

Abstract
Background:
Primary mutations in the KIT gene are the driving force for gastrointestinal stromal tumors (GIST) tumorigenesis. Predictive role of KIT mutation status aids oncologists in patient management. There is a paucity of comprehensive data on the frequency of mutations in the KIT gene in GIST affecting Indian patients. The aims of this study were to determine the frequency and spectrum of molecular alterations affecting the KIT gene and assess their association with clinicopathologic features in a cohort of patients of GIST.
Materials and Methods:
Morphological and immunohistochemically confirmed GIST cases (n = 114) accessioned from August 2014-June 2015 were analyzed for mutations in KIT exons 9, 11, 13, and 17 and subjected to Sanger sequencing onto the ABI 3500 Genetic Analyzer. The sequences were analyzed using sequence analysis software: SeqScape® and Chromas Lite.
Results:
KIT mutations were seen in 70% of cases and the majority of KIT mutations involved exon 11 (57%), followed by exon 9 (10%), exon 13 (3%), and exon 17 (1%). Most common exon 11 mutations were in-frame deletions (61.4%) followed by substitution mutations (19.3%). Exon 9 mutations showed identical duplication of Ala-Tyr at codons 502–503. Simultaneous mutations affecting exon 11 and 13 were discovered. Novel variations, namely, p.Q556E (c.1666C>G), p.Q556dup (c.1666_1668dupCAG), p.K558_V559delinsS (c.1672_1677delAAGGTTinsAGT), p.Y503_F504insTY (c.1509_1510insACCTAT), and p.K642R (c.1925A>G) involving exons 11, 9, and 13, respectively, were observed.
Interpretation and Conclusions:
First study with complete analysis of all 4 exons of KIT (exons 9, 11, 13, and 17) in Indian GIST patients. Along with well-described KIT mutations, several rare double mutations as well as novel alterations were reported in this series.
Keywords: Exon11, exon9, gastrointestinal stromal tumors, KIT, wild type
Introduction
Gastrointestinal stromal tumors (GIST) are the most common gastrointestinal mesenchymal tumors.[1] GISTs are immunoreactive to CD117 and DOG1 in >95% of cases.[2] Molecular studies have shown that ~85% GISTs possess mutually exclusive gain-of-function mutations in either the KIT or PDGFRA gene leading to the pathogenesis of GIST through the activation of KIT downstream signals pathways.[2,3]
KIT, a member of the receptor tyrosine kinases where exon 11 encodes for the Juxtamembrane region, exon 9 for part of the extracellular domain, exon 13 and exon 17 for the intracellular kinases, ATP binding domain and phosphotransferase domain, respectively.[3] The range of mutations reported in the literature with the most common mutations identified in exon 11 is 20%–92%[4] followed by exon 9 in 3%–18%[5,6] with a lower frequency of around 0.6%–4% in exons 13 and 17.[6]
Currently, treatment with Imatinib (Gleevec®; Novartis, Basel, Switzerland) is the standard first-line of therapy for patients with GIST. Patients with exon 11 mutations exhibit maximal benefit from prolonged adjuvant treatment, exon 9 mutations may benefit with the escalation of Imatinib doses and wild-type GISTs demonstrate a limited response to Imatinib.[7] Therefore, genetic testing is emerging as a predictive biological marker that will aid clinicians in decision making while treating different subsets of GIST.
There is a paucity of molecular data on Indian GISTs as case reports,[8] short series[9] our clinical data.[10,11] The primary aim of this study was to determine the frequency of KIT mutations in a large cohort of GIST patients. This is the largest study from India covering all hot spot regions (exons 11, 9, 13, and 17) of the KIT gene.
Materials and Methods
Histopathologic and immunohistochemistry review
Patients with histologically confirmed GIST cases accessioned in the Divisions of Surgical Pathology and Molecular Pathology of Tata Memorial Center from August 2014 to June 2015. Hematoxylin and eosin (H and E) stained sections and the respective formalin-fixed paraffin embedded (FFPE) blocks of the tumor tissue were retrieved.
The study was approved by the Institutional Ethics Committee of Tata Memorial Center. All cases were reviewed, and the diagnosis was confirmed in accordance with the WHO classification.[12] Representative sections with >50% tumor were selected for molecular testing. Demographic, clinical, and immunohistochemistry (IHC) details were recorded from electronic medical records. Antibodies for IHC included CD117 (1:1000 dilution, Rabbit polyclonal, Dako, USA) and DOG1 (1:200 dilution, DOG1.1, Biocare, USA).
Molecular analysis
Genomic DNA extraction
Sections (4 μm × 10 μm) from FFPE blocks were deparaffinized using limonene, digested using Proteinase K at 65°C followed by DNA extraction using the QIAamp DNA mini kit (Qiagen, Inc., Valencia, CA) and quantified using Nanodrop (Thermoscientific, USA).
Polymerase chain reaction for ACTNB
Multiplex polymerase chain reaction (PCR) for beta-actin (ACTNB) housekeeping gene comprising two primer pairs was performed to check the integrity of DNA. Samples amplifiable for both the primer pairs were selected for KIT PCR [Supplementary Table 1 (579.4KB, tif) ].
ACTNB and KIT exon primer sequences
KIT polymerase chain reaction
In brief, PCR reaction was performed in 20 μl containing 100 ng of template DNA, 4 μL of × 5 PCR buffer, 0.7 μL (10 pmol/μL) of each primer,[13] 1.6 μL of 10 mM deoxynucleoside triphosphates, and 0.4 μL of GXL Taq polymerase (Takara, Clontech). The PCR conditions were: 95°C for 15 min followed by 40 cycles of 95°C for 30 s, 56°C for 1 min, 72°C for 1 min, and final extension at 72°C for 10 min. PCR products were analyzed in 1.5% agarose gel and subjected to purification using EXO-SAP IT (USB, Affymetrix).
Gene sequencing on 3500 genetic analyzer
The purified products were bidirectionally sequenced using Big Dye version 3.1 cycle sequencing kit (Applied Biosystems, USA). Big Dye X-Terminator kit was used for purification followed by loading on ABI 3500 Genetic Analyzer for capillary electrophoresis (Applied Biosystems, Foster City, CA, USA).
Sequence analysis
SeqScape® (Applied Biosystems) and Chromas Lite software were used for sequence analysis using KIT reference sequence (Gene ID 3815). Mutations were reported as per Human Genome Variation Society (www.hgvs.org) recommendations. The dbSNP, COSMIC, and Ensembl genome databases were referred.
Statistical analysis
SPSS statistical software version 16.0 (IBM, NY, USA) was used for correlation of the KIT mutation status with clinicopathological features. Chi-square test with the value of P < 0.05 was considered statistically significant.
Results
Among 114 confirmed GIST cases 100 were subjected to molecular analysis.
Clinicopathological features of the entire cohort (n = 100)
Median age: 55 years (range: 23–79 years) with 60% being >50 years. Male to female ratio was 2.8:1. Primary tumors site wise distribution: stomach (47%), small intestine (36%) (jejunum [19%], duodenum [9%], ileum [5%], duodenojejunum [1%], jejuno-ileum [1%] and in one case exact small intestinal location was unknown), colon (2%), and rectum (6%). Extra-gastrointestinal GIST was observed in 9% of cases at following sites: retroperitoneum (3%), mesentery (3%), omentum (2%), and pelvic cavity (1%). On histopathological evaluation, spindle, epithelioid, and mixed subtypes accounted for 75%, 19%, and 6% of the tumors, respectively. IHC evaluation revealed CD117 positivity in 98%, among CD117 negative cases one was negative for DOG-1; positive for CD34 [Table 1].
Table 1.
Correlation of clinicopathological features with KIT mutational status
Genotype analysis
The tumor samples represented primary (73%), metastatic (21%), and local recurrence (5%) sites. In one case, primary and metastatic tumors both were analyzed.
KIT mutations were seen in 70 (70%) cases involving exons 11 (57%), 9 (10%), 13 (3%), and 17 (1%). One case with double mutations of exons 11 and 13. A majority of mutations were heterozygous (n = 64, 91.4%). Homozygous mutations in 6 (8.6%); which was in exon 11 in 5 cases. (In-frame deletion [n = 3]; substitution [n = 2]) and exon 13 in 1 case with substitution mutation [Table 2].
Table 2.
Summary of the spectrum of KIT mutations observed in gastrointestinal stromal tumors cases (n=70)

Exon 11 mutations
Exon 11 mutations were in 57% of cases [Table 2]. In-frame deletions in 35 (61.4%), 11 substitutions (19.3%), 9 double mutations (15.7%), 1 insertion and duplication (1.8%), respectively. Common mutation was p.W557_K558 del (c.1669_1674delTGGAAG) [Figure 1a] in 13 cases (22.8%); 3 cases each (5.3%) with p.K550_V555del (c.1649_1666AACCCATGTATGAAGTAC), p.V559D (c.1676T>A); p.V560D (c.1679T>A) [Figure 2].
Figure 1.
Partial electropherograms of KIT gene mutations (a) Exon 11 heterozygous deletion c.1669_1674delTGGAAG. (b and c) Double mutations involving exon 11 (novel variations) (b) KIT exon 11 c.1666_1668dupCAG; 1669_1674delTGGAAG mutation (c) KIT exon 11 c.1672_1677delAAGGTTinsAGT mutation. (d) Heterozygous novel variation in exon 13 c.1925A >G (red arrow)
Figure 2.
Partial electropherograms with substitution mutations involving exon 11 (a) heterozygous mutation (c.1679T>A) (black arrow) (b) homozygous mutation (c.1679T>G) (arrowhead)
Exon 11 mutations were heterogeneous with in-frame deletion of 3–51 nucleotides (codons 550–576) in classic hot-spot region at the 5’ end of the exon (codons 550–560). Double mutations were identified in 9 cases (16%), 8 within exon 11 (14.3%), exon 11 and 13 were involved in one case (1.8%). In all double mutations, one mutation was consistently a deletion, whereas the 2nd mutation represented substitutions (n = 5), insertions (n = 2) and duplication (n = 1). Interestingly, 3 novel mutations were unraveled, i.e., p.Q556E (c.1666C>G), p.Q556dup (c.1666_1668dupCAG) [Figure 1b] and p.K558_V559delinsS (c.1672_1677delAAGGTTinsAGT) [Figure 1c] as partner mutations among the cases with double mutations [Supplementary Table 2 (8.9MB, tif) ].
Clinicopathological details in KIT mutated cases (n=70)
One case with simultaneous mutations in exons 11 and 13 harbored in-frame deletion in exon 11; p.M552_K558del (c.1654_1674delATGTATGAAGTACAGTGGAAG) and a novel substitution mutation; p.K642R (c.1925A>G) [Figure 1d] in exon 13 in a treatment-naive young male (35 years) GIST at gastric location, spindle morphology, and immunoreactivity to CD117 and DOG1.
The substitution mutations were p.V559D (3/57; 5%), p.V560D (3/57; 5%), p.V559A (2/57; 3.5%), and 1 (1.8%) cases each with p.V560G, p.T574I, and p.L576P among this 9 were homozygous and 2 heterozygous.
Insertion of 3 nucleotides, p.K558delinsBP (c.1673_1674insTCC), and duplication p.Y577_K580dup (c.1731_1742dupTTATGATCACAA) was seen 1 case (1.8%) each, respectively.
The KIT exon 11 mutation was predominant in men (n = 40, 70.1%), in the stomach (n = 28, 49%) spindle histology (n = 46, 80.7%), and immunoreactivity to CD117 (n = 55, 98.2%) [Table 2]. Gastric tumors showed deletions (54.5%) and duplications (100%), whereas small intestinal tumors mostly displayed substitution (40%) and insertion (100%) mutations.
Exon 9 mutations
Mutations were identified in 10 cases located in the small intestine with significant association (P = 0.004). One was located in the retroperitoneum. Ninety percent (9/10) tumors revealed internal tandem duplications (ITD) of 6 nucleotides (c.1504_1509 dup GCCTAT) causing duplication of Ala-Tyr at codons 502–503 [Figure 3a]. All were male predominant (8/10) with spindle cell morphology (6/10). One case with the insertion of 6 nucleotides (c.1509_1510insACCTAT) causing p.Y503_F504insTY [Figure 3b], was a novel mutation in a 70 years old man with duodenal GIST, epithelioid morphology, immunoreactive for CD117 and DOG1.
Figure 3.
Partial electropherograms showing exon9 mutations (a) internal tandem duplication mutation (c.1504_1509insGCCTAT) (black arrow) (b) novel variation with insertion mutation (c.1509_1510insACCTAT) (arrow head)
Exon 13 mutations
Three cases involving p.K642E mutation (c.1924A>G) [Figure 4a], 2/3 were in elderly men, at gastric, an anorectal site with mixed morphology. One was a double mutation in association with exon 11 [Table 2].
Figure 4.
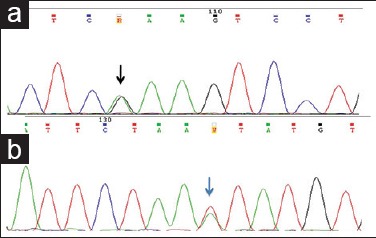
Partial electropherograms with exon13 and 17 mutations (a) heterozygous mutation involving exon 13 (c.1924A>G) (black arrow). (b) Heterozygous mutation involving exon 17 (c.2466T>A) (blue arrow)
Exon 17 mutation
A single case with p.N822K (c.2466T>A) [Figure 4b] was identified. The tumor originated in jejunum in an elderly man with spindle morphology.
Clinicopathological features of KIT-mutated cases(n = 70)
Age range was 23–79 (median-55 years) 61.4% (n = 43) cases were >50 years. Sex ratio was 2.7:1 (M-51; F-19). Small intestine (30/70; 42.8%) was the most common primary site followed by the stomach (29/70; 41.4%). Spindle, epithelioid, and mixed subtypes in 53 (75.7%), 13 (18.6%), and 4 (5.7%) cases, respectively. On IHC, 69 cases were positive for CD117 and/or DOG1, and in 1 case both were negative. However, CD34 was positive. Clinicopathological features had no significant correlation with the KIT mutation status.
Discussion
The frequency of KIT mutations in GIST is variable with 50%–92.9% in the Western countries[14,15,16] to 38.5%–84.2% in Asian regions.[5,17,18,19] There are only 2 studies, and few case reports from India.[5,8,9] Cyriac et al.[9] retrospectively analyzed KIT exons 9 and 11 on 19 cases of metastatic GIST. Ahmad et al.[5] evaluated KIT exons 9, 11, and 13 in 70 patients diagnosed with GIST. The frequency of KIT mutations in our cohort was 70% (70/100) is the highest reported from India comparable to Western data.[15,20,21] An interesting observation in the present study is the documentation of mutations in exons 13, 17, and simultaneous mutations in exons 11 and 13. The variation in mutations frequency across literature stems from referral center bias, tumor tissue types, methodology, and analysis.[22]
In concordance with published studies, the majority of exon 11 mutations were in the classic “hot-spot” region (codons 550–560), with remarkable heterogeneity. The incidence of ITD affecting 3’ portion of exon 11 in our series was 1.8% lower than the reported rate of 11%–14%.[4] The primary double KIT mutations which are quite rare may involve nucleotides within the same hotspot exon, which is considered to have an origin in a single mutagenic event or is characterized by losses or frameshift/stop codon mutations in the wild-type KIT allele of KIT-mutated GISTs.[23] Only 4 examples of double primary mutations involving different hotspot exons of KIT in treatment-naive GISTs are available in literature till date, of which 3 involved exons 9 and 11,[24,25] one in exons 13 and 17.[26] A novel occurrence of primary double mutation in exons 11 and 13, in a therapy-naive patient with gastric GIST was reported in this study.
Prognostic value of KIT mutations has conflicting reports. Patients with in-frame deletions (codons 557–558), Indels have been associated with aggressive behavior as compared to substitutions.[4] Lasota et al.[27] have reported a risk of progressive disease and malignant clinical behavior with homozygous exon 11 mutations as compared to heterozygous and are considered as an adverse prognostic marker in GIST. Duplications at 3’ portion of exon 11 have been linked to gastric GISTs which are relatively indolent.[28] Prognostic significance of primary double KIT mutations is not fully recognized on account of their rarity; however, cases involving diverse hotspot exons seem to confer an aggressive behavior or a high-risk phenotype.[23]
The frequency of exon 9 mutation was 10% which is in the range of 5%–18% in the literature[5] with a distinct subset of GIST localized to small intestine characterized by an identical duplication (codons 502–503). A novel variant Y503_F504insTY (1509_1510insACCTAT) in a duodenal GIST was reported in this study.
The incidence of exons 13 (p.K642E) and 17 (p.N822K) mutations in our series were in the range as previously published in the literature. A few reports indicate dismal prognosis for exon 13 mutated gastric tumors and exon 17 mutations implications are unknown.
Conclusions
The present study is the first largest comprehensive analysis of all 4 exons of KIT in Indian patients with GIST with important caveats. Not only common, well-described KIT mutations identified but also rare double mutations, novel alterations were reported in this cohort. The clinical implications associated with these mutations have already been published by this group.[10,11]
Financial support and sponsorship
The support from Molecular Pathology, Surgical Pathology and Medical Oncology GI Unit is acknowledged.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
The support from Molecular Pathology, Surgical Pathology and Medical Oncology GI Unit is acknowledged.
References
- 1.ESMO/European Sarcoma Network Working Group. Gastrointestinal stromal tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):iii21–6. doi: 10.1093/annonc/mdu255. [DOI] [PubMed] [Google Scholar]
- 2.Corless CL. Gastrointestinal stromal tumors: What do we know now? Mod Pathol. 2014;27(Suppl 1):S1–16. doi: 10.1038/modpathol.2013.173. [DOI] [PubMed] [Google Scholar]
- 3.Lasota J, Miettinen M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs) Semin Diagn Pathol. 2006;23:91–102. doi: 10.1053/j.semdp.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Andersson J, Bümming P, Meis-Kindblom JM, Sihto H, Nupponen N, Joensuu H, et al. Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology. 2006;130:1573–81. doi: 10.1053/j.gastro.2006.01.043. [DOI] [PubMed] [Google Scholar]
- 5.Ahmad F, Lad P, Bhatia S, Das BR. Molecular spectrum of c-KIT and PDGFRA gene mutations in gastro intestinal stromal tumor: Determination of frequency, distribution pattern and identification of novel mutations in Indian patients. Med Oncol. 2015;32:424. doi: 10.1007/s12032-014-0424-7. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto H, Oda Y, Kawaguchi K, Nakamura N, Takahira T, Tamiya S, et al. c-KIT and PDGFRA mutations in extragastrointestinal stromal tumor (gastrointestinal stromal tumor of the soft tissue) Am J Surg Pathol. 2004;28:479–88. doi: 10.1097/00000478-200404000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Foo WC, Liegl-Atzwanger B, Lazar AJ. Pathology of gastrointestinal stromal tumors. Clin Med Insights Pathol. 2012;5:23–33. doi: 10.4137/CPath.S9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sen A, Gangavatiker R. EXON 11, C KIT mutation in a ‘CD 117’ & ‘DOG 1’ negative colonic gastrointestinal tumor. Med J Armed Forces India. 2014;70:186–8. doi: 10.1016/j.mjafi.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cyriac S, Rajendranath R, Sagar TG. Gastrointestinal stromal tumor: Analysis of outcome and correlation with c-KIT status in Indian population. Indian J Cancer. 2014;51:35–9. doi: 10.4103/0019-509X.134616. [DOI] [PubMed] [Google Scholar]
- 10.Ramaswamy A, Ostwal V, Shetty O, Sahu A, Paul D, Pai T, et al. Neoadjuvant imatinib in locally advanced gastrointestinal stromal tumours, will KIT mutation analysis be a pathfinder? J Gastrointest Cancer. 2016;47:381–8. doi: 10.1007/s12029-016-9835-3. [DOI] [PubMed] [Google Scholar]
- 11.Ramaswamy A, Pande N, Shetty O, Shetty N, Gupta S, Ostwal V. Pazopanib in metastatic multiply treated progressive gastrointestinal stromal tumors: Feasible and efficacious. J Gastrointest Oncol. 2016;7:638–43. doi: 10.21037/jgo.2016.03.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC; 2010. [Google Scholar]
- 13.Debiec-Rychter M, Dumez H, Judson I, Wasag B, Verweij J, Brown M, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40:689–95. doi: 10.1016/j.ejca.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 14.Wasag B, Debiec-Rychter M, Pauwels P, Stul M, Vranckx H, Oosterom AV, et al. Differential expression of KIT/PDGFRA mutant isoforms in epithelioid and mixed variants of gastrointestinal stromal tumors depends predominantly on the tumor site. Mod Pathol. 2004;17:889–94. doi: 10.1038/modpathol.3800136. [DOI] [PubMed] [Google Scholar]
- 15.Kern A, Görgens H, Dittert DD, Krüger S, Richter KK, Schackert HK, et al. Mutational status of KIT and PDGFRA and expression of PDGFRA are not associated with prognosis after curative resection of primary gastrointestinal stromal tumors (GISTs) J Surg Oncol. 2011;104:59–65. doi: 10.1002/jso.21905. [DOI] [PubMed] [Google Scholar]
- 16.Tryggvason G, Hilmarsdottir B, Gunnarsson GH, Jónsson JJ, Jónasson JG, Magnússon MK. Tyrosine kinase mutations in gastrointestinal stromal tumors in a nation-wide study in Iceland. APMIS. 2010;118:648–56. doi: 10.1111/j.1600-0463.2010.02643.x. [DOI] [PubMed] [Google Scholar]
- 17.Kang HJ, Nam SW, Kim H, Rhee H, Kim NG, Kim H, et al. Correlation of KIT and platelet-derived growth factor receptor alpha mutations with gene activation and expression profiles in gastrointestinal stromal tumors. Oncogene. 2005;24:1066–74. doi: 10.1038/sj.onc.1208358. [DOI] [PubMed] [Google Scholar]
- 18.Teong YT, Teo ST, Tan LP, Wu BQ, Peh SC. An immunohistochemical and molecular study of gastrointestinal stromal tumours. Med J Malaysia. 2006;61:526–33. [PubMed] [Google Scholar]
- 19.Du CY, Shi YQ, Zhou Y, Fu H, Zhao G. The analysis of status and clinical implication of KIT and PDGFRA mutations in gastrointestinal stromal tumor (GIST) J Surg Oncol. 2008;98:175–8. doi: 10.1002/jso.21104. [DOI] [PubMed] [Google Scholar]
- 20.Emile JF, Brahimi S, Coindre JM, Bringuier PP, Monges G, Samb P, et al. Frequencies of KIT and PDGFRA mutations in the MolecGIST prospective population-based study differ from those of advanced GISTs. Med Oncol. 2012;29:1765–72. doi: 10.1007/s12032-011-0074-y. [DOI] [PubMed] [Google Scholar]
- 21.Baker G, Babb C, Schnugh D, Nayler S, Louw M, Goedhals J, et al. Molecular characterisation of gastrointestinal stromal tumours in a South African population. Oncol Lett. 2013;5:155–60. doi: 10.3892/ol.2012.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saponara M, Urbini M, Astolfi A, Indio V, Ercolani G, Del Gaudio M, et al. Molecular characterization of metastatic exon 11 mutant gastrointestinal stromal tumors (GIST) beyond KIT/PDGFRa genotype evaluated by next generation sequencing (NGS) Oncotarget. 2015;6:42243–57. doi: 10.18632/oncotarget.6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricci R, Martini M, Cenci T, Antinori A, Cassano A, Larocca LM. Case of rectal GI stromal tumor demonstrating that KIT and PDGFRA mutations are not always mutually exclusive. J Clin Oncol. 2016;34:e107–9. doi: 10.1200/JCO.2013.49.1258. [DOI] [PubMed] [Google Scholar]
- 24.Lee JH, Zhang X, Jung WY, Chae YS, Park JJ, Kim I. DNA ploidy and C-KIT mutation in gastrointestinal stromal tumors. World J Gastroenterol. 2004;10:3475–9. doi: 10.3748/wjg.v10.i23.3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakurai S, Oguni S, Hironaka M, Fukayama M, Morinaga S, Saito K. Mutations in c-KIT gene exons 9 and 13 in gastrointestinal stromal tumors among Japanese. Jpn J Cancer Res. 2001;92:494–8. doi: 10.1111/j.1349-7006.2001.tb01121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–74. doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- 27.Lasota J, vel Dobosz AJ, Wasag B, Wozniak A, Kraszewska E, Michej W, et al. Presence of homozygous KIT exon 11 mutations is strongly associated with malignant clinical behavior in gastrointestinal stromal tumors. Lab Invest. 2007;87:1029–41. doi: 10.1038/labinvest.3700628. [DOI] [PubMed] [Google Scholar]
- 28.Lasota J, Dansonka-Mieszkowska A, Stachura T, Schneider-Stock R, Kallajoki M, Steigen SE, et al. Gastrointestinal stromal tumors with internal tandem duplications in 3’ end of KIT juxtamembrane domain occur predominantly in stomach and generally seem to have a favorable course. Mod Pathol. 2003;16:1257–64. doi: 10.1097/01.MP.0000097365.72526.3E. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ACTNB and KIT exon primer sequences
Clinicopathological details in KIT mutated cases (n=70)