point with arbitrary units within a two-dimensional space reflecting the similarity of one compound to another.
A novel approach based on the ABC (“aggregating bundles of clusters”) technique was implemented [3]. The premise of this method is that most of the genes do not contribute to the organization of the compounds but the analysis should tilt towards the genes that do. Because the three selected “focal” non-redundant genes Cyp7a1, Mrp3 and Bsep are implicated with cholestasis, other gene responses could be ranked in importance by how much they correlated to responses of these three genes. Application of the ABC procedure yielded a display of the compounds that was interpretable in the context of cholestasis. Use of Dij as a dissimilarity measure allowed for generation of a two-dimensional representation of similar and dissimilar compounds (Fig. 5). Compounds that are similar to each other according to (Dij) are oriented close to one another while compounds that are dissimilar according to (Dij) are spaced further apart. Many of the best-characterized cholestatic compounds (depicted in red) fell into a tight cluster on the lower right-hand side of Fig. 5, and their gene responses after selection were almost identical. An expanded view of this cholestatic cluster is provided in Fig. 6. A few compounds with weaker responses, or those that act via a different mechanism, displayed as a “bridge” in this plot between the tight cluster of known cholestatic (red) compounds on the lower right and the clearly non-cholestatic (black) compounds at bottom left in Fig. 5. Additionally there was some separation of compounds falling in the non-cholestatic left region of Fig. 5: while a number of the compounds on the top left and those closest to the cluster on the lower right are reportedly cholestatic in man, separation from non-cholestatic compounds was minimal and not investigated further in this study.
Fig. 5.

Two-dimensional projection of the similarities and dissimilarities of training set pharmaceuticals and model toxicants based on multi-dimensional scaling following the ABC clustering procedure. Non-cholestatic compounds are noted in black and cholestatic compunds are noted in red. The applied statistical methods generate a composite representative point with arbitrary units within a two-dimensional space reflecting the similarity of one compound to another. A strong cholestatic (red) cluster can be noted on the lower right.
Fig. 6.

Expanded view of the cholestatic (red) compounds in the right-hand “arm” of Fig. 1. Two-dimensional projection of the similarities and dissimilarities of training set pharmaceuticals and model toxicants based on multi-dimensional scaling following the ABC clustering procedure. The applied statistical methods generate a composite representative point with arbitrary units within a two-dimensional space reflecting the similarity of one compound to another.
Cholestatic potential of compounds on the lower right (cholestatic) and bottom left (non-cholestatic) were confirmed via literature references. From this exercise, a ten-compound cholestatic (cholestatic in rat, and excluding LPS and gastrointestinally toxic NSAIDs) and four-compound non-cholestatic (non-cholestatic in rat, but cholestatic in human) compound training set (Table 2) was used to derive an improved gene expression signature for rat-specific cholestasis. We applied a boosting procedure and elastic net methodology to select a signature set of Codelink IDs [16], [41]. The top set of 100 genes derived using an arbitrary cutoff with highest frequencies (i.e., those with highest statistical significance) is recorded as our final rat cholestasis signature and presented in Table 3.
Table 2.
ABC technique-derived prediction set of rat cholestatic and non-cholestatic pharmaceuticals and model toxicants. Note the selected non-cholestatic compounds in rat are all cholestatic in human patients.
| Cholestatic compounds from the right arm | Non-cholestatic compounds from the left arm |
|---|---|
| Ethinyl Estradiol | ErythroMC Estolate |
| Methylenedianiline | Rifampin |
| ANIT | Captopril |
| Disulfiram | Niacin |
| Piperonyl butoxide | |
| Methapyrilene | |
| Ketoconazole | |
| Bromobenzene | |
| Flutamide | |
| Nimesulide |
Table 3.
Rat specific cholestasis gene signature. The top 100 genes associated with rat cholestasis, as derived by a boosting procedure with the elastic net approach. Genes were selected by comparing compounds (all known cholestatic in patients) from the two training sets in Table 2.
| Codelink | Accession # | Rat gene |
|---|---|---|
| GE1266870 | BE115850 | Gstcd |
| GE20245 | NM_013215 | Akr7a3 |
| GE1298481 | BM391646 | Nars |
| GE21882 | NM_031588 | Nrg1 |
| GE14709 | AI230228 | Psat1 |
| GE1147805 | CB578405 | Zfp57 |
| GE12632 | AI704698 | Tubb2c |
| GE16026 | AW252110 | Rexo2 |
| GE1130267 | BI296275 | Mmd2 |
| GE20928 | NM_031828 | Kcnma1 |
| GE16596 | AW915559 | Pprc1 |
| GE20102 | NM_012991 | Npap60 |
| GE1206415 | BI297088 | Golt1b |
| GE15377 | BI296275 | Mmd2 |
| GE15995 | AW251324 | Mthfd2-201 |
| GE18028 | BI273851 | Hmgn1 |
| GE1287407 | BI282107 | Pbdc1 |
| GE1262741 | BE110134 | Similar to ATPase inhibitory factor 1 |
| GE1259381 | NM_138515 | Cyp2d4v1 |
| GE19894 | NM_012674 | Spink1 |
| GE1173529 | CA507720 | Pgs1 |
| GE16306 | BQ206238 | Ankhd1 |
| GE1168184 | NM_031318 | Tctex1 |
| GE13574 | NM_053800 | Txn1 |
| GE1185733 | BF552430 | NosipGE1116501 BF565830 |
| GE17633 | BE110630 | Fam98a |
| GE19200 | BF550623 | Cad |
| GE1128152 | AF507943 | GTPase Ran |
| GE1150923 | BF398625 | Yars |
| GE17038 | H33863 | Mrps27 |
| GE1106781 | BU760106 | Tubb6 |
| GE20964 | NM_133539 | Mrpl17 |
| GE1287274 | BF405056 | |
| GE1249888 | BG380189 | Map3k6 |
| GE16626 | BI302502 | Fkbp11 |
| GE1167706 | NM_173102 | Tubb5 |
| GE1261576 | BF416050 | |
| GE20940 | NM_024390 | Hpgd |
| GE18776 | BF406522 | Cdr2 |
| GE17115 | BE096047 | Ran |
| GE1174448 | BC062045 | similar to lipase, member H |
| GE1181134 | BM389611 | |
| GE19529 | NM_134449 | Prkcdbp |
| GE1101245 | BF401159 | |
| GE15329 | AI412180 | Gsr |
| GE1170908 | BE096969 | Slc35e3 |
| GE1195574 | BG373486 | Lsm4 |
| GE20274 | NM_017014 | Gstm1 |
| GE14242 | BI291621 | Coq6 |
| GE1305121 | AI235475 | EST232037 |
| GE12903 | AA946350 | Gnai1 |
| GE1206195 | BI274696 | similar to Fam48a protein |
| GE19392 | BG374448 | TLOAEA57YJ04 |
| GE1166787 | BM391708 | |
| GE1291343 | BQ202678 | Xpo1 |
| GE1219528 | BF562712 | Trmt10a |
| GE1135723 | BM384823 | Ppp1r3b |
| GE1237072 | BQ202004 | Gstm4 |
| GE1277644 | NM_031039 | Gpt1 |
| GE1236134 | AW433632 | |
| GE22230 | NM_053962 | Sds |
| GE17670 | BE113943 | Tigar |
| GE1216317 | BU759545 | Trmt10a |
| GE1210818 | NM_024148 | Apex1 |
| GE1125325 | X78847 | GstYc2 |
| GE1262042 | BI296358 | Apmap |
| GE1100230 | NM_138831 | Slc16a10 |
| GE12644 | NM_175707 | Ppil3 |
| GE1297200 | BF412090 | Xkr8 |
| GE1168155 | BF288225 | EST452816 |
| GE17771 | BE113165 | Nek6 |
| GE1205310 | NM_133558 | Cml1 |
| GE22027 | NM_053902 | Kynu |
| GE1197342 | BQ194442 | Krt23 |
| GE15769 | AA956532 | |
| GE14364 | BG379780 | Mcee |
| GE20988 | NM_031051 | Mif |
| GE1158473 | BF521726 | Srxn1 |
| GE16476 | BM388044 | |
| GE16012 | H35647 | Tnfrsf12a |
| GE18057 | BF281848 | Mthfd1l |
| GE1284234 | AI169152 | Mafb |
| GE1130388 | AI137440 | |
| GE1167776 | BF420077 | |
| GE16853 | AW918024 | Nif3l1 |
| GE1119096 | CA505586 | |
| GE1274350 | BF388173 | Ptprd |
| GE1201399 | BF415461 | |
| GE15155 | AI409108 | Nup205_predicted |
| GE13570 | NM_053439 | Ran |
| GE15126 | AI408727 | Lurap1l |
| GE1296800 | NM_184048 | Taf9 |
| GE16570 | AW915407 | EST346711 |
| GE12527 | BM384489 | Hmgn3 |
| GE1228263 | BE098757 | |
| GE20373 | NM_017147 | Cfl1 |
| GE1236758 | NM_198771 | Fam3c |
| GE19575 | NM_053962 | Sds |
| GE1210173 | BF394858 |
Using the ABC approach, our results indicate that separation of paradigm human cholestasis compounds into cholestatic/non-cholestatic classes using rat experimental data gives as good or better separation than that observed with our proprietary Johnson & Johnson/Janssen compound-derived signature.
4. Discussion
In the present study the ABC statistical approach [3] was used with three non-redundant cholestasis indicator genes, Cyp7a1, Mrp3 and Bsep, to separate and visualize human cholestatic and non-cholestatic compounds in the rat. Although these three genes are well known to be regulated by the nuclear bile acid receptor (FXR), their different responses across the drugs tested (their non-redundant responses) suggest more complicated regulation in rats, consistent with integrated bile salt, cholesterol, sterols and other hepatic lipid metabolism pathways and the host of transcription factors involved (such as LXR, SREBP, PPAR, PXR, CAR and others, in addition to FXR and SHP; reviewed by Karagianni and Talianidis [9]. These three starting genes are recognized to be important in cholestasis, and were important in our previous in-house cholestasis signature based on multiple doses of proprietary compounds and histopathological evidence of cholestasis (Alex Nie, unpublished data). The separation by the ABC approach of paradigm human cholestasis compounds into cholestatic/non-cholestatic compounds in the rat was as good or better than observed with the proprietary Johnson & Johnson/Janssen compound derived signature. The ABC approach [3] was used to statistically weigh additional genes according to their similarity/dissimilarity to the three pivotal cholestasis genes and to derive predictions for potential cholestatic propensities of our paradigm compounds. The most robust cholestatic and non-cholestatic compounds in rat (all cholestatic in human) derived from this approach were used as a training set, along with a boosting procedure/elastic net approach [41], to select a broad 100-gene signature set associated with rat-specific cholestasis.
The chief advantage of the ABC approach is that it allows tight clustering of similar toxicants of interest (in this case rat cholestatic compounds) and displays the compounds along a continuum. Thus the most active rat cholestatic compounds essentially behaved identically by gene expression: cholestatic model compounds ANIT, LPS, and ethinyl estradiol, many high dose NSAIDs which can cause gut bleeding and endotoxin entry (phenacetin, flurbiprofen, sulindac, flufenamic acid, nimesulide, indomethacin, diclofenac, and ibuprofen [2], [18], flutamide, disulfiram, methapyrilene, and the tool compounds piperonyl butoxide, bromobenzene, butylated hydroxytoluene, and methylene dianiline clustered together). Naltrexone, 2-acetamidoflourene, amsacrine, hydrazine and tannic acid were slightly outside of the tight cholestatic cluster, and dipyridamole, ketoconazole and cyproterone acetate were further removed from the cluster, showing different gene expression although quite distant from the non-cholestatic compounds.
Aniline was the only known rat cholestatic compound that fell in the “bridging” area between the cholestatic and non-cholestatic compounds. Carbamazepine, clozapine and dieldrin also fell within this bridging area, suggesting pronounced differences in gene expression but also shared features with cholestatic compounds. Erythromycin, glibenclamide, chlorpromazine, simvastatin, sulfamethoxazole, and tamoxifen (all known cholestatic drugs in human) were the closest of the non-cholestatic cluster to the cholestatic compounds, but clearly non-cholestatic in the rat, at least with acute dosing at high dose.
A number of pharmaceuticals that cause cholestasis in humans do not appear to induce cholestasis in rats: chlorpromazine and erythromycin-induced cholestasis appears to be human-specific [5], [8]. Other human cholestatic compounds such as furosemide, rifampicin, niacin, captopril and dapsone showed no cholestatic gene expression response in the rat which may reflects species differences in cholestatic mechanisms such as binding and activation/inactivation of PXR, FXR, LXR and bile salt transporters [40]. Rifampin and glibenclamide reportedly block both rat BSEP and human BSEP transport in vitro and produce cholestasis in patients [25], [33] but the rat appears to be much better at processing these compounds and many other human cholestasis-inducing drugs [8]. Species differences in drug effects on metabolic regulatory pathways, drug-induced changes in bile salt solubility, interference with clearance of other endogenous substrates (particularly steroid hormones and bilirubin), and precipitation of compound in the bile ducts, septicemia and localized inflammation, such as pancreatitis, and idiosyncratic/hypersensitivity and inflammation may also modulate cholestasis differently in man versus the rat [38].
The regulation of the three cholestasis indicator genes Cyp7a1, Bsep and Mrp3 used as the starting point for the ABC approach, and the responses of these three genes across Janssen toxicogenomics database compounds is different enough that several components of the rat cholestatic response are captured in our analysis. The approach allows for improved characterization of cholestasis in the rat than would be obtained using Cyp7a1 and other redundant, identically regulated genes, such as Cyp8b1.
Importantly, the 100 genes comprising our derived rat cholestasis signature would not have been obvious from most biochemical pathway analyses. There were, however, several duplicate genes with different accession numbers that were selected by the algorithm (monocyte to macrophage differentiation associated, Mmd2, tRNA methyltransferase 10a, Trmt10a, serine dehydratase, Sds, glutathione transferases, Ran, high mobility group nucleosome-binding proteins, Hmgns, and Methylenetetrahydrofolate dehydrogenases, Mthfd), confirming the gene selections. Other transporters selected were for aromatic amino acids and nucleotide sugars, and another Cyp selected, Cyp2d4, is well known for 21-hydroxylation of steroid hormones rather than of bile salts or cholesterol. The robustness of the three starting genes and selection against strongly redundant responses means there are a limited number of tightly linked and/or well-known cholestatic signature genes.
In terms of in vivo study design, our experimental protocol was designed for tissue collection at an optimal time point for detection of robust gene expression changes. The non-proprietary paradigm compounds used in this work were generally administered as single dose, with animals necropsied 24 h later. Histopathology was not performed, as these compounds were typically well-characterized for their cholestatic potential in rat and human. Additionally, this acute treatment regimen was not expected to induce clinical manifestation of cholestasis after such a short dosing period. Exploration of dose-response and identification of additional cholestatic agents with multiple or chronic dosing regimens was not within the scope of this study: such investigations may yield additional insight and improved predictivity.
While other microarray studies of cholestasis exist for the rat, they have generally employed a single cholestatic agent. Such signatures are appropriate for the individual particular cholestatic model studied but may be more limited when applied to the variety of structural classes encountered in drug discovery and development. Comparison to ethinyl estradiol EE-treatment (24 h sampling time) showed minimal overlap with our >80 named genes (aflatoxin aldehyde reductase, FK506 binding protein, guanine nucleotide binding protein alpha inhibiting, high mobility group nucleosomal binding domain 1, NIMA-related kinases, nucleoporins, RAN, member RAS oncogene family, Serine protease inhibitor, Kazal type, and several tubulins) [7]. Comparison with a 15 named gene for cholestasis derived by the supervised learning approach SVM (support vector machine) showed only one gene overlap—protein kinase c-binding protein [32]. Comparison with a signature for hepatobiliary injury showed pronounced differences, with dysregulated genes reflecting immune activation and changes in integrins and extracellular matrix markers [6], which were missing in our 24-h cholestasis gene signature.
The data mining approach used in the present study provides an innovative analysis of pharmaceuticals and model toxicants that induce cholestasis in the rat, and an improved, more predictive 100-gene signature beyond the original three-gene panel comprised of Cyp7a1, Mrp3 and Bsep. While applicability of this statistical approach remains to be demonstrated with other data sets, similar data mining approaches with future or existing data may yield insights into other toxicological properties, using comprehensive gene expression as surrogates for important biological responses.
Transparency document
Acknowledgments
The authors thank Komal Singh Sandu and co-workers at Strand Life Sciences, and Jennifer Fostel, Carolyn Favaro and Xiang Yao for enabling public access of this project data.
Contributor Information
Yauheniya Cherkas, Email: ycherka@its.jnj.com.
Michael K. McMillian, Email: m_mcmillian@invitrocue.com.
Dhammika Amaratunga, Email: damaratu@its.jnj.com.
Nandini Raghavan, Email: nraghava@its.jnj.com.
Jennifer C. Sasaki, Email: jennifer.sasaki@azneuro.com.
References
- 1.Abou-El-Makarem M.M., Millburn P., Smith R.L., Williams R.T. Biliary excretion in foreign compounds. Species difference in biliary excretion. Biochem. J. 1967;105:1289–1293. doi: 10.1042/bj1051289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aitken A.E., Richardson T.A., Morgan E.T. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu. Rev. Pharmacol. Toxicol. 2006;46:123–149. doi: 10.1146/annurev.pharmtox.46.120604.141059. [DOI] [PubMed] [Google Scholar]
- 3.Amaratunga D., Cabrera J., Kovtun V. Microarray learning with ABC. Biostatistics. 2008;9:128–136. doi: 10.1093/biostatistics/kxm017. [DOI] [PubMed] [Google Scholar]
- 4.Dawson S., Stahl S., Paul N., Barber J., Kenna J.G. In vitro inhibition of the bile salt export pump correlates with risk of cholestatic drug-induced liver injury in humans. Drug Metab. Dispos.: Biol. Fate Chem. 2012;40:130–138. doi: 10.1124/dmd.111.040758. [DOI] [PubMed] [Google Scholar]
- 5.Drew R., Priestly B.G. Choleretic and cholestatic effects of infused bile salts in the rat. Experientia. 1979;35:809–811. doi: 10.1007/BF01968265. [DOI] [PubMed] [Google Scholar]
- 6.Ellinger-Ziegelbauer H., Adler M., Amberg A., Brandenburg A., Callanan J.J., Connor S., Fountoulakis M., Gmuender H., Gruhler A., Hewitt P., Hodson M., Matheis K.A., McCarthy D., Raschke M., Riefke B., Schmitt C.S., Sieber M., Sposny A., Suter L., Sweatman B., Mally A. The enhanced value of combining conventional and omics analyses in early assessment of drug-induced hepatobiliary injury. Toxicol. Appl. Pharmacol. 2011;252:97–111. doi: 10.1016/j.taap.2010.09.022. [DOI] [PubMed] [Google Scholar]
- 7.Henriquez-Hernandez L.A., Flores-Morales A., Santana-Farre R., Axelson M., Nilsson P., Norstedt G., Fernandez-Perez L. Role of pituitary hormones on 17alpha-ethinylestradiol-induced cholestasis in rat. J. Pharmacol. Exp. Ther. 2007;320:695–705. doi: 10.1124/jpet.106.113209. [DOI] [PubMed] [Google Scholar]
- 8.Jemnitz K., Veres Z., Vereczkey L. Contribution of high basolateral bile salt efflux to the lack of hepatotoxicity in rat in response to drugs inducing cholestasis in human. Toxicol. Sci. 2010;115:80–88. doi: 10.1093/toxsci/kfq044. [DOI] [PubMed] [Google Scholar]
- 9.Karagianni P., Talianidis I. Transcription factor networks regulating hepatic fatty acid metabolism. Biochim. Biophys. Acta. 2015;1851:2–8. doi: 10.1016/j.bbalip.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Kararli T.T. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm. Drug Dispos. 1995;16:351–380. doi: 10.1002/bdd.2510160502. [DOI] [PubMed] [Google Scholar]
- 11.Kojima K., Hosokawa M., Kobayashi K., Tainaka H., Chiba K. Microarray analysis of hepatic gene expression during long-term cholestasis induced by common bile duct ligation in rats. Res. Commun. Mol. Pathol. Pharmacol. 2004;115–116:63–75. [PubMed] [Google Scholar]
- 12.Kullak-Ublick G.A., Stieger B., Meier P.J. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology. 2004;126:322–342. doi: 10.1053/j.gastro.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Lai Y. Identification of interspecies difference in hepatobiliary transporters to improve extrapolation of human biliary secretion. Expert Opin. Drug Metab. Toxicol. 2009;5:1175–1187. doi: 10.1517/17425250903127234. [DOI] [PubMed] [Google Scholar]
- 14.Leone A., Nie A., Brandon Parker J., Sawant S., Piechta L.A., Kelley M.F., Mark Kao L., Jim Proctor S., Verheyen G., Johnson M.D., Lord P.G., McMillian M.K. Oxidative stress/reactive metabolite gene expression signature in rat liver detects idiosyncratic hepatotoxicants. Toxicol. Appl. Pharmacol. 2014;275:189–197. doi: 10.1016/j.taap.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 15.Leone A.M., Kao L.M., McMillian M.K., Nie A.Y., Parker J.B., Kelley M.F., Usuki E., Parkinson A., Lord P.G., Johnson M.D. Evaluation of felbamate and other antiepileptic drug toxicity potential based on hepatic protein covalent binding and gene expression. Chem. Res. Toxicol. 2007;20:600–608. doi: 10.1021/tx600351g. [DOI] [PubMed] [Google Scholar]
- 16.Li H., Luan Y. Boosting proportional hazards models using smoothing splines, with applications to high-dimensional microarray data. Bioinformatics. 2005;21:2403–2409. doi: 10.1093/bioinformatics/bti324. [DOI] [PubMed] [Google Scholar]
- 17.Lin J.H. Species similarities and differences in pharmacokinetics. Drug Metab. Dispos.: Biol. Fate Chem. 1995;23:1008–1021. [PubMed] [Google Scholar]
- 18.McMillian M., Nie A.Y., Parker J.B., Leone A., Kemmerer M., Bryant S., Herlich J., Yieh L., Bittner A., Liu X., Wan J., Johnson M.D. Inverse gene expression patterns for macrophage activating hepatotoxicants and peroxisome proliferators in rat liver. Biochem. Pharmacol. 2004;67:2141–2165. doi: 10.1016/j.bcp.2004.01.029. [DOI] [PubMed] [Google Scholar]
- 19.Meier P.J., Stieger B. Bile salt transporters. Annu. Rev. Physiol. 2002;64:635–661. doi: 10.1146/annurev.physiol.64.082201.100300. [DOI] [PubMed] [Google Scholar]
- 20.Meyers M., Slikker W., Pascoe G., Vore M. Characterization of cholestasis induced by estradiol-17 beta-d-glucuronide in the rat. J. Pharmacol. Exp. Ther. 1980;214:87–93. [PubMed] [Google Scholar]
- 21.Morgan R.E., Trauner M., van Staden C.J., Lee P.H., Ramachandran B., Eschenberg M., Afshari C.A., Qualls C.W., Jr., Lightfoot-Dunn R., Hamadeh H.K. Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol. Sci. 2010;118:485–500. doi: 10.1093/toxsci/kfq269. [DOI] [PubMed] [Google Scholar]
- 22.Morgan R.E., van Staden C.J., Chen Y., Kalyanaraman N., Kalanzi J., Dunn R.T., Afshari C.A., 2nd, Hamadeh H.K. A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol. Sci. 2013;136:216–241. doi: 10.1093/toxsci/kft176. [DOI] [PubMed] [Google Scholar]
- 23.Moseley R.H., Wang W., Takeda H., Lown K., Shick L., Ananthanarayanan M., Suchy F.J. Effect of endotoxin on bile acid transport in rat liver: a potential model for sepsis-associated cholestasis. Am. J. Physiol. 1996;271:G137–146. doi: 10.1152/ajpgi.1996.271.1.G137. [DOI] [PubMed] [Google Scholar]
- 24.Nie A.Y., McMillian M., Parker J.B., Leone A., Bryant S., Yieh L., Bittner A., Nelson J., Carmen A., Wan J., Lord P.G. Predictive toxicogenomics approaches reveal underlying molecular mechanisms of nongenotoxic carcinogenicity. Mol. Carcinog. 2006;45:914–933. doi: 10.1002/mc.20205. [DOI] [PubMed] [Google Scholar]
- 25.Pauli-Magnus C., Meier P.J. Hepatobiliary transporters and drug-induced cholestasis. Hepatology. 2006;44:778–787. doi: 10.1002/hep.21359. [DOI] [PubMed] [Google Scholar]
- 26.Pauli-Magnus C., Stieger B., Meier Y., Kullak-Ublick G.A., Meier P.J. Enterohepatic transport of bile salts and genetics of cholestasis. J. Hepatol. 2005;43:342–357. doi: 10.1016/j.jhep.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 27.Raghavan N., Nie A.Y., McMillian M., Amaratunga D. A linear prediction rule based on ensemble classifiers for non-genotoxic carcinogenicity. Stat. Biopharm. Res. 2012;4:185–193. [Google Scholar]
- 28.Rodriguez-Garay E.A. Cholestasis: human disease and experimental animal models. Ann. Hepatol. 2003;2:150–158. [PubMed] [Google Scholar]
- 29.Russell D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 30.Sandhu K.S., Veeramachaneni V., Yao X., Nie A., Lord P., Amaratunga D., McMillian M.K., Verheyen G.R. Release of (and lessons learned from mining) a pioneering large toxicogenomics database. Pharmacogenomics. 2015:1–23. doi: 10.2217/pgs.15.38. [DOI] [PubMed] [Google Scholar]
- 31.Schaap F.G., van der Gaag N.A., Gouma D.J., Jansen P.L. High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology. 2009;49:1228–1235. doi: 10.1002/hep.22771. [DOI] [PubMed] [Google Scholar]
- 32.Steiner G., Suter L., Boess F., Gasser R., de Vera M.C., Albertini S., Ruepp S. Discriminating different classes of toxicants by transcript profiling. Environ. Health Perspect. 2004;112:1236–1248. doi: 10.1289/txg.7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stieger B., Fattinger K., Madon J., Kullak-Ublick G.A., Meier P.J. Drug- and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology. 2000;118:422–430. doi: 10.1016/s0016-5085(00)70224-1. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka A., Tsuneyama K., Mikami M., Uegaki S., Aiso M., Takikawa H. Gene expression profiling in whole liver of bile duct ligated rats: VEGF-A expression is up-regulated in hepatocytes adjacent to the portal tracts. J. Gastroenterol. Hepatol. 2007;22:1993–2000. doi: 10.1111/j.1440-1746.2006.04629.x. [DOI] [PubMed] [Google Scholar]
- 35.Trauner M., Boyer J.L. Bile salt transporters: molecular characterization, function, and regulation. Physiol. Rev. 2003;83:633–671. doi: 10.1152/physrev.00027.2002. [DOI] [PubMed] [Google Scholar]
- 36.Trauner M., Fickert P., Stauber R.E. Inflammation-induced cholestasis. J. Gastroenterol. Hepatol. 1999;14:946–959. doi: 10.1046/j.1440-1746.1999.01982.x. [DOI] [PubMed] [Google Scholar]
- 37.Tsai C.A., Lee T.C., Ho I.C., Yang U.C., Chen C.H., Chen J.J. Multi-class clustering and prediction in the analysis of microarray data. Math. Biosci. 2005;193:79–100. doi: 10.1016/j.mbs.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Yang K., Kock K., Sedykh A., Tropsha A., Brouwer K.L. An updated review on drug-induced cholestasis: mechanisms and investigation of physicochemical properties and pharmacokinetic parameters. J. Pharm. Sci. 2013;102:3037–3057. doi: 10.1002/jps.23584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zidek N., Hellmann J., Kramer P.J., Hewitt P.G. Acute hepatotoxicity: a predictive model based on focused illumina microarrays. Toxicol. Sci. 2007;99:289–302. doi: 10.1093/toxsci/kfm131. [DOI] [PubMed] [Google Scholar]
- 40.Zollner G., Marschall H.U., Wagner M., Trauner M. Role of nuclear receptors in the adaptive response to bile acids and cholestasis: pathogenetic and therapeutic considerations. Mol. Pharm. 2006;3:231–251. doi: 10.1021/mp060010s. [DOI] [PubMed] [Google Scholar]
- 41.Zou H., Hastie T. Regularization and variable selection via the elastic net. J. R. Stat. Soc.: Ser. B (Stat. Methodol.) 2005;67:301–320. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.