

Summary
Many viral RNAs are modified by methylation of the N6 position of adenosine (m6A). m6A is thought to regulate RNA splicing, stability, translation and secondary structure. Influenza A virus (IAV) expresses m6A-modified RNAs but the effects of m6A on this segmented RNA virus remain unclear. We demonstrate that global inhibition of m6A addition inhibits IAV gene expression and replication. In contrast, overexpression of the cellular m6A “reader” protein YTHDF2 increases IAV gene expression and replication. To address whether m6A residues modulate IAV RNA function in cis, we mapped m6A residues on the IAV plus (mRNA) and minus (vRNA) strands and used synonymous mutations to ablate m6A on both strands of the hemagglutinin (HA) segment. These mutations inhibited HA mRNA and protein expression, while leaving other IAV mRNAs and proteins unaffected, and also resulted in reduced IAV pathogenicity in mice. Thus, m6A residues in IAV transcripts enhance viral gene expression.
Keywords: m6A, influenza A virus, post-transcriptional regulation, mRNA function, viral pathogenesis
eTOC blurb
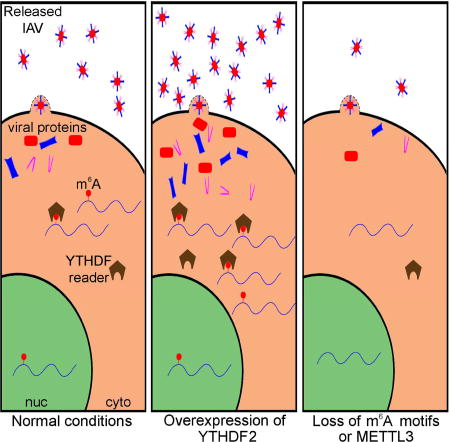
Influenza A virus (IAV) transcripts bear numerous epitranscriptomic m6A modifications. Courtney et al. map these modifications on both the IAV mRNA and vRNA strands and demonstrate that m6A increases viral RNA expression in cis. Moreover, IAV mutants lacking HA sites on the viral HA segment show reduced pathogenicity in vivo.
Introduction
The covalent modification of individual bases on mRNA transcripts has recently emerged as a potentially critical mechanism for the post-transcriptional regulation of gene expression (Li and Mason, 2014; Meyer and Jaffrey, 2014). Analysis of the cellular epitranscriptome, defined as internal single nucleotide modifications that do not alter the mRNA sequence, has identified at least ten different modifications, of which the most prevalent is the addition of a methyl group to the N6 position of adenosine, referred to as m6A. It has been reported that the average ~2.2 kb cellular mRNA contains 3 internal m6A residues, and highly regulated mRNAs may contain 10 or more m6As (Desrosiers et al., 1975; Linder et al., 2015). Moreover, addition of m6A has been proposed to regulate mRNA function at multiple steps, including splicing, stability, translation and secondary structure (Li and Mason, 2014; Meyer and Jaffrey, 2014).
The cellular machinery that adds m6A to mRNAs and detects its presence is highly conserved in multicellular eukaryotes and the global loss of m6A results in profoundly deleterious phenotypes, including embryonic lethality, in species ranging from plants to vertebrate animals (Batista et al., 2014; Geula et al., 2015; Hongay and Orr-Weaver, 2011; Zhong et al., 2008). m6A is added co-transcriptionally to nuclear pre-mRNAs by a protein complex consisting minimally of the methyltransferase METTL3 and two co-factors, METTL14 and WTAP, known collectively as m6A “writers” (Ke et al., 2017; Li and Mason, 2014; Meyer and Jaffrey, 2014). Once mRNAs have entered the cytoplasm, they encounter three m6A “reader” proteins called YTHDF1, YTHDF2 and YTHDF3, which are thought to mediate many of the phenotypic effects exerted by m6A.
In addition to the important but still emerging role played by m6A in regulating cellular mRNA function, m6A has also been detected on every viral mRNA transcript examined so far including, perhaps unexpectedly, mRNAs encoded by several cytoplasmic RNA viruses (Gonzales-van Horn and Sarnow, 2017; Kennedy et al., 2017). The first virus found to express mRNAs bearing internal m6A residues was influenza A virus (IAV), which was reported to contain ~24 m6A residues on the various viral mRNAs, of which the highest number, eight m6As, was detected by biochemical analysis of the HA mRNA segment (Krug et al., 1976; Narayan et al., 1987). However, these m6A residues were not mapped and no examination of how m6A affects IAV gene expression or replication has been reported. Nevertheless, it is known that the drug 3-deazaadenosine (DAA), which inhibits m6A addition by depleting intracellular levels of the methyl donor S-adenosylmethionine (SAM) (Bader et al., 1978; Fustin et al., 2013) is a potent inhibitor of IAV replication at doses that do not exert any evident cytotoxic effect (Fischer et al., 1990), thus suggesting that m6A plays a positive effect in the IAV life cycle.
While few reports as yet exist addressing how m6A affects viral gene expression and replication, this has been examined in the case of HIV-1. Three groups have reported the mapping of multiple m6A residues on the HIV-1 RNA genome and these groups have also examined the effect of overexpressing or knocking down the cellular reader and/or writer proteins on HIV-1 replication (Kennedy et al., 2016; Lichinchi et al., 2016a; Tirumuru et al., 2016). Two groups reported that m6A exerted a positive effect on HIV-1 replication (Kennedy et al., 2016; Lichinchi et al., 2016a), while a third group reported that m6A exerted an inhibitory effect (Tirumuru et al., 2016). As m6A is added to A residues in a well-established sequence consensus (minimally 5’-RAC-3’, where R is purine), it is unclear why HIV-1, which rapidly escapes from other cis-acting inhibitory sequence elements, such as siRNA targets (Boden et al., 2003; Das et al., 2004), should retain m6A sites if these are indeed inhibitory. Another group looked at m6A residues present on the genome of the cytoplasmic RNA virus hepatitis C virus (HCV) and concluded these did not significantly affect viral RNA replication but did reduce the production of progeny viral particles (Gokhale et al., 2016). Similarly, inhibition of m6A addition was also reported to enhance the production of infectious viral particles in cells infected with the cytoplasmic RNA virus Zika virus (ZKV) (Lichinchi et al., 2016b).
In this manuscript, we examine how m6A addition affects IAV gene expression and replication. We first demonstrate that mutational inactivation of the key m6A writer METTL3 in the human lung epithelial cell line A549 inhibits IAV replication, while ectopic overexpression of the m6A reader YTHDF2, but not YTHDF1 or YTHDF3, increases IAV replication and infectious particle production. We then used RNA:protein crosslinking and immunoprecipitation techniques to map the location of m6A residues on both the IAV mRNA/cRNA (plus) strands and vRNA (minus) strands and observed a high level of m6A addition on the viral mRNAs encoding the structural proteins HA, NA, M1/M2 and NP but lower levels on the mRNAs encoding the viral polymerase proteins PB2, PB1 and PA. Finally, we generated mutant forms of IAV in which eight prominent m6A sites on the HA mRNA/cRNA plus strand, or nine m6A sites on the HA vRNA minus strand, were silently mutated, and we observed that both these IAV mutants selectively expressed lower levels of HA mRNA and protein while expression of the other IAV mRNAs and proteins was unaffected. Moreover, these m6A-deficient IAV mutants were found to be significantly less pathogenic when introduced into mice. These data demonstrate that silent mutations that ablate m6A sites present on a viral RNA can significantly reduce viral gene expression in cis, therefore provide a potential explanation for the apparently ubiquitous presence of m6A residues on transcripts encoded by nuclear DNA and RNA viruses (Kennedy et al., 2017).
Results
As noted above, it has previously been reported that IAV replication can be effectively inhibited using non-toxic does of the drug DAA, which has been reported to inhibit the addition of m6A residues to mRNA transcripts by inducing the depletion of SAM, the methyl donor for METTL3 (Bader et al., 1978; Fischer et al., 1990; Fustin et al., 2013). Although DAA is therefore not a highly specific inhibitor of m6A addition, this result, which is reproduced in Fig. 1A, is nevertheless consistent with the hypothesis that m6A addition exerts a positive effect on IAV replication. To extend these data, we generated knock-outs of the key m6A writer METTL3 in the human lung epithelial cell line A549, using gene editing with CRISPR/Cas. We obtained two independent A549 cell clones that were defective for METTL3 expression based on both Western blot (Fig. 1B) and sequencing of all three copies of the METTL3 gene present in A549 cells (Fig. S1). Analysis of the ability of these cells to support the replication of the IAV isolate PR8 revealed a substantial, ~8-fold reduction in IAV gene expression measured by both Western blot for the IAV NS1, NP and M2 proteins (Fig. 1C) and qRT-PCR analysis of the expression of the spliced IAV M2 RNA (Fig. 1D). Similar data were obtained when expression of the IAV NP mRNA was quantified (data not shown). Moreover, we observed a highly significant (p<0.01) reduction in the production of infectious IAV by the two METTL3 knockout cell lines M3.1 and M3.2, when compared to wild type A549 cells or cells transduced with a lentiviral vector expressing green fluorescent protein (GFP) (Fig. 1E). In conclusion, these data argue that m6A addition exerts a positive effect on IAV gene expression and virion production.
Figure 1. IAV gene expression is greatly reduced in METTL3 knockout cell lines.

(A) Treatment of cells with a non-toxic dose of DAA, an inhibitor of m6A addition, reduced expression of the IAV proteins NS1 and M2 in A549 cells, as determined by Western blot. A549 cells were infected at an MOI of 1.0. (B) Two clonal A549 METTL3 knockout cell lines, M3.1 and M3.2, were established using gene editing and confirmed by Western blot and genomic sequencing. See Fig. S1 for genomic sequences and Table S1 for sgRNA sequences. (C) Two A549 METTL3 knockout lines and two control cell lines, including the parental A549 cell line and a GFP-Flag expressing A549 cell line, were infected with IAV-PR8 at an MOI of 0.01 and NP, NS1 and M2 protein expression determined by Western blot at 24, 48 and 72 hours post infection (hpi). Quantification of the band intensities for NS1 and M2 at 72 hpi revealed expression levels of 0.12 ± 0.01 and 0.12 ± 0.07 for M3.1 and 0.09 ± 0.03 and 0.08 ± 0.03 for M3.2, respectively, when expression in wild type cells was normalized to 1.0. Actin was used as the loading control. (D) Quantitative RT-PCR (qRT-PCR) was performed to determine the levels of the spliced IAV M2 mRNA at the same time points post-infection. See Table S2 for primer sequences. (E) Viral production from the wild type and METTL3 KO cells at 72 hpi was quantified by plaque assay on MDCK cells These data represent the average of three biological replicates with SD indicated (*=p<0.05, **=p<0.01).
Potent activation of IAV gene expression by ectopic YTHDF2
As noted above, m6A residues are bound by one of the three cytoplasmic reader proteins YTHDF1, YTHDF2 and YTHDF3 (Kennedy et al., 2017; Meyer and Jaffrey, 2014) and we have previously reported that overexpression of these proteins, in particular YTHDF2, significantly enhances HIV-1 gene expression and replication (Kennedy et al., 2016). To test whether this is also true for IAV, we generated clonal cell lines derived from A549 by transduction with lentiviral vectors expressing either YTHDF1 or YTHDF2 (Fig. 2A). Following infection of these cell lines with IAV-PR8 at an MOI of 0.01, we observed an obvious increase in the level of expression of the IAV NS1, NP and M2 proteins when assayed at at 24 h, 48 h and 72 h post-infection (Fig. 2B). We also detected a higher level of M2 mRNA (Fig. 2C) and NP mRNA (data not shown) expression in the infected A549 cultures overexpressing YTHDF2, as measured by qRT-PCR, as well as a significant (p<0.05) increase in the production of infectious progeny IAV virions (Fig. 2D), when compared to wild type A549 cells or A549 cells transduced with a control lentiviral vector expressing GFP. While both clonal A549 cell lines overexpressing YTHDF2 displayed this remarkable positive effect on IAV gene expression, we did not see any significant effect, either positive or negative, upon overexpression of YTHDF1 (Fig. 2). As shown in Fig. S2D, IAV also gave rise to significantly larger plaques when assayed on YTHDF2-overexpressing A549 cells, when compared to control cells or cells overexpressing YTHDF1, which argues that YTHDF2 overexpression not only increases IAV gene expression but also facilitates viral spread in culture.
Figure 2. Overexpression of YTHDF2 increases all aspects of viral gene expression.

(A) A549 cell lines overexpressing YTHDF1-Flag (Y1), YTHDF2-Flag (Y2.1 and Y2.2) or GFP were generated by lentiviral transduction followed by single cell cloning. (B) A549 cell lines were infected with IAV-PR8 at an MOI of 0.01 and expression of the viral proteins NP, NS1 and M2 assessed by Western blot at 24, 48 and 72 hpi. GFP, YTHDF1 and YTHDF2 were detected using anti-Flag. The parental A549 cell line, and A549 cells expressing GFP-Flag, were used as controls. Quantification of the band intensities for NS1 and M2 at 72 hpi revealed expression levels of 6.28 ± 1.28 and 5.19 ± 2.13 for Y2.1 and 5.70 ± 1.65 and 5.84 ± 1.73 for Y2.2, respectively, when the expression in wild type cells was normalized to 1.0. Actin was used as the loading control. Single round infections are described in Fig. S2. (C) qRT-PCR was used to determine the mRNA levels of the spliced M2 IAV mRNA at the same time points post-infection. See Table S2 for primer sequences. (D) The viral titer produced from these cell lines at 72hpi was determined by plaque assay on MDCK cells. These data represent the average of three biological replicates with SD indicated (*=p<0.05, **=p<0.01).
To confirm and extend these data looking at a spreading IAV infection (Fig. 2), we also performed a single cycle IAV replication assay using an MOI of 1.0 and in the absence of trypsin, which is required for IAV spread in culture. As shown in Fig. S2A, we again detected a dramatic, ~16-fold increase in the expression of the IAV NS1 and M2 proteins in the A549 cells overexpressing YTHDF2, while YTHDF1 exerted at most a weak positive effect. In addition, we also performed single cycle replication assays, again in the absence of trypsin, using an MOI of 3.0, which should result in the immediate infection of ~93% of the cells, as was indeed confirmed by immunofluorescence (Fig. S2C). This high MOI experiment (Fig. S2B) again revealed a strong enhancement in the expression of the IAV NS1, M2 and NP proteins in the A549 cells overexpressing YTHDF2, when compared to control cells. These data (Fig. S2A and 2B) therefore demonstrate that YTHDF2 can significantly increase the expression of IAV gene products in the absence of viral spread and thus argue that YTHDF2 is not acting solely by increasing the production of progeny IAV virions. Finally, as we had been unable to generate YTHDF3 overexpressing A549 cells using lentiviral transduction, we decided to generate YTHDF1, YTHDF2 and YTHDF3 overexpressing A549 cells using an entirely different technique, i.e., a dCas-based synergistic activation module (SAM) targeted to the promoter of the endogenous YTHDF1, YTHDF2 or YTHDF3 gene (Konermann et al., 2015). The resultant clonal A549 cell lines only modestly overexpressed YTHDF1 or YTHDF2 relative to wild type A549 cells, but we were able to derive two clonal cell lines overexpressing YTHDF3, which is normally almost undetectable in A549 cells (Fig. S3). Analysis of the ability of these A549 cell clones to support IAV gene expression, by Western blot for the IAV NS1 and M2 proteins, again revealed a strong induction by ectopic YTHDF2 but no obvious positive effect on IAV gene expression upon overexpression of YTHDF1 or, in this case, YTHDF3.
Mapping m6A sites on IAV mRNAs and vRNAs
While the data presented so far clearly demonstrate that m6A addition and detection by YTHDF2 strongly enhances IAV replication, they do not demonstrate that this effect is direct. As noted above, IAV mRNAs have previously been reported to be highly m6A modified (Krug et al., 1976; Narayan et al., 1987) and we therefore decided to map these m6A sites using a previously described photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) approach (Hafner et al., 2010; Kennedy et al., 2016) to precisely define the binding sites of ectopically expressed YTHDF1, YTHDF2 or YTHDF3 in IAV-infected 293T cells that had been pulsed with 4-thiouridine (4SU), followed by RNase treatment of the immunoprecipitated RNA:protein complex, cDNA synthesis and deep sequencing of the resultant YTHDF protein binding sites. As previously noted, a key advantage of the PAR-CLIP technique is that reverse transcription of the recovered short RNAs results in the insertion of characteristic T to C mutations at sites of protein crosslinking to 4SU residues, thus allowing the exclusion from the subsequent bioinformatics analysis of all background reads resulting from non-crosslinked RNAs (Kennedy et al., 2016; Konermann et al., 2015). As shown in Fig. 3, we detected a large number of YTHDF protein binding sites on the viral mRNAs/cRNAs encoding the HA, NP, NA and M open reading frames but far fewer on the mRNAs/cRNAs encoding the PB2, PB1 and PA open reading frames. This apparent dichotomy is interesting as it suggests that m6A residues are selectively present on IAV mRNAs encoding the major viral structural proteins, which are expressed at high levels in IAV infected cells, and less prevalent on the mRNAs encoding the three IAV RNA-dependent RNA polymerase (RdRP) subunits, which are required at lower levels for optimal viral replication. Interestingly, this does not reflect the prevalence of potential 5’-RAC-3’ m6A addition sites, which are evenly distributed across the IAV genome (data not shown).
Figure 3. Identification of m6A sites on IAV-PR8 plus sense mRNA.

PAR-CLIP and PA-m6A-seq were performed using 293T cell lines 24 hpi with IAV-PR8 at an MOI of 10 (A) Concatenated map of the IAV-PR8 transcriptome that reads were aligned to. (B) Complete transcriptome coverage tracks are shown for PAR-CLIP performed on Flag-GFP, Flag-YTHDF1, Flag-YTHDF2 and Flag-YTHDF3 expressing 293T cells, while PA-m6A-seq was performed using wild type 293T cells. The PA-m6A-seq lane has a Y axis of 0–500 reads, and all others are depicted with Y axes of 0–200 reads. (C) An expanded view of the HA segment of IAV-PR8, with 8 prominent m6A sites numbered. PA-m6A-seq has a Y axis of 0–250 reads, and all others are depicted with Y axes of 0–100 reads. Reverse transcription of crosslinked 4SU residues results in characteristic T>C mutations and the level of T>C conversion at specific residues is indicated by red/blue bars.
In addition to using PAR-CLIP with the three YTHDF proteins to map m6A residues in living, IAV-infected cells, we also used the distinct PA-m6A-seq technique (Chen et al., 2015), which involves isolation of poly(A)+ mRNA from IAV-infected, 4SU-pulsed cells followed by binding of an m6A-specific antibody in vitro, crosslinking, RNase treatment, cDNA synthesis and deep sequencing. These data, shown on the bottom row of Figs. 3B and 3C, again confirmed that m6A residues were present on multiple locations of the viral mRNAs encoding HA, NP, M and NA and at lower levels on the mRNAs encoding PB2, PB1 and PA.
The mRNA encoding the IAV HA protein has previously been reported to contain eight internal m6A residues (Narayan et al., 1987) and we decided to focus on this unspliced RNA for our further research. As shown in Fig. 3C, there is good concordance between the three PAR-CLIP experiments performed using the three YTHDF proteins and the data generated using the PA-m6A-seq technique in terms of the mapped location of m6A residues on the HA mRNA, and in fact our data identified eight major m6A peaks on the HA mRNA that were all located in the HA open reading frame (ORF), almost all of which could be inactivated by silent mutagenesis of the m6A consensus addition site 5’-RAC-3’ (Fig. S3).
In addition to mapping m6A sites present on the IAV mRNA/cRNA segments, we were also able to map m6A residues on the eight IAV vRNA minus strands using both PAR-CLIP with the YTHDF proteins and using PA-m6A-seq (Fig. 4). The latter was unexpected, as PA-m6A-seq uses highly purified poly(A)+ RNA as the input RNA and vRNAs are not polyadenylated. We hypothesize that viral vRNAs may have annealed to complementary poly(A)+ viral mRNAs during their isolation and, hence, been recovered along with the total cellular poly(A)+ RNA population. Strand origin was assigned based on the detection of A to G, versus T to C, mutations that arise from sites of protein crosslinking to incorporated 4SU residues. Regardless, the PAR-CLIP and PA-m6A-seq data obtained again revealed multiple m6A sites on the vRNA segments encoding the viral structural proteins HA, NP, NA and M and a lower level of m6A sites on the vRNA segments specific for PB2, PB1 and PA (Fig. 4B). Importantly, these data again showed a strong concordance in the mapped locations of m6A residues on the HA vRNA segment and identified nine prominent m6A peaks (Fig. 4C), all of which could be inactivated by mutagenesis of coincident m6A consensus m6A addition sites (5’-RAC-3’) in the HA vRNA segment. These nine introduced mutations were again designed to not affect the predicted coding capacity of the cognate HA mRNA (Fig. S3).
Figure 4. Identification of m6A sites on IAV-PR8 minus strand vRNA.

PAR-CLIP and PA-m6A-seq were performed on RNA from 293T cell lines 24 hpi with IAV-PR8 at MOI of 10. (A) Concatenated map of the IAV-PR8 genome used for read alignment. (B) Complete genomic coverage tracks are shown, similar to Fig. 2. The PA-m6A-seq lane has a Y axis of 0–2000 reads, and all others are depicted with Y axes of 0–300 reads. (C) An expanded view of the HA segment of IAV-PR8, with 9 prominent m6A sites numbered. PA-m6A-seq has a Y axis of 0–1000 reads, and all others are depicted with Y axes of 0–150 reads. Green/brown bars indicate the level of A>G conversion at specific A residues.
Loss of m6A sites present on the HA segment of IAV selectively reduces HA gene expression
Based on the m6A mapping data for the IAV HA RNA segment, we generated a full length version of the HA gene segment lacking the plus-sense m6A sites mapped in Fig. 3C as well as a second HA gene segment lacking all nine minus-sense m6A sites mapped in Fig. 4C (see Fig. S3 for the precise point mutations introduced into HA). As noted above, these mutations, a total of 14 point mutations on the HA plus strand and 15 on the HA minus strand, all introduced synonymous codons into the encoded HA mRNA. Replication competent IAV-PR8 mutants lacking m6A sites mapped on the HA plus-sense RNA (+Mut) or on the minus-sense RNA (−Mut) were then rescued by transfection of 293T cells and amplified in embryonated chicken eggs. We recovered comparable levels of the wild type, +Mut and −Mut IAV viruses from eggs, as confirmed by assay of neuraminidase activity (Fig. S4A) and these contained comparable levels of the HA vRNA segment (Fig.5A). However, analysis of purified IAV virions by Western blot revealed a modest, 20–40% drop in the incorporation of the HA protein into the –Mut and +Mut virions compared to wild type IAV-PR8 (Fig. 5B). Quantification of HA mRNA expression using qRT-PCR after infection of A549 cells at an MOI of 0.01 also revealed a significantly lower level of HA mRNA expression (p<0.01) for both the −Mut and +Mut IAV mutant viruses when compared to wild type IAV-PR8 (Fig. 5C). In contrast, the observed levels of other IAV mRNAs, typified here by the M2 mRNA, were not affected (Fig. 5D). This demonstrates that the effect of these mutations on the expression of the HA mRNA is specific. An even more striking phenotype was observed when the A549 cells infected with wild type and mutant IAV were analyzed for viral protein expression by Western blot (Fig. 5E). In this experiment, NS1 and M2 protein expression was similar for the wild type and both mutant IAVs at 48 h and 72 h post-infection and these two viral proteins therefore serve as internal controls. In sharp contrast, HA protein expression was down almost 2-fold for the +Mut virus and down ~3-fold for the −Mut virus and this difference was significant (p<0.05). Therefore, we can conclude that the presence of m6A residues on both the IAV mRNA plus strands and vRNA minus strands has a direct, positive effect on IAV HA mRNA and protein expression. Our results also indicate that the observed reduction in HA protein expression does not inhibit the spread of IAV in embryonated chicken eggs (Fig. S4A) and only modestly affects IAV spread in cultured A549 cells (Fig. S4B).
Figure 5. Mutagenesis of m6A motifs on the IAV HA segment inhibits HA mRNA and protein expression.

Mutations introduced into the HA segment are described in Fig. S3. (A) Viral RNA extracted from purified IAV-PR8 virions grown in embryonated chicken eggs was separated on a TBE-urea gel and stained. (B) Protein was extracted from purified IAV and Western blotting used to determine HA protein levels in wild type or mutant virions. Band intensity quantification revealed HA0 and HA1 levels of 0.8 ± 0.63 and 0.8 ± 0.54 for the +Mut virions and 0.5 ± 0.53 and 0.6 ± 0.48 for the −Mut virions, respectively, when HA levels in wild type virions was normalized to 1.0. The viral protein M1 was used as a loading control. (C) A multicycle spreading infection was initiated by infection of A549 at an MOI of 0.01 using all 3 IAV-PR8 variants. Total RNA was extracted at 24, 48 and 72 hpi and qRT-PCR then used to determine HA mRNA levels, relative to a GAPDH mRNA internal control. See Table S2 for primer sequences. (D) The same total RNA samples used in panel C were used to quantify M2 mRNA levels, again using GAPDH mRNA as an internal control. (E) Similar to panel C except that Western blotting was performed to evaluate the level of expression of the IAV HA, NS1 and M2 proteins at 24, 48 and 72 hpi. Quantification of HA band intensity at 72 hpi revealed expression levels of 0.69 ± 0.11 for the +Mut virus and 0.32 ± 0.13 for the −Mut virus, when normalized to M2 expression levels. Actin was used as the loading control. Viral spread was also determined by flow cytometry (see Fig. S4). (F) A549 cells were infected at an MOI of 1.0 with the three IAV-PR8 variants and total RNA extracted 12 hpi. qRT-PCR was used to determine the levels of the cellular mRNAs encoding RIG-I, MDA5 and IFNβ1. All data are drawn from three biological replicates with SD indicated (**=p<0.01).
While the mechanism underlying the positive effect of m6A on HA expression is currently unknown, we considered the possibility that m6A residues on IAV transcripts might inhibit cellular innate immune responses to viral infection, as has indeed been proposed (Durbin et al., 2016; Kariko et al., 2005). If this were the case, one would predict that infection with an IAV mutant lacking m6A sites would induce a higher level of expression of innate immune response genes such as RIG-I, MDA5 and/or interferon β (IFN β). Analysis of the expression of the mRNAs encoding these three immune effectors instead failed to reveal any positive effect of the m6A mutations introduced into the IAV HA segment (Fig. 5E). Therefore, we conclude that the m6A sites present on the IAV HA mRNA and vRNA do not detectably downregulate the innate immune response to IAV infection.
Loss of m6A sites present on the HA segment of IAV reduces viral pathogenicity in vivo
The modestly reduced level of HA mRNA and protein expression in the two m6A-deficient IAV mutants analyzed in culture in Fig. 5 suggested that these mutants might also display attenuated pathogenicity in vivo. To test this possibility, we infected mice with either 10 plaque forming units (PFU) or 50 PFU of either the parental IAV-PR8 isolate, which is highly pathogenic in mice, or of the −Mut and +Mut IAV-PR8 mutants, which have reduced m6A levels on the HA vRNA or mRNA strand, respectively. As may be observed (Fig. 6), we saw reduced pathogenicity for both IAV mutants, but especially for the −Mut variant, at both the 50 PFU (Fig. 6B) and 10 PFU (Fig. 6C) dose, with only the WT virus causing lethality at the lower dose, and this reduction was statistically significant at both infectious doses. In addition, we also saw reduced weight loss in the surviving mice infected with the two mutant viruses (Figs. 6A and 6C).
Figure 6. IAV mutants depleted for m6A on the viral HA strand show reduced pathogenicity in vivo.

Wildtype C57BL/6 mice were infected with 50 plaque forming units (PFU) of WT PR8, +Mut or −Mut and monitored for (A) body weight with 80% of the starting body weight as a humane endpoint, as indicated by the dashed line and (C) mortality. Similarly, mice were infected with 10 PFU of WT PR8, +Mut or −Mut and monitored for (B) body weight and (D) mortality. Five mice were used per group. Significance is indicated on the graph based on the log-rank Mantel-Cox test.
Discussion
Although work from several groups has demonstrated that a number of different DNA and RNA viruses express mRNAs that are modified by addition of m6A (Gonzales-van Horn and Sarnow, 2017; Kennedy et al., 2017), the role of m6A in regulating viral gene expression and replication remains largely unclear. We and others have reported that m6A addition to viral transcripts enhances the replication of HIV-1 (Kennedy et al., 2016; Lichinchi et al., 2016a), though a third group has disputed this finding (Tirumuru et al., 2016). In the case of the cytoplasmic RNA virus HCV, m6A has been proposed to reduce the production of progeny virions without affecting viral RNA replication or protein expression (Gokhale et al., 2016) and, in the case of ZKV, enhanced virion production has also been observed upon knockdown of m6A addition (Lichinchi et al., 2016b). However, given that m6A has been detected on transcripts expressed by a wide range of viral species (Kennedy et al., 2017) and as sites of m6A addition are, at least in the case of HIV-1, evolutionarily conserved (Kennedy et al., 2016) we would argue that m6A must facilitate some aspect(s) of the viral life cycle. Especially for rapidly evolving viruses, such as HIV-1, IAV or ZKV, one would expect the speedy selection of viral variants that had lost the consensus sites required for m6A addition if these indeed exerted an inhibitory effect in cis.
In this manuscript, we have sought to address whether m6A affects the replication and pathogenicity of IAV, which was the first virus reported to bear multiple m6A residues (Krug et al., 1976). We initially addressed whether the global perturbation of m6A addition or recognition in the human lung epithelial cell line A549 would affect IAV replication and we observed an ~5-fold decrease in IAV protein expression and replication when the gene encoding the m6A methyltransferase METTL3 was inactivated by gene editing (Fig.1) and a similar ≥5-fold increase in IAV gene expression and virion production in A549 cells overexpressing the m6A reader protein YTHDF2 (Figs. 2 and S2). In contrast, overexpression of YTHDF1 and YTHDF3 did not greatly affect IAV replication. To our knowledge, this is the largest increase in IAV replication ever observed upon simple overexpression of a single human protein in infected cells and argues that addition and detection of m6A plays a critical role in promoting IAV replication. Importantly, this marked positive effect was also observed under conditions where IAV spread was blocked (Fig. S2A and B), thus arguing that YTHDF2 overexpression is not promoting IAV replication by, for example, enhancing the production of progeny virions but rather must be acting to promote viral gene expression and/or viral RNA replication. We note that the strong positive effect of YTHDF2 on IAV virion production (Fig. 2D) may have practical importance given that difficulties with IAV vaccine production in embryonated chicken eggs have led to efforts to instead produce IAV vaccine strains in cultured mammalian cells, resulting in the recent licensing of the IAV vaccine Flucelvax (Houser and Subbarao, 2015). However, the production of IAV by cultured cells is relatively inefficient and the ability to substantially enhance this process by simple overexpression of YTHDF2 might therefore represent a significant technical advance.
While the negative effect of METTL3 inactivation and the positive effect of YTHDF2 overexpression on IAV replication are both readily detectable (Figs. 1 and Fig. 2), this could clearly result from the effect of m6A residues on the expression of cellular, rather than IAV, RNA transcripts. To address this concern, we therefore mapped m6A residues of both the plus sense, mRNA/cRNA transcripts encoded by IAV (Fig. 3) and on the minus sense, vRNAs (Fig. 4). We then selected an unspliced, highly m6A modified IAV gene segment, encoding the IAV HA protein, and generated mutant IAV-PR8 viral stocks in which m6A sites on the HA mRNA, or m6A sites on the IAV HA vRNA, were inactivated by mutation of 5’-RAC-3’ m6A consensus sequences found coincident with the mapped sites of m6A addition (Figs. 3 and Fig. 4). Importantly, although all 17 mapped m6A addition sites were located in the HA ORF, we were able to inactivate almost all of the 5’-RAC-3’ consensus sequences by using synonymous mutations that do not affect the coding capacity of the HA segment. It should be noted that these mutations do not remove all potential m6A addition sites found on the IAV HA segment (Figs. 3 and Fig. 4), including two potential m6A sites on the viral mRNA strand that could not be silently mutated, so these mutant viruses are likely to be m6A hypomethylated on the HA segment, rather than totally devoid of m6A.
Initial analysis showed that while these two viral mutants replicated in embryonated chicken eggs to similar levels as the wild type IAV-PR8 virus (Fig. 5A and Fig. S4A), the resultant IAV virions bore slightly lower levels of the HA protein (Fig. 5B). When used to infect wild type A549 cells in culture, the mutant IAVs lacking m6A on HA mRNAs (+Mut) or vRNA (−Mut) both expressed lower levels of the HA mRNA (Fig.5C) while the expression levels of other IAV mRNAs was unaffected (Fig. 5D). Consistent with these RNA data, the IAV mutants lacking m6A residues on the HA transcripts also expressed lower levels of the viral HA protein relative to the IAV M2 and NS1 proteins expressed in the same infected cells, which again were unaffected (Fig. 5E). These data are therefore consistent with the hypothesis that m6A editing of the HA mRNA and vRNA transcripts directly enhances HA RNA replication and/or expression, which is consistent with the finding that YTHDF2 overexpression also enhances IAV gene expression under conditions where viral spread cannot occur (Fig. S2). Importantly, when we analyzed the pathogenic potential of these IAV-PR8 mutants, which bear reduced levels of m6A on the HA strand but are otherwise wildtype, we observed a statistically significant reduction in the pathogenic potential of both the +Mut and −Mut viruses (Fig. 6). This suggests that an IAV variant lacking m6A residues on all eight gene segments would likely be highly attenuated and also suggests that the addition of m6A might be a good target for the potential development of a broad spectrum antiviral that inhibits not only IAV replication but also the replication of some of the other viruses reported to bear m6A residues on their transcripts. Indeed, the drug DAA, which has been reported to inhibit m6A addition by depleting intracellular SAM levels is a potent inhibitor of not just IAV replication (Fig. 1A), but also a range of other viruses at doses that are nontoxic in animals (Bray et al., 2000; Wyde et al., 1990).
Because m6A and other epitranscriptomic changes have been proposed to inhibit the detection of foreign RNA molecules by cellular innate immune effectors (Durbin et al., 2016; Kariko et al., 2005), we addressed whether the reduced addition of m6A residues to HA transcripts might result in the enhanced activation of RIG-I, MDAS or IFNβ mRNA expression; however, this was not observed. Similarly, inactivation of METTL3 or overexpression of YTHDF2 did not exert an obvious effect on the basal level of expression of innate immune effectors in A549 cells (data not shown). It remains possible that elimination of all m6A residues on the IAV genome might give a different result.
The data presented in this manuscript do not define the mechanism by which m6A enhances IAV gene expression and replication, but do eliminate some possibilities. Because m6A addition enhances the expression of both spliced (NS1 and M2) and unspliced (NP) IAV gene segments equivalently, it is unlikely that m6A acts by regulating splicing. Also, because the increase in IAV protein expression closely matches the increase in mRNA expression (Figs. 1 and Fig. 2), it is unlikely that the positive effect of m6A on IAV gene expression is due to enhanced translation. Finally, as m6A residues present in cis enhance the function of not only the HA mRNA but also the HA vRNA, which is confined to the cell nucleus until packaged into progeny virion particles late in the viral life cycle, an effect of m6A on nuclear RNA export also seems unlikely. Instead, m6A seems to be acting by increasing IAV RNA levels by either enhancing viral replication or by enhancing viral RNA stability. Interestingly, while m6A has been reported to destabilize RNAs in some settings (Ke et al., 2017; Wang et al., 2014), others have reported that m6A can act to stabilize mRNAs (Fry et al., 2017). Of note, we have previously reported that m6A residues enhance the expression level of HIV-1 mRNAs as well as indicator plasmid mRNAs bearing m6A residues in cis, which may suggest that m6A is used by both IAV and HIV-1 to increase the steady state level of viral mRNA expression by a similar, post-transcriptional mechanism. Certainly, it seems very unlikely that these viruses would have retained m6A residues on their transcripts if these indeed exerted a destabilizing effect.
In conclusion, our data argue that m6A residues act in cis to promote the expression of IAV transcripts and that inhibition of m6A addition to IAV RNAs either indirectly, by elimination of METTL3, or directly, by elimination of sites of m6A addition on the IAV HA transcript, therefore results in a drop in not only viral gene expression and replication but also pathogenicity. Precisely how m6A exerts this strong positive effect on the replication of the nuclear RNA virus IAV, and how the divergent data obtained with the cytoplasmic RNA viruses HCV and ZKV, suggesting inhibition of viral spread by m6A (Gokhale et al., 2016; Lichinchi et al., 2016b), can be reconciled remains to be determined.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the Lead Contact, Bryan R. Cullen (bryan.cullen@duke.edu).
Experimental Model and Subject Details
Cell lines
The human 293T and A549 cell lines, and canine MDCK cells, were obtained from the ATCC. Wildtype and modified versions of these cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and antibiotics at 37°C in 5% CO2. The 293T cell lines expressing ectopic, FLAG-tagged YTHDF proteins were previously published (Kennedy et al., 2016). The 293T and MDCK cell lines are of female origin, while the A549 cell line is of male origin.
YTHDF overexpressing A549 cell lines
Lentiviral vectors, based on pLEX, expressing FLAG-tagged cDNAs encoding human YTHDF1 and YTHDF2, or gfp (Kennedy et al., 2016), were used to generate A549 cells that overexpress these proteins. 293T cells (2.5 × 106) were transfected using the PEI method with a lentiviral vector (15 µg), the packaging plasmid ΔCR8.74 (10 µg), and the VSV-G envelope expression plasmid pMD2.G (5 µg) to generate lentiviral particles that were used to transduce A549 cells. After puromycin (2 µg/ml) selection, the transduced cells were single cell cloned and cell lines expressing high levels of FLAG-tagged YTHDF1, YTHDF2 or gfp identified by Western blot. One clonal YTHDF1 and two clonal YTHDF2 overexpressing A549 cell lines were generated, termed A549/Y1, A549/Y2.1 and A549/Y2.2.
METTL3 knock-out A549 cell lines
CRISPR/Cas9 was used to knock-out the METTL3 gene in A549 cells. An sgRNA specific for exon 4 of the METTL3 gene (Table S1) was obtained from the GECKO sgRNA library (Sanjana et al., 2014) and cloned into the lentiviral vector lentiCRISPRv2. 293T cells (2.5 × 106) were transfected using the PEI method with the lentiviral vector expressing both the sgRNA and Cas9 (15 µg), the packaging plasmid ΔCR8.74 (10 µg), and the VSV-G envelope expression plasmid pMD2.G (5 µg) to generate lentiviral particles that were used to transduce A549 cells. After puromycin (2 µg/ml) selection, the transduced cells were single cell cloned and cells lacking METTL3 expression identified by Western blot. Genome editing of the targeted region was also confirmed by PCR of exon 4 of METTL3 from genomic DNA followed by sequencing (Fig. S1). Two METTL3 knockout A549 cell lines were generated, termed A549/M3.1 and A549/M3.2.
CRISPR SAM expressing cell lines
293T cells (2.5 × 106) were transfected using the PEI method with a lentiviral vector expressing MS2-p65-HSF1 (15 µg) (Konermann et al., 2015), the packaging plasmid ΔCR8.74 (10 µg), and the VSV-G envelope expression plasmid pMD2.G (5 µg) to generate lentiviral particles used to transduce A549 cells. After hygromicin B (25 µg/ml) selection, the transduced cells were single cell cloned and a cell line expressing a high level of MS2-p65-HSF1 identified. This A549/MS2-p65-HSF1 cell line was then transduced with lentiviral particles generated in wildtype 293T cells transfected with lentiSAMv2, ΔCR8.74, and pMD2.G, as described above. LentiSAMv2 expresses both a dCAS9/VP64 fusion protein and an sgRNA expressed from a U6 promoter. Transduced cells were then selected for both blasticidin (10 µg/mL) and hygromycin B (25 µg/mL) resistance to generate polyclonal cell lines expressing dCas9/VP64, and MS2-p65-HSF1 and an sgRNA specific for the endogenous human promoter driving expression of YTHDF1, YTHDF2 or YTHDF3 (Table S1). A cell line transduced with a lentiviral vector lacking any sgRNA was used as a no guide control. A549-based cell lines overexpressing each YTHDF protein were then obtained by single cell cloning and overexpression of the YTHDF proteins verified by Western blot. The sgRNA sequences used were obtained from Table S6 from Konermann et al., 2015.
Animal subjects
Female wildtype C57BL/6J mice were purchased (000664, Jackson Labs) and infected at 6 weeks old. All mice were maintained on a 12 h light/ dark cycle with continuous access to food and water. Experiments were performed at biosafety level 2 at Duke University. All procedures involving animals were performed as approved by the Duke University IACUC.
Method Details
Western blots
All Western blots were performed using the same protocol. Protein samples were extracted using Laemmli buffer, sonicated and denatured at 95°C for 10 min before being loaded onto a Tris-Glycine SDS polyacrylamide gel. After electrophoresis, proteins were transferred to a nitrocellulose membrane, and then blocked in 5% milk in PBS-Tween. Membranes were incubated in primary and secondary antibodies diluted in 5% milk/PBS-Tween, with membranes washed 3 times for 15 min each with PBS-Tween. Dilutions for each antibody are listed in the resource table. Chemiluminescence was visualized using WesternBright ECL.
IAV infections
Both 293T and A549 cells were infected with wild type and mutant IAV-PR8 in Optimem supplemented with 1% BSA. Single round infections were performed at an MOI of 1.0 or MOI of 3.0 where indicated, while multicycle, spreading infections were performed at an MOI of 0.01. Cells were incubated with infectious media at 37°C for 2 h before this was replaced with DMEM supplemented with 1% bovine serum albumin (BSA) and antibiotics. For multicycle infections, post-infection media were also supplemented with TPCK-treated Trypsin.
Quantitative RT-PCR
qRT-PCR was performed to determine the mRNA levels for IAV genes M2 and HA, and cellular immune response genes RIG-I, MDA5 and IFNβ1. The level of GAPDH mRNA was used to normalize all qRT-PCR experiments. All primer sequences are listed in Table S2. For multicycle IAV infections, RNA was collected at 0, 24, 48 and 72 hpi. For the evaluation of immune response genes, RNA was collected at 12 hpi. RNA was extracted using the TRIzol method. cDNA was generated using the Ambion cDNA synthesis kit following the manufacturer’s protocol. All cDNAs were generate with random primers, except for detection of IAV HA mRNA, which utilized an HA specific RT primer (HA.RT; Table S2). This primer was designed to reverse transcribe only HA mRNA, using the methodology published by Kawakami et al. (2011), by adding a non-viral tag CCAGATCGTTCGAGTCGT to the mRNA sequence, which was then used to specifically amplify HA mRNA. All qPCR was performed using Sybr Select Master Mix following the manufacturer’s instructions. All qRT-PCR was quantified using the ΔΔCT method.
IAV plaque assays
Wild type and mutant IAV-PR8 was grown in embryonated chicken eggs using standard protocols, and the titer determined by plaque assay on MDCK cells. MDCK cells were grown in 6-well plates to 80–90% confluency. Serial dilutions of the IAV-PR8 stock were then overlaid on the MDCK cells and incubated at 37°C for 1h. Infect ious media were then removed and replaced with plaque media, consisting of MEM, 5% NaHCO3, 1% BSA, 1µg/µl TPCK-treated Trypsin and 1% agar. MDCK cells were then incubated at 37°C for 72 h. Cells were fixed with 4% Paraformaldehyde (PFA) for 1h and the agar was washed off the wells. Cells were incubated with a polyclonal anti-PR8 antibody in 5% milk and PBS-Tween for 1 h, then washed 3 times for 5 min each with PBS-Tween and incubated with anti-mouse HRP in 5% milk and PBS-Tween for 1 h. Cells were then washed 3 times for 5 min each with PBS-Tween before being incubated with True Blue HRP Substrate for 15 min. The viral titer was ascertained by multiplying the number of plaques by the dilution factor. Plaque diameter was determined using ImageJ.
PAR-CLIP and PA-m6A-seq analysis
PAR-CLIP was performed as previously described (Hafner et al., 2010). 293T-based cell lines expressing FLAG-tagged YTHDF proteins, or FLAG-tagged GFP as a control (Kennedy et al., 2016), were pulsed with 100 µM 4SU in fresh media for 4 h. Cells were then infected with IAV-PR8 in 4SU-supplemented media for 4 h before media were replaced with 4SU-supplemented fresh media and incubated for a further 20 h. The cells were then UV irradiated, harvested and the PAR-CLIP protocol performed using an antibody specific for the FLAG epitope tag.
For PA-m6A–seq, 293T cells were again pulsed with 4SU as described above. At the end of the 4SU pulse, total RNA was extracted using TRIzol, and poly(A)+ purified using oligo-dT. 10 µg of poly(A)+ RNA was then used for the standard PA-m6A-seq protocol (Chen et al., 2015) using an m6A specific polyclonal antibody.
Small RNA sample preparation and deep sequencing
RNA isolated from either PAR-CLIP or PA-m6A-seq was processed with the TruSeq Small RNA Sample Preparation Kit. Adapter-ligated RNA was reverse-transcribed using SuperScript III and then amplified with GoTaq green PCR master mix with the TruSeq 3′ indices. PCR products were separated by gel electrophoresis on 10% (wt/vol) polyacrylamide gels and DNA bands corresponding to the expected ~145 bp libraries isolated.
PAR-CLIP and PA-m6A-seq libraries were sequenced on a HiSeq 2000. Base calling was performed with CASAVA and was processed with the fastx toolkit. Reads with a length greater than 14 bp were used for downstream bioinformatic analysis.
Bioinformatics
Read alignments were performed using Bowtie (Langmead et al., 2009). Reads were first aligned to the human genome build hg19 allowing up to 1 mismatch, then unaligned reads were aligned to the IAV-PR8 transcriptome, again allowing up to 1 mismatch. Characteristic T>C mutations, resulting from 4SU incorporation and crosslinking, were present among the viral aligned reads. In addition, A>G mutations were also present, corresponding to negative strand alignments. Reads containing T>C mutations were grouped as positive strand mRNA reads, while those containing A>G mutations were grouped as negative strand vRNA reads. All data was processed using in-house Perl scripts and Samtools (Li et al., 2009), and visualized with IGV, as previously described (Kennedy et al., 2016).
Rescue of IAV PR8 mutants
A/Puerto Rico/8/1934 was used with a sequence corresponding to those deposited under accession numbers AF389115-389122 with the following changes. Point mutation T>C.1082 in segment 5. Segments were cloned into the ambisense pDZ vector by Gibson cloning, as previously described (Quinlivan et al., 2005).
Mutant PR8 viruses lacking m6A sites were generated using the same sequences. Eight, on the plus strand, or nine, on the minus strand, silent mutations were introduced into the IAV-PR8 HA segment to remove DRACH motifs shown to be methylated by PAR-CLIP and PA-m6A-seq (Fig. S3). Two gene blocks containing the full length IAV-PR8 HA segment containing these mutations, either on the plus or minus strand, were cloned into the ambisense pDZ vector by Gibson cloning, as previously described (Quinlivan et al., 2005). These IAV-PR8 mutants were then rescued using previously described methods (Heaton et al., 2016). Briefly, pDZ clones for each IAV segment and either the wild type, plus strand mutant or minus strand mutant pDZ-HA clone were transfected into 293T cells. Virus was amplified in embryonated chicken eggs. All wild type and mutant IAV-PR8 variants were plaque-purified, and HA segments sequenced to confirm the introduction of mutations, prior to phenotypic analysis. Mutant PR8 variants were titered using a Neuraminidase assay (Sigma), following the manufacturer’s instructions.
To visualize packaged vRNA segments, IAV virions were isolated from embryonated eggs, overlaid on a 30% sucrose gradient and centrifuged at 27,500rpm for 90 min. Viral RNA was extracted using TRIzol and the vRNAs resolved on a 4% TBE Urea gel and then stained with ethidium bromide.
Immunofluorescent cell staining
A549 cell lines were grown on coverslips and infected with IAV-PR8 at an MOI of 3 for a single round infection, as described above. After 24 hpi media was removed and cells were fixed in 4% PFA at room temperature for 20 min. PFA was removed and the cells were washed in PBS. Cells were permeabilized in 0.1% Triton-X before being blocked in 1% BSA/PBS for 30 min under gentle agitation at room temperature. Cells were then probed with anti-NP at a 1:100 dilution in 1% BSA/PBS for 1 h under gentle agitation. Cells were washed for 10 min 3 times with PBS before being probed with anti-mouse Alexa Fluor 488 at a dilution of 1:1000 in 3% BSA/PBS for 1 h under gentle agitation. Then cells were again washed for 10 min 3 times in PBS before the coverslips were mounted on slides using Vectashield mounting media and imaged.
A549 multicycle infection flow cytometry
A549 lung epithelial cells were infected at an MOI of 0.01 and then incubated at 37°C in serum-free media containing 0.3 µg/mL trypsin to facilitate a multicycle infection. Samples were collected at the indicated timepoints, stained for live cells (ThermoFisher Live/Dead fixable violet stain), then fixed in 2% paraformaldehyde for 10 min at room temperature. After all samples were collected, they were stained using PY102 (provided by Tom Moran at the Experimental Therapeutics Institute at the Icahn School of Medicine at Mount Sinai) at 1 µg/mL in PBS/BSA. Samples were analyzed on a Fortessa X20 (BectonDickinson) then processed using FlowJo software.
Animal experiments
At 6 weeks old, female wild type C57BL/6J mice were infected intranasally at the indicated doses of the wildtype IAV-PR8 isolate, +Mut or −Mut viruses in 40 mL of pharmaceutical grade PBS under anesthesia (ketamine/xylazine). Mouse morbidity was monitored via daily weighing, and a loss of >20% of the starting weight was defined as the humane endpoint. For all experimental conditions, 5 mice were used per dose.
Quantification and statistical analysis
Band intensities for Western blots were quantified using ImageJ. For qRT-PCR assays and titering of IAV-PR8, a Student’s t-test was performed to determine significance with p<0.05 deemed as statistically significant. For all qRT-PCR experiments 3 independent biological samples were collected and analyzed, with 3 experimental replicates for each sample. For Western blotting 3 independent biological samples were collected and analyzed. For immunofluorescent cell staining 3 independent biological replicates were stained and imaged. For flow cytometry 3 independent biological samples replicates were collected and analyzed, with 2 experimental replicates for each sample.
Data and software availability
The deep sequencing datasets generated in this study have been deposited in the GEO database under accession number GSE98033.
Supplementary Material
Highlights.
m6A sites on influenza A virus (IAV) mRNAs and vRNAs were mapped.
High levels of m6A modification increase IAV RNA expression.
IAV mutants lacking m6A sites on the HA segment are attenuated in culture.
These same IAV HA m6A mutants show reduced pathogenicity in mice.
Acknowledgments
This research was supported by National Institutes of Health grant R21-AI130574. D.G.C. was funded by Marie-Skłodowska Curie Global Fellowship MSCA-IF-GF:747810. K.T. was supported by National Institutes of Health grant T32-CA009111 and R.E.D. was supported by T32-GM007184.
The authors thank Feng Zhang for reagents used in this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
D.G.C., E.M.K., H.P.B., R.E.D., N.S.H. and B.R.C designed experiments, analyzed the data and wrote the manuscript. D.G.C., E.M.K., K.T., H.P.B. and R.E.D. performed the experiments.
References
- Bader JP, Brown NR, Chiang PK, Cantoni GL. 3-Deazaadenosine, an inhibitor of adenosylhomocysteine hydrolase, inhibits reproduction of Rous sarcoma virus and transformation of chick embryo cells. Virology. 1978;89:494–505. doi: 10.1016/0042-6822(78)90191-5. [DOI] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–719. doi: 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden D, Pusch O, Lee F, Tucker L, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J Virol. 2003;77:11531–11535. doi: 10.1128/JVI.77.21.11531-11535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M, Driscoll J, Huggins JW. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-L-homocysteine hydrolase inhibitor. Antiviral Res. 2000;45:135–147. doi: 10.1016/s0166-3542(00)00066-8. [DOI] [PubMed] [Google Scholar]
- Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N, Han D, Dominissini D, Dai Q, Pan T, et al. High-resolution N(6) -methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew Chem Int Ed Engl. 2015;54:1587–1590. doi: 10.1002/anie.201410647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AT, Brummelkamp TR, Westerhout EM, Vink M, Madiredjo M, Bernards R, Berkhout B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol. 2004;78:2601–2605. doi: 10.1128/JVI.78.5.2601-2605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers RC, Friderici KH, Rottman FM. Characterization of Novikoff hepatoma mRNA methylation and heterogeneity in the methylated 5’ terminus. Biochemistry. 1975;14:4367–4374. doi: 10.1021/bi00691a004. [DOI] [PubMed] [Google Scholar]
- Durbin AF, Wang C, Marcotrigiano J, Gehrke L. RNAs containing modified nucleotides fail to trigger RIG-I conformational changes for innate immune signaling. MBio. 2016;7:e00833–00816. doi: 10.1128/mBio.00833-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AA, Muller K, Scholtissek C. Specific inhibition of the synthesis of influenza virus late proteins and stimulation of early, M2, and NS2 protein synthesis by 3-deazaadenosine. Virology. 1990;177:523–531. doi: 10.1016/0042-6822(90)90517-u. [DOI] [PubMed] [Google Scholar]
- Fry NJ, Law BA, Ilkayeva OR, Holley CL, Mansfield KD. N6-methyladenosine is required for the hypoxic stabilization of specific mRNAs. RNA. 2017 doi: 10.1261/rna.061044.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013;155:793–806. doi: 10.1016/j.cell.2013.10.026. [DOI] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- Gokhale NS, McIntyre AB, McFadden MJ, Roder AE, Kennedy EM, Gandara JA, Hopcraft SE, Quicke KM, Vazquez C, Willer J, et al. N6-methyladenosine in Flaviviridae viral RNA genomes regulates infection. Cell Host Microbe. 2016;20:654–665. doi: 10.1016/j.chom.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales-van Horn SR, Sarnow P. Making the mark: The role of adenosine modifications in the life cycle of RNA viruses. Cell Host Microbe. 2017;21:661–669. doi: 10.1016/j.chom.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton NS, Moshkina N, Fenouil R, Gardner TJ, Aguirre S, Shah PS, Zhao N, Manganaro L, Hultquist JF, Noel J, et al. Targeting Viral Proteostasis Limits Influenza Virus, HIV, and Dengue Virus Infection. Immunity. 2016;44:46–58. doi: 10.1016/j.immuni.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongay CF, Orr-Weaver TL. Drosophila Inducer of MEiosis 4 (IME4) is required for Notch signaling during oogenesis. Proc Natl Acad Sci U S A. 2011;108:14855–14860. doi: 10.1073/pnas.1111577108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser K, Subbarao K. Influenza vaccines: challenges and solutions. Cell Host Microbe. 2015;17:295–300. doi: 10.1016/j.chom.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariko K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Kawakami E, Watanabe T, Fujii K, Goto H, Watanabe S, Noda T, Kawaoka Y. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J Virol Methods. 2011;173:1–6. doi: 10.1016/j.jviromet.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vagbo CB, Geula S, Hanna JH, Black DL, Darnell JE, Jr, Darnell RB. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017;31:990–1006. doi: 10.1101/gad.301036.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EM, Bogerd HP, Kornepati AV, Kang D, Ghoshal D, Marshall JB, Poling BC, Tsai K, Gokhale NS, Horner SM, et al. Posttranscriptional m(6)A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe. 2016;19:675–685. doi: 10.1016/j.chom.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EM, Courtney DG, Tsai K, Cullen BR. Viral epitranscriptomics. J Virol. 2017;91:e02263–02216. doi: 10.1128/JVI.02263-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug RM, Morgan MA, Shatkin AJ. Influenza viral mRNA contains internal N6-methyladenosine and 5’-terminal 7-methylguanosine in cap structures. J Virol. 1976;20:45–53. doi: 10.1128/jvi.20.1.45-53.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing, S The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mason CE. The pivotal regulatory landscape of RNA modifications. Annu Rev Genomics Hum Genet. 2014;15:127–150. doi: 10.1146/annurev-genom-090413-025405. [DOI] [PubMed] [Google Scholar]
- Lichinchi G, Gao S, Saletore Y, Gonzalez GM, Bansal V, Wang Y, Mason CE, Rana TM. Dynamics of the human and viral m6A RNA methylomes during HIV-1 infection of T cells. Nature Microbiology. 2016a;1:16011. doi: 10.1038/nmicrobiol.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichinchi G, Zhao BS, Wu Y, Lu Z, Qin Y, He C, Rana TM. Dynamics of human and viral RNA methylation during Zika virus infection. Cell Host Microbe. 2016b;20:666–673. doi: 10.1016/j.chom.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767–772. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan P, Ayers DF, Rottman FM, Maroney PA, Nilsen TW. Unequal distribution of N6-methyladenosine in influenza virus mRNAs. Mol Cell Biol. 1987;7:1572–1575. doi: 10.1128/mcb.7.4.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlivan M, Zamarin D, Garcia-Sastre A, Cullinane A, Chambers T, Palese P. Attenuation of equine influenza viruses through truncations of the NS1 protein. J Virol. 2005;79:8431–8439. doi: 10.1128/JVI.79.13.8431-8439.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumuru N, Zhao BS, Lu W, Lu Z, He C, Wu L. N(6)-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. Elife. 2016;5 doi: 10.7554/eLife.15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyde PR, Ambrose MW, Meyer HL, Zolinski CL, Gilbert BE. Evaluation of the toxicity and antiviral activity of carbocyclic 3-deazaadenosine against respiratory syncytial and parainfluenza type 3 viruses in tissue culture and in cotton rats. Antiviral Res. 1990;14:215–225. doi: 10.1016/0166-3542(90)90003-p. [DOI] [PubMed] [Google Scholar]
- Zhong S, Li H, Bodi Z, Button J, Vespa L, Herzog M, Fray RG. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008;20:1278–1288. doi: 10.1105/tpc.108.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.