

Abstract
Alzheimer's disease (AD) and osteoporosis are both common degenerative diseases in the elderly population. The incidence of both diseases increases with age and will be posing enormous societal burden worldwide. It may appear that AD and osteoporosis are two distinct diseases although many risk factors are shared. Previous observational studies have shown that patients with osteoporosis have higher risks of developing AD than those who do not have osteoporosis. Although osteoporosis, falls, and fractures are more often seen in patients with AD than other older adults, the association between these two diseases may be due to a pathophysiological link rather than one condition causing the other. Several in vitro and in vivo studies lend support to this notion. Patients with AD have excessive amyloid plaques in the brain, and the pathology may extend to peripheral organs and cause skeletal amyloid deposition, which would enhance receptor activator nuclear factor-kappa B ligand signaling and lead to greater osteoclast activities. Patients with osteoporosis may have Vitamin D deficiency or lower levels of Vitamin D binding protein, which protects against amyloid aggregation, thus linking Vitamin D deficiency and AD or osteoporosis and AD. Osteoporosis coexisting with AD provides a window to examine the amyloid hypothesis from peripheral tissues. Future studies are warranted to clarify the role of genetic background regarding Vitamin D levels, exposure to sunlight, estrogen replacement therapy, and physical activity in patients with both chronic diseases.
KEYWORDS: Alzheimer's disease, Amyloid, Osteoporosis, Vitamin D
INTRODUCTION
The number of dementia cases is growing as the global population ages. Dementia brings great burden to society. A recent study estimated that the total cost of dementia worldwide was $604 billion in 2010 [1]. Alzheimer's disease (AD) is the leading cause of neurodegenerative dementia, and the prevalence has continued to grow over the past few decades [2,3,4]. AD is characterized by progressive cognitive decline which interferes with daily activities. The pathologic hallmarks of AD are cerebral extracellular amyloid plaques and intracellular neurofibrillary tangles, but not all people with amyloid plaques or neurofibrillary tangles develop AD. The underlying pathophysiology of AD remains elusive, particularly for older adults, in whom chronic diseases are prevalent and likely contribute to cognitive aging through different routes which may or may not include the amyloid cascade.
Osteoporosis is defined as a skeletal disorder with compromised bone strength that increases the risk of fracture. Osteoporosis is a degenerative disorder often seen in the elderly population and osteoporotic fracture leads to high rates of morbidity and mortality in old age [5,6]. Although osteoporosis and AD appear to be two distinct diseases, many risk factors are shared such as alcohol consumption and smoking [7,8,9]. It is not known whether osteoporosis per se is a risk factor for AD, a complication caused by AD, shares common etiologies with AD, or simply coexists with AD as a result of statistical coincidence. We herein review the link between osteoporosis and AD from the epidemiological perspective and propose the underlying molecular pathomechanism.
COEXISTENCE OF ALZHEIMER'S DISEASE AND OSTEOPOROSIS
The risk of Alzheimer's disease in osteoporosis
The risk of AD in osteoporosis and the risk of osteoporosis in AD are two different aspects to view the relationship between these two conditions. In the Framingham study, Tan et al. found that lower bone mineral density (BMD) was associated with the risk of developing AD [10]. Dual-energy X-ray absorptiometry (DXA) of the femoral neck, femoral trochanter, and distal radius was measured for BMD analysis in the study and the results showed that the lowest quartile of the femoral neck BMD was associated with an increased risk of AD in women (relative risk [RR] = 2.04, 1.11–3.75) but not in men (RR = 1.33, 0.46–3.86), suggesting the protective role of estrogen. Compared with the general population, postmenopausal women were found to have higher risks of developing AD and other types of dementia [11]. However, the lower BMD as a risk factor for AD was replicated in a Chinese cohort for both men and women and then in the National Health Insurance Research database of Taiwan, showing that ethnic differences may modify the gender effect in the association between osteoporosis and AD [12,13]. Estrogen has the potential to influence both brain aging and bone metabolism. The prevalence of AD is slightly higher in women than men and postmenopausal women are predisposed to develop osteoporosis. However, more clinical studies are warranted to determine the role of estrogen in older adults with regard to the risk of osteoporosis and AD [14,15,16].
The risk of osteoporosis in Alzheimer's disease
AD is also considered a risk factor for osteoporosis. Previous studies have shown that patients with AD have a reduced level of hip BMD and a nearly 2-fold risk of hip fracture [17,18]. The circadian rhythm is often poorly regulated in patients with AD, and thus, they may wander at night but be sleepy during the day. These symptoms are more pronounced as cognitive function deteriorates [19]. The condition increases caregiver burden and also limits outdoor physical activity, which may, in turn, reduce sunlight exposure and subsequently cause Vitamin D deficiency. The lack of physical activity and deficiency in Vitamin D may explain the higher risk of osteoporosis in AD. Bone density and bone quality are two major components of bone strength [20]. The association between osteoporosis and AD is not simply about BMD reduction measured by DXA but also about the decline of bone quality, which often results in fragility fractures. Fragility fractures are fractures due to a fall from a standing height or less. This type of minor trauma normally does not cause fractures, indicating that skeletal abnormality in AD may be more than loss of bone density.
The association between osteoporosis and AD is further supported by the correlation between BMD values measured by DXA and cerebral volume measured by magnetic resonance imaging. These two measurements are fine biomarkers to monitor neuronal loss in the brain and mineral loss in bone [21,22]. In addition, BMD correlates well with cognitive function, suggesting a parallel course for AD and osteoporosis. Although all physiological functions decline with age, comparable changes in cerebral and peripheral pathologies have more implications than biological aging. A more than statistical coincidence of AD and osteoporosis has been shown in observational studies and likely results from common etiologies rather than one condition causing the other. The underlying mechanism is of great interest as it may direct the future study design for AD and osteoporosis prevention.
PATHOPHYSIOLOGICAL LINK BETWEEN ALZHEIMER'S DISEASE AND OSTEOPOROSIS
Skeletal amyloidosis
Accumulation of abnormally folded amyloid beta peptide (Aβ) in cerebral amyloid plaques is the pathologic hallmark of AD. Aβ originates from the amyloid precursor protein (APP), a membrane protein expressed in many tissues and synapses of neurons with unknown function. A group of specific enzymes named secretases cleave APP into distinct fragments. APP cleavage by β-secretase and then γ-secretase leads to pathological Aβ. Aβ40 and Aβ42 are two subtypes of Aβ which aggregate and form oligomers. Oligomers are the units which form protofibrils and later fibrils and plaques. Genetic models of AD are typically established by excessively expressing Aβ protein and the current hypothesis of AD etiology centers around amyloid plaques [23,24].
Unlike the complexity and controversy in AD, the pathogenesis of osteoporosis is known as an imbalance between bone formation and mineralization. Hyperparathyroidism, Vitamin D deficiency, and steroid use are common causes of osteoporosis. Osteoporosis is mostly asymptomatic until minor trauma or falls lead to fractures. Bone formation involves bone matrix production and mineralization, whereas bone resorption is a biological erosive process mediated by osteoclasts. When the balance leans toward bone resorption, BMD decreases and osteoporosis develops [20]. Bone resorption is driven by the receptor activator nuclear factor-kappa B ligand (RANKL)-receptor activator nuclear factor-kappa B (RANK) signaling network, a signaling complex with multiple downstream pathways [25]. The binding of RANKL to RANK triggers the cascade beginning with tumor necrosis factor receptor-associated factor-6 (TRAF-6) [Figure 1]. TRAF-6 phosphorylates the inhibitor of nuclear factor-kappa B (IκB) kinase and causes the degradation of the IκB, which releases nuclear factor-kappa B (NF-κB) to translocate to the nucleus and initiate osteoclast-specific gene transcription. Extracellular signal-regulated kinase (ERK) is also activated along the RANK signaling pathway by the upstream mitogen-activated protein kinase-1. The phosphorylation and activation of ERK are related to osteoclast survival, motility, and cytoskeletal rearrangement. Another important RANKL signaling pathway is the costimulation of osteoclast-associated receptor. This signaling stimulates intracellular calcium ion release and eventually leads to nuclear translocation of the master signaling molecule nuclear factor of activated T-cells-cytoplasmic 1 (NFATc1) to express osteoclast-specific genes [25,26,27,28,29,30].
Figure 1.
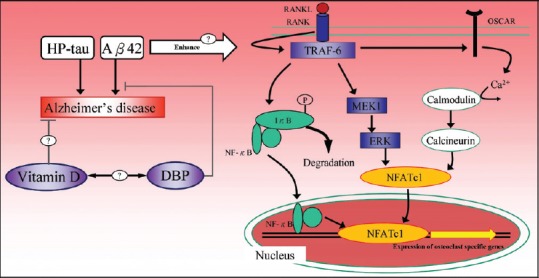
The proposed mechanism of amyloid deposition and Vitamin D in the pathogenesis of Alzheimer's disease and osteoporosis. NF-κB: Nuclear factor kappa B, Aβ: Amyloid beta peptide, RANK: Receptor activator NF-κB, RANKL: Receptor activator NF-κB ligand; IκB: Inhibitor of NF-κB, TRAF-6: Tumor necrosis factor receptor-associated factor-6, MEK1: Mitogen-activated protein kinase kinase-1, ERK: Extracellular signal-regulated kinase, OSCAR: Osteoclast-associated receptor, NFATc1: Nuclear factor of activated T-cells-cytoplasmic 1, HP-tau: Hyperphosphorylated tau, DBP: Vitamin D binding protein
Amyloid deposition in the brain and RANKL signaling are two seemingly independent pathways leading to AD and osteoporosis. The possible linkage between these two pathways has been investigated by measuring osteoclast activities in a transgenic mice model of AD. Both in vitro and in vivo examinations showed enhanced Aβ expression in bone, together with increased adipose tissue formation in the marrow space, analogous to osteoporotic bones [31,32]. The abnormally expressed amyloid deposition appears to interfere with the RANKL signaling cascade and in turn the balance between bone formation and bone resorption. Similar findings extend to human studies. Li et al. examined vertebral trabecular bone specimens and revealed a remarkable increase in messenger RNA and protein expression of Aβ42 and APP [33]. Furthermore, the immunohistochemistry of osseous specimens showed an inverse correlation between BMD and amyloid expression (r = −0.617 and −0.531 for Aβ42 and APP, respectively). The enhanced expression of Aβ42 and APP plays as a shared etiology for both AD and osteoporosis.
The effect of Aβ42 on the RANKL signaling network has been studied in vitro. Aβ42 enhances RANKL-induced osteoclastic bone resorption by upregulating IκB degradation and enhancing NF-κB activity, activating ERK phosphorylation, and stimulating the calcium oscillation signaling pathway. The Aβ42-enhanced activation of these three cytoplasmic signaling pathways eventually leads to increased NFATc1 expression in a dose-dependent manner [34]. Albeit Aβ42 alone exerted a less pronounced effect than cotreatment with RANKL in their experiment, the ability of stimulation and activation of signaling pathways by Aβ42 can be independent of RANKL as well. The role of amyloid deposition is important not only for AD in the brain but also for osteoporosis in the bone. It may be of great value if we could examine bone from AD patients with comorbid osteoporosis at the population level to test the amyloid hypothesis as the common etiology. The expression of Aβ42 and APP could even serve as a biomarker for AD if the above hypothesis is proven correct. We summarized the mechanism of skeletal amyloid-induced osteoporosis and its association with AD in Figure 1.
Vitamin D deficiency
Vitamin D deficiency is a risk factor for osteoporosis [20]. Given the association between AD and osteoporosis, it is not known if Vitamin D deficiency is on the common pathway for both diseases. Although less well recognized, Vitamin D deficiency is attracting more attention as a risk factor for AD. Observational studies have found that lower levels of plasma 25-hydroxy Vitamin D (25(OH)D) are significantly associated with increased risks of developing all-cause dementia and AD [35,36,37,38]. However, there were several limitations in these observational studies. For example, the two large prospective studies from the US to Denmark used blood samples collected from participants decades earlier. How the plasma level of 25(OH)D might have changed over this long period is uncertain [35,36]. It is also very difficult to control intrapersonal variations in plasma Vitamin D concentrations during the long follow-up period. Although there were adjustments in these studies for the timing and season of Vitamin D sampling and even physical activity, actual sunlight exposure could not be measured accurately. Lifestyle changes over time, Vitamin D measurement errors, comorbid heart disease and other chronic conditions, and medication use would all contribute to confound the causal inference.
Ideally, the association between Vitamin D and AD should be addressed in a randomized controlled trial, but it is essentially unrealistic. An alternative study design, Mendelian randomization, was employed to assess the contribution of 25(OH)D in the occurrence of AD [39]. The rationale of this study design was based on the fact that single nucleotide polymorphisms (SNPs) are inherited and randomly assorted at conception. This obviates the confounders from the modifiable exposures that observational studies would usually encounter [40]. In one study, the association between AD and four previously identified SNPs related to Vitamin D deficiency was investigated [41]. The authors found an increased risk of developing AD when these four SNPs were all shown (odds ratio = 1.25, 1.03–1.51) and it seemed that one single SNP, rs2282679 in GC (encodes Vitamin D binding protein [DBP]), was the key player as the whole effect was no longer significant if this particular SNP was not factored into the model. This finding suggested that DBP might be involved in the pathomechanism of AD [39].
In fact, the interaction between DBP and Aβ has been shown in both in vitro and in vivo studies [42]. DBP appeared to directly interact with Aβ and inhibit its aggregation and oligomerization. DBP also attenuated Aβ-related neuronal injury and protected against Aβ-related memory impairment in mice. DBP contains binding sites for several proteins including Vitamin D, C5a, actin, fatty acids, and endotoxins, and thus, it is a multifunctional protein. One of the major roles of DBP is to scavenge extracellular toxic actin, which is released into the extracellular space after cell death. DBP functions to sequester monomeric actin and prevent it from polymerization. Therefore, it is assumed that the role of DBP in AD may have to do with Aβ metabolism. The association between Vitamin D deficiency and AD might be through the function of DBP rather than Vitamin D itself. Multiple sclerosis, a demyelinating neurological disorder, is known to be associated with lower levels of Vitamin D. The incidence of multiple sclerosis increases along with geographic latitude or levels of sunlight exposure [43]. The geographic distribution is not, however, consistent in AD [44]. Further studies are warranted to examine whether Vitamin D itself is causally related to AD or merely secondary to the effect of DBP.
FUTURE DIRECTIONS
All the above evidences seem compelling in explaining the association between AD and osteoporosis. Comorbid osteoporosis or Vitamin D deficiency is neither the consequence nor the cause of AD. They more likely share a common cause with AD at the genetic or molecular level concerned with amyloid deposition in the bone or DBP function in the brain. There are still puzzles to be solved. For example, the specific step in which Aβ42 interferes with the RANKL signaling pathway and the interaction of Aβ42 with other molecules is basically unknown; the biological function of 25(OH)D is independent of DBP and whether supplemental Vitamin D would affect the cognitive outcome in AD patients or older adults in general is not well understood [45].
The current therapeutic direction for AD focuses on amyloid clearance [23]. Preliminary data from anti-amyloid antibody clinical trials has shown great promise. Amyloid positron emission tomography (PET) showed that cerebral amyloid plaques were cleared after a series of injections of aducanumab for 12 months, along with stabilization of cognitive deterioration [46]. Disease-specific immunotherapy has become the mainstream in drug development for AD. Since antibody therapy enters the central nervous system and targets amyloid plaques, it has the potential to clear amyloid deposition in peripheral organs too, such as skeletal tissues. Amyloid PET has been proven to accurately label cerebral amyloid plaques and even detect cardiac amyloidosis [47,48]. The application of amyloid PET will be extended to further clarify the role of amyloid deposition in different organs and serve as an outcome marker for clinical trials.
CONCLUSION
Previous observational studies have reported the increased frequency of comorbid osteoporosis in AD. The relationship between these two diseases is more likely one of shared etiology than one condition causing the other. There are two major pathophysiological links for osteoporosis and AD. The overexpression of Aβ may take place in both brain and bone, interfering with the RANKL signaling cascade, enhancing osteoclast activities, and leading to osteoporosis. DBP plays a pivotal role in the alternative explanation of the cause of reduced levels of Vitamin D and increased Aβ aggregation. There is a growing body of evidence from in vitro and in vivo studies that the AD pathology in the brain can be reflected by examining the bone. Future investigation will focus on assessing biomarkers of cognitive aging in patients with osteoporosis and looking into the bone microstructure of patients with AD.
Financial support and sponsorship
The study was supported by the Ministry of Science and Technology (MOST103-2314-B-303-008).
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Wimo A, Jönsson L, Bond J, Prince M, Winblad B, et al. Alzheimer Disease International. The worldwide economic impact of dementia 2010. Alzheimers Dement. 2013;9:1–11.e3. doi: 10.1016/j.jalz.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Brookmeyer R, Evans DA, Hebert L, Langa KM, Heeringa SG, Plassman BL, et al. National estimates of the prevalence of Alzheimer's disease in the United States. Alzheimers Dement. 2011;7:61–73. doi: 10.1016/j.jalz.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rocca WA, Petersen RC, Knopman DS, Hebert LE, Evans DA, Hall KS, et al. Trends in the incidence and prevalence of Alzheimer's disease, dementia, and cognitive impairment in the United States. Alzheimers Dement. 2011;7:80–93. doi: 10.1016/j.jalz.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP, et al. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement. 2013;9:63–75.e2. doi: 10.1016/j.jalz.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Bliuc D, Nguyen ND, Milch VE, Nguyen TV, Eisman JA, Center JR, et al. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA. 2009;301:513–21. doi: 10.1001/jama.2009.50. [DOI] [PubMed] [Google Scholar]
- 6.Bliuc D, Nguyen ND, Nguyen TV, Eisman JA, Center JR. Compound risk of high mortality following osteoporotic fracture and refracture in elderly women and men. J Bone Miner Res. 2013;28:2317–24. doi: 10.1002/jbmr.1968. [DOI] [PubMed] [Google Scholar]
- 7.Peters R, Peters J, Warner J, Beckett N, Bulpitt C. Alcohol, dementia and cognitive decline in the elderly: A systematic review. Age Ageing. 2008;37:505–12. doi: 10.1093/ageing/afn095. [DOI] [PubMed] [Google Scholar]
- 8.Tysiewicz-Dudek M, Pietraszkiewicz F, Drozdzowska B. Alzheimer's disease and osteoporosis: Common risk factors or one condition predisposing to the other? Ortop Traumatol Rehabil. 2008;10:315–23. [PubMed] [Google Scholar]
- 9.Cataldo JK, Prochaska JJ, Glantz SA. Cigarette smoking is a risk factor for Alzheimer's disease: An analysis controlling for tobacco industry affiliation. J Alzheimers Dis. 2010;19:465–80. doi: 10.3233/JAD-2010-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan ZS, Seshadri S, Beiser A, Zhang Y, Felson D, Hannan MT, et al. Bone mineral density and the risk of Alzheimer disease. Arch Neurol. 2005;62:107–11. doi: 10.1001/archneur.62.1.107. [DOI] [PubMed] [Google Scholar]
- 11.Amouzougan A, Lafaie L, Marotte H, Dẻnariẻ D, Collet P, Pallot-Prades B, et al. High prevalence of dementia in women with osteoporosis. Joint Bone Spine. 2016 doi: 10.1016/j.jbspin.2016.08.002. pii: S1297-319X(16)30139-7. doi: 10.1016/j.jbspin.2016.08.002. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 12.Zhou R, Deng J, Zhang M, Zhou HD, Wang YJ. Association between bone mineral density and the risk of Alzheimer's disease. J Alzheimers Dis. 2011;24:101–8. doi: 10.3233/JAD-2010-101467. [DOI] [PubMed] [Google Scholar]
- 13.Chang KH, Chung CJ, Lin CL, Sung FC, Wu TN, Kao CH. Increased risk of dementia in patients with osteoporosis: A population-based retrospective cohort analysis. Age (Dordr) 2014;36:967–75. doi: 10.1007/s11357-013-9608-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brenner DE, Kukull WA, Stergachis A, van Belle G, Bowen JD, McCormick WC, et al. Postmenopausal estrogen replacement therapy and the risk of Alzheimer's disease: A population-based case-control study. Am J Epidemiol. 1994;140:262–7. doi: 10.1093/oxfordjournals.aje.a117245. [DOI] [PubMed] [Google Scholar]
- 15.Depypere H, Vierin A, Weyers S, Sieben A. Alzheimer's disease, apolipoprotein E and hormone replacement therapy. Maturitas. 2016;94:98–105. doi: 10.1016/j.maturitas.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Henderson VW. Estrogen-containing hormone therapy and Alzheimer's disease risk: Understanding discrepant inferences from observational and experimental research. Neuroscience. 2006;138:1031–9. doi: 10.1016/j.neuroscience.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 17.Wang HK, Hung CM, Lin SH, Tai YC, Lu K, Liliang PC, et al. Increased risk of hip fractures in patients with dementia: A nationwide population-based study. BMC Neurol. 2014;14:175. doi: 10.1186/s12883-014-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y, Shen L, Ji HF. Alzheimer's disease and risk of hip fracture: A meta-analysis study. Scientific World Journal. 2012;2012:872173. doi: 10.1100/2012/872173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JH, Bliwise DL, Ansari FP, Goldstein FC, Cellar JS, Lah JJ, et al. Daytime sleepiness and functional impairment in Alzheimer disease. Am J Geriatr Psychiatry. 2007;15:620–6. doi: 10.1097/JGP.0b013e3180381521. [DOI] [PubMed] [Google Scholar]
- 20.Nanes MS, Kallen CB. Osteoporosis. Semin Nucl Med. 2014;44:439–50. doi: 10.1053/j.semnuclmed.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 21.Loskutova N, Honea RA, Vidoni ED, Brooks WM, Burns JM. Bone density and brain atrophy in early Alzheimer's disease. J Alzheimers Dis. 2009;18:777–85. doi: 10.3233/JAD-2009-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loskutova N, Honea RA, Brooks WM, Burns JM. Reduced limbic and hypothalamic volumes correlate with bone density in early Alzheimer's disease. J Alzheimers Dis. 2010;20:313–22. doi: 10.3233/JAD-2010-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, et al. Alzheimer's disease. Lancet. 2016;388:505–17. doi: 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- 24.Vallabhajosula S. Positron emission tomography radiopharmaceuticals for imaging brain Beta-amyloid. Semin Nucl Med. 2011;41:283–99. doi: 10.1053/j.semnuclmed.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–42. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 26.Nakashima T, Takayanagi H. New regulation mechanisms of osteoclast differentiation. Ann N Y Acad Sci. 2011;1240:E13–8. doi: 10.1111/j.1749-6632.2011.06373.x. [DOI] [PubMed] [Google Scholar]
- 27.Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251–64. doi: 10.1016/j.bone.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 28.Kular J, Tickner J, Chim SM, Xu J. An overview of the regulation of bone remodelling at the cellular level. Clin Biochem. 2012;45:863–73. doi: 10.1016/j.clinbiochem.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Q, Shao J, Chen W, Li YP. Osteoclast differentiation and gene regulation. Front Biosci. 2007;12:2519–29. doi: 10.2741/2252. [DOI] [PubMed] [Google Scholar]
- 30.Baron R. Arming the osteoclast. Nat Med. 2004;10:458–60. doi: 10.1038/nm0504-458. [DOI] [PubMed] [Google Scholar]
- 31.Cui S, Xiong F, Hong Y, Jung JU, Li XS, Liu JZ, et al. APPswe/Aß regulation of osteoclast activation and RAGE expression in an age-dependent manner. J Bone Miner Res. 2011;26:1084–98. doi: 10.1002/jbmr.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia WF, Jung JU, Shun C, Xiong S, Xiong L, Shi XM, et al. Swedish mutant APP suppresses osteoblast differentiation and causes osteoporotic deficit, which are ameliorated by N-acetyl-L-cysteine. J Bone Miner Res. 2013;28:2122–35. doi: 10.1002/jbmr.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, Liu B, Zhang L, Rong L. Amyloid beta peptide is elevated in osteoporotic bone tissues and enhances osteoclast function. Bone. 2014;61:164–75. doi: 10.1016/j.bone.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 34.Li S, Yang B, Teguh D, Zhou L, Xu J, Rong L, et al. Amyloid β peptide enhances RANKL-induced osteoclast activation through NF-κB, ERK, and calcium oscillation signaling. Int J Mol Sci. 2016;17:pii: E1683. doi: 10.3390/ijms17101683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Littlejohns TJ, Henley WE, Lang IA, Annweiler C, Beauchet O, Chaves PH, et al. Vitamin D and the risk of dementia and Alzheimer disease. Neurology. 2014;83:920–8. doi: 10.1212/WNL.0000000000000755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Afzal S, Bojesen SE, Nordestgaard BG. Reduced 25-hydroxyvitamin D and risk of Alzheimer's disease and vascular dementia. Alzheimers Dement. 2014;10:296–302. doi: 10.1016/j.jalz.2013.05.1765. [DOI] [PubMed] [Google Scholar]
- 37.Shen L, Ji HF. Vitamin D deficiency is associated with increased risk of Alzheimer's disease and dementia: Evidence from meta-analysis. Nutr J. 2015;14:76. doi: 10.1186/s12937-015-0063-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Annweiler C, Llewellyn DJ, Beauchet O. Low serum Vitamin D concentrations in Alzheimer's disease: A systematic review and meta-analysis. J Alzheimers Dis. 2013;33:659–74. doi: 10.3233/JAD-2012-121432. [DOI] [PubMed] [Google Scholar]
- 39.Mokry LE, Ross S, Morris JA, Manousaki D, Forgetta V, Richards JB, et al. Genetically decreased Vitamin D and risk of Alzheimer disease. Neurology. 2016;87:2567–574. doi: 10.1212/WNL.0000000000003430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith GD, Ebrahim S. 'Mendelian randomization': Can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 41.Wang TJ, Zhang F, Richards JB, Kestenbaum B, van Meurs JB, Berry D, et al. Common genetic determinants of Vitamin D insufficiency: A genome-wide association study. Lancet. 2010;376:180–8. doi: 10.1016/S0140-6736(10)60588-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moon M, Song H, Hong HJ, Nam DW, Cha MY, Oh MS, et al. Vitamin D-binding protein interacts with aβ and suppresses aβ-mediated pathology. Cell Death Differ. 2013;20:630–8. doi: 10.1038/cdd.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pozuelo-Moyano B, Benito-León J. Vitamin D and multiple sclerosis. Rev Neurol. 2013;56:243–51. [PubMed] [Google Scholar]
- 44.Russ TC, Murianni L, Icaza G, Slachevsky A, Starr JM. Geographical variation in dementia mortality in italy, new zealand, and chile: The impact of latitude, Vitamin D, and air pollution. Dement Geriatr Cogn Disord. 2016;42:31–41. doi: 10.1159/000447449. [DOI] [PubMed] [Google Scholar]
- 45.Dastani Z, Berger C, Langsetmo L, Fu L, Wong BY, Malik S, et al. In healthy adults, biological activity of Vitamin D, as assessed by serum PTH, is largely independent of DBP concentrations. J Bone Miner Res. 2014;29:494–9. doi: 10.1002/jbmr.2042. [DOI] [PubMed] [Google Scholar]
- 46.Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature. 2016;537:50–6. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 47.Mallik A, Drzezga A, Minoshima S. Clinical amyloid imaging. Semin Nucl Med. 2017;47:31–43. doi: 10.1053/j.semnuclmed.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 48.Law WP, Wang WY, Moore PT, Mollee PN, Ng AC. Cardiac amyloid imaging with 18F-florbetaben PET: A pilot study. J Nucl Med. 2016;57:1733–9. doi: 10.2967/jnumed.115.169870. [DOI] [PubMed] [Google Scholar]