

Abstract
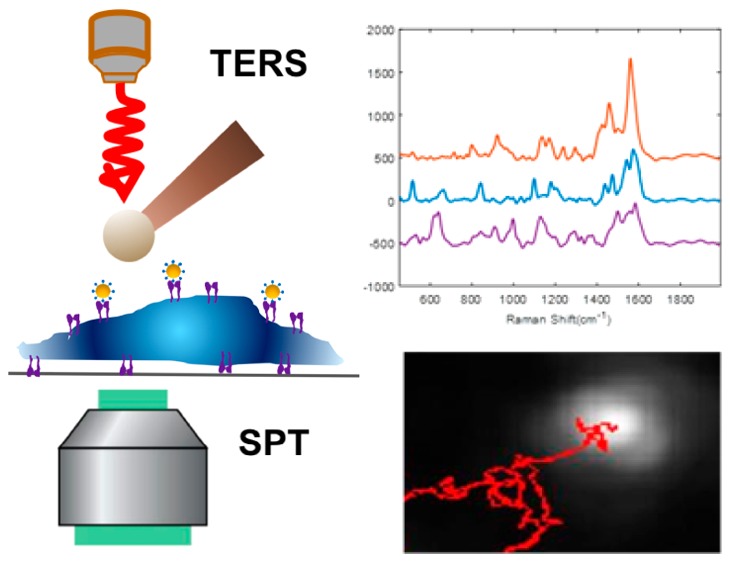
The specific interaction between a ligand and a protein is a key component in minimizing off-target effects in drug discovery. Investigating these interactions with membrane protein receptors can be quite challenging. In this report, we show how spectral variance observed in surface-enhanced Raman scattering (SERS) and tip-enhanced Raman scattering (TERS) can be correlated with ligand specificity in affinity-based assays. Variations in the enhanced Raman spectra of three peptide ligands (i.e., cyclic-RGDFC, cyclic-isoDGRFC, and CisoDGRC), which have different binding affinity to αvβ3 integrin, are reported from isolated proteins and from receptors in intact cancer cell membranes. The SERS signal from the purified proteins provides basis spectra to analyze the signals in cells. Differences in the spectral variance within the SERS and TERS data for each ligand indicate larger variance for nonspecific ligand–receptor interactions. The SERS and TERS results are correlated with single particle tracking experiments of the ligand-functionalized nanoparticles with purified receptors on glass surfaces and living cells. These results demonstrate the ability to elucidate protein–ligand recognition using the observed vibrational spectra and provide perspective on binding specificity for small-molecule ligands in intact cell membranes, demonstrating a new approach for investigating drug specificity.
The recognition of a ligand by a protein receptor is a key interaction that triggers biological processes ranging from intercellular communication to intracellular signaling. Understanding ligand–receptor binding is crucial for both the regulation of these biological processes and their manipulation in drug development research. Membrane receptors are common targets for therapeutic drugs.1 However, monitoring how a drug interacts with a protein receptor on a molecular level is quite challenging. Ligand–receptor binding assays are often based on affinity or use purified receptors without the environmental constraints of the cellular membrane.
Among various ligand–receptor binding assays, surface plasmon resonance (SPR) is the most common label-free ligand-binding assay for protein receptors. In SPR the isolated and purified receptor is immobilized on the SPR sensor chip, and the interaction with ligands is monitored to provide both binding affinity and kinetics.2 Recently, microscopic techniques including fluorescence-based super resolution microscopy3,4 and atomic force microscopy5,6 have been developed to visualize the interactions between ligands and receptors in the cell membrane with spatial resolution below the diffraction limit. Though all of the above-mentioned techniques are able to reveal the biophysical properties like binding affinity and kinetics, they lack the capability to provide molecular information about the ligand–receptor binding complexes. Structure-based ligand binding assays, such as nuclear magnetic resonance7,8 and X-ray crystallography,9,10 are able to analyze the structure of receptors and the molecular details of ligand–receptor interactions. However, these methods assay isolated receptors, which can be difficult and time-consuming. A technique that can investigate the molecular nature of ligand–receptor binding in intact cell membranes may significantly facilitate the process of drug development.
Raman spectroscopy is an intriguing method to investigate the structures of biomolecules in cell membranes by directly measuring their vibrational modes.11,12 The chemical specific information encoded in the Raman spectra reveals the identities of the molecules. Furthermore, signal enhancements by plasmonic nanoparticles (so-called “surface-enhanced Raman scattering”, SERS) significantly improve the sensitivity of the technique and enable detection at the single-molecule level.13,14 By attaching a plasmonic nanostructure at the apex of a scanning probe microscope (SPM) tip, tip-enhanced Raman scattering (TERS) combines the chemical sensitivity of SERS and nanoscale spatial resolution of SPM, making it an attractive approach to study the molecular composition of biomembranes.15,16 Our lab has previously demonstrated that Raman signals from immobilized receptors binding with specific ligands attached to a gold nanoparticle (GNP) can be detected through plasmonic coupling between ligand-functionalized GNPs and a TERS tip.17−19 Through protein mutation experiments, we have shown that amino acids near the ligand-binding site are responsible for the observed TERS signal.19 Recently, we reported that this targeted-TERS methodology can selectively detect ligand–receptor binding in intact cell membranes20,21 and differentiate similar receptors binding with the same ligand, due to differences in their ligand binding sites.22 These results indicate that TERS can provide chemical insights into the structure of specific membrane receptors and demonstrate the potential of TERS to investigate ligand–receptor binding chemistry within cell membranes. In addition, single particle tracking (SPT) has been used to study the dynamics of nanoparticles with respect to the internalization and trafficking of cell surface receptors.23,24 The ligand-functionalized nanoparticles in the TERS experiments can also be monitored by SPT, providing valuable information about dynamic processes and interactions, such as intracellular transport, nanoparticle entry, and binding to the cell membrane.25,26
To investigate this approach, we chose integrin receptors. Integrin receptors are important membrane receptors that regulate cellular migration, invasion, and proliferation in tumors and are therefore an appealing target for cancer therapy.27 The known affinity of certain integrins toward peptidomimetic ligands containing Arg-Gly-Asp (RGD) and isoAsp-Gly-Arg (isoDGR) sequences28,29 has resulted in efforts to develop efficient and specific integrin antagonists drugs based on these peptidomimetics.28 However, challenges remain as evidenced by the recent failure of a cyclic-RGD based integrin inhibitor in a phase 3 trial for the treatment of glioblastoma.30 The ability to specify the chemical interactions between potential drug candidates and the targeted membrane receptors in cell membranes could facilitate the drug validation process and help avoid costly late stage trial failure. We have previously demonstrated the ability of TERS to chemically characterize cell membrane receptor binding with a ligand-conjugated nanoparticle.21,22 Here we combine TERS measurements with SPT experiments to investigate the binding interactions between αvβ3 integrin and three cyclic-peptides in a human metastatic colon cancer (SW620) cell line. By correlating TERS and SPT experiments, we are able to assign molecular information from the ligand–receptor interaction with the ligand binding affinity.
Experimental Section
Materials
Gold nanoparticles (80 nm citrate NanoXact_ gold) were purchased from nanoComposix (San Diego, CA). Cyclic-RGDFC, cycli-isoDGRFC, and CisoDGRC peptides were synthesized by Peptide 2.0 Inc. (purity 90%, Chantilly, VA). Purified human integrin αvβ3 protein was purchased from EMD Millipore Corporation (>95%, Temecula, CA). Poly-d-lysine-coated coverslips were purchased from BD BioCoat Cellware. Cell culture reagents were purchased from Thermo Fisher Scientific (Waltham, MA). Ultrapure water (18.2 MΩ cm) from a Barnstead Nanopure filtration system was used for all experiments.
Nanoparticle Functionalization and Characterization
Cysteine-containing peptides were conjugated to the gold nanoparticles through a ligand exchange reaction (Scheme 1). All three peptides were prepared at the same concentration. Briefly, 10 μL of 0.05 mM peptide was mixed with 1 mL of citrate-GNP (0.05 mg mL–1 or 16.6 pM) colloid solution. The molar ratio of peptide to GNP was calculated to be 5.2 × 104:1. After 24 h incubation, the colloidal solution was centrifuged (10 000 rcf, 12 min) and resuspended in pure water to remove the excessive and unbound peptides. The peptide-conjugated gold nanoparticles (peptide-GNPs) were reconstituted in 1 mL of water and stored at 4 °C for later use.
Scheme 1. Schematic Illustration of Gold Nanoparticles Conjugated with Three Different Peptide Ligands (cyclic-RGDFC, cylic-isoDGRFC, and CisoDGRC).

Nanoparticle characterization was carried out by UV–vis absorption, dynamic light scattering, and zeta-potential measurements. UV–vis measurements were performed using a UV-3100PC spectrophotometer (VWR International, Radnor, PA), coupled with a Deuterium-Tungsten halogen lamp. Dynamic light scattering (DLS) and zeta potential measurements were performed using a Zetasizer Nano-ZS system (Malvern, Worcestershire, U.K.).
SERS Detection of Purified Integrin αvβ3 Bound with Peptide-GNPs
A total of 10 μL of peptide (cyclic-RGDFC, cyclic-isoDGRFC, and CisoDGRC) functionalized GNPs were mixed with 4 μL of integrin αvβ3 (0.25 mg mL–1), in 100 μL of 0.1× PBS solution. After 2 h of vortex mixing, the mixture was concentrated to ∼20 μL through centrifugation, then dropped onto a clean glass slide, and sealed with a coverslip. Raman spectra of the αvβ3-bound peptide-GNPs and peptide-GNPs were acquired using a home-built Raman spectrometer consisting of a 660 nm diode laser, Isoplane-320 spectrograph, and ProEM EMCCD (Princeton Instruments, Trenton, NJ). Consecutive Raman spectra of the nanoparticles were collected with 0.9 mW laser power and 1 s acquisition time.
Cell Sample Preparation
SW620 cells were seeded on poly-d-lysine-coated coverslips to enhance attachment. After 24-h attachment, cells were rinsed with 0.1× PBS and incubated with 100 μL of peptide-GNPs for 2 h. The unadsorbed nanoparticles were removed by rinsing with 0.1× PBS several times. Cells were then fixed with paraformaldehyde (4% in PBS) for 10 min, rinsed with 0.1× PBS and water, and dried before TERS experiments. The SW620 cells were cultured following a previously published procedure.31
TERS Imaging
TERS measurements were carried out with a combined AFM-Raman system that has been previously reported.21,22 The system incorporates a commercial AFM microscope (Nanonics MV4000) and a home-built Raman spectrometer containing a Horiba Jobin Yvon monochromator. A 633 nm HeNe excitation laser was used to illuminate the sample. Radial polarization of the laser was achieved using a liquid-crystal mode converter (ArcOptix), producing a longitudinal mode at the focus that results in increased enhancement and better spatial resolution from the TERS tip.32 The TERS tip is a transparent glass tip embedded with gold nanoparticles (Nanonics Imaging Ltd. Israel). The collected TERS signal was filtered by a 633 nm dichroic beamsplitter and a 633 nm long pass filter (RazorEdge, Semrock), dispersed by a 600 g mm–1 grating, and collected by a CCD camera cooled at −70 °C. TERS maps were obtained by scanning the sample stage under the TERS tip positioned in the laser focus. The acquisition time was 1s per pixel and laser power was measured to be 0.9–1.0 mW to avoid damaging the samples.
Raman data analysis
SERS and TERS spectra and maps were plotted using Matlab R2015b (Mathworks). Raw SERS spectra of the peptide-GNPs (with or without αvβ3 binding) were preprocessed through a weighted least-squares (WLS, Whittaker filter, fifth order polynomial) automatic baseline subtraction to remove differences due only to the background in each spectrum. These spectra were used to decompose the pure components of the SERS data using multivariate curve resolution (MCR), and further used to classify the TERS data in order to determine the class of each spectrum in the TERS map. TERS maps and MCR maps were reconstructed in MATLAB according to single-peak intensities and MCR scores, respectively. Individual SERS and TERS spectra of three different peptides bound with αvβ3 were analyzed by principal component analysis (PCA) and hierarchical cluster analysis (HCA). MCR, PCA, and HCA were performed using PLS toolbox (eigenvector).
Dark Field Imaging and Single Particle Tracking
Experiments were performed on an Olympus BX51 microscope. A 565 nm mounted light emitting diode (Thorlabs, M565L3) was used for bottom illumination. A condenser focused the excitation light onto the sample. All scattered light was collected by a water immersion objective (Olympus LUMPLFLN 40XW 0.8 NA) and directed onto a complementary metal-oxide semiconductor (CMOS) camera (Hamamatsu C11440).
Dark-field time-lapse videos were recorded for 1–2 min each with a cycle time of 10 ms acquisition per frame. Minimal intensity projection images and single particle trajectories from each time-lapse video were calculated in Nikon NIS Elements Advanced Research software and data analysis was performed in Matlab 2015b (Mathworks) and Origin 9.0 (OriginLab Corp.).
Results and Discussion
Investigation of Peptide-GNPs Binding with Purified Integrin αvβ3
While peptides containing RGD and isoDGR sequences are known to bind with integrins, the flanking sequences are reported to affect the binding selectivity and affinity.29 In this work, we use the three cyclic peptides with similar structures (cyclic-RGDFC, cyclic-isoDGRFC, and CisoDGRC), as shown in Scheme 1, to study their interactions with αvβ3 integrin. The ligands were conjugated onto the GNPs through a covalent Au–S (Cys) bond. Characterization data of the functionalized GNPs are presented in Supporting Information (Figure S1 and Table S1).
SERS measurements of purified integrin αvβ3 mixed with three peptide-GNPs were performed to identify the distinct spectral features associated with each ligand binding to the integrin. Consecutive SERS spectra of αvβ3-bound cyclic-RGDFC-GNPs were acquired (1s acquisition) where GNPs formed plasmon-enhanced clusters. Figure 1a shows a heat map constructed from the sequential SERS spectra. Temporal fluctuations of the SERS signals, including intensity and frequency variations of vibrational bands, were observed (Figure 1b). Similar spectral variations were also observed in time-resolved SERS spectra of cyclic-isoDGRFC-GNPs and CisoDGRC-GNPs bound with αvβ3 (Figure S2). These temporal fluctuations are often seen in SERS spectra of large single molecules such as proteins,14,33 and are recognized to arise from changes in conformation and orientation of the molecules interacting with the nanostructures. Indeed, the temporal fluctuations in SERS signals have been used to access conformational information on single protein and lipid molecules.34,35 In our experiment, the SERS signal fluctuation is hypothesized to originate from transient binding between the peptide-GNPs and the αvβ3 receptor. It is reported that ligand binding changes the conformation of the αvβ3 integrin receptor.36
Figure 1.

(a) Consecutive SERS acquisitions of αvβ3-bound cyclic-RGDFC-GNPs (n = 100). (b) Selected SERS spectra corresponding to dotted lines in panel a. SERS spectra of cyclic-RGDFC-GNPs and αvβ3-bound cyclic-RGDFC-GNPs were analyzed with multivariate curve resolution (MCR) to construct a two-component model. (c) MCR score distribution plot. (d) Two pure spectral components generated by MCR were compared with the average SERS spectra of cyclic-RGDFC-GNPs.
In order to analyze the variance and extract the pure spectral components, the SERS data were analyzed using multivariate curve resolution (MCR). The SERS data set, composed of the spectra of αvβ3-bound cyclic-RGDFC-GNPs (n = 100) and cyclic-RGDFC-GNPs (n = 300), was analyzed with a two-component MCR model. The MCR score plot exhibited two separate clusters of data points, providing a clear classification of two samples (Figure 1c). The two corresponding spectral components generated by MCR are shown in Figure 1d. Component 1 resembles the SERS spectra from cyclic-RGDFC-GNPs; while component 2 reflects the average SERS spectra of αvβ3-bound cyclic-RGDFC-GNPs. The observed Raman bands are attributable to the amino acids reported at the ligand-binding site of integrin αvβ3,36,37 including Tyr (641 cm–1, 845 cm–1), Lys (912 cm–1), Ser (1127 cm–1), Trp/Phe (999 cm–1, 1582 cm–1), and some protein backbone bands. More band assignments can be found in Supporting Information (Table S2). It is noted that the two-component MCR model captured about 70% of the variance in the SERS data. The residuals were mainly from the spectra of αvβ3-bound cyclic-RGDFC-GNPs (Figure S3). The above-mentioned changes in conformation affect the SERS signal of αvβ3-bound cyclic-RGDFC-GNPs. Additionally the signal fluctuations suggest a low number of receptors give rise to each individual spectrum. These changes in conformation and stochastic fluctuation make it difficult to perfectly describe the protein with a single component. However, the 30% variance captured by component 2 from the data is a significant portion and correlates with previous studies of ligand binding.36,37
The same MCR analysis was performed on ligand–receptor complexes of αvβ3-bound cyclic-isoDGRFC-GNPs and CisoDGRC-GNPs, respectively (Figure 2). Similar to cyclic-RGDFC, MCR analysis of cyclic-isoDGRFC binding with integrin αvβ3 generated two spectral components: component 1 is similar to the average SERS spectra of cyclic-isoDGRFC-GNPs and component 2 still shows multiple bands associated with amino acids (e.g., Ser, Tyr, Lys, and Trp) at the ligand-binding site of integrin αvβ3 (Figure 2b). The MCR score plot (Figure 2a) showed that part of the data points of the αvβ3-bound cyclic-isoDGRFC-GNPs (red dots) clustered along component 2, while the other part of data points clustered together with the data of cyclic-isoDGRFC-GNPs (gray dots) along component 1, suggesting these SERS spectra arise mainly from the cyclic-isoDGRFC ligand rather than αvβ3 receptor. This suggests the protein diffuses more readily from the aggregated nanoparticles in the laser focus or may have lower affinity.
Figure 2.

Two-component MCR models of SERS spectra of αvβ3-bound cyclic-isoDGRFC-GNPs (a, b) and CisoDGRC-GNPs (c, d). MCR Distribution plots (a, c) and pure spectral components (b, d) are displayed.
The MCR results observed for the CisoDGRC peptide were distinct from the previous peptides. A 2-component model again accounted for >70% of the variance, dividing the SERS data of αvβ3-bound CisoDGRC-GNPs into two separate clusters as as shown in Figure 2c. In contrast to the previous peptides, neither of the spectral components shows similarity to the SERS spectra of CisoDGRC-GNPs. The two derived components show vibrational modes attributable to Tyr, Lys, and Trp (Figure 2d). The increased variance of the SERS data relative to the other two peptides suggests a higher level of heterogeneity in the αvβ3-CisoDGRC-GNPs binding interaction.
A comparison of the spectra associated with the peptide-integrin binding from all three ligands further shows increased variance for the αvβ3-bound CisoDGRC-GNPs. To more clearly illustrate the observed variance, Figure 3 shows a 3-component PCA model of the spectra associated with integrin binding. SERS data of cyclic-RGDFC and cyclic-isoDGRFC show comparable clustering sizes while data points of CisoDGRC had a much wider dispersion. The difference in variance suggests heterogeneity in the receptor–ligand binding interaction. The cyclic-isoDGRFC and cyclic-RGDFC show a single cluster, indicating a more specific interaction; while the CisoDGRC appears to form 3 distinct clusters suggestive of multiple different binding interactions. These differences in binding specificity can be tested against binding affinity.
Figure 3.
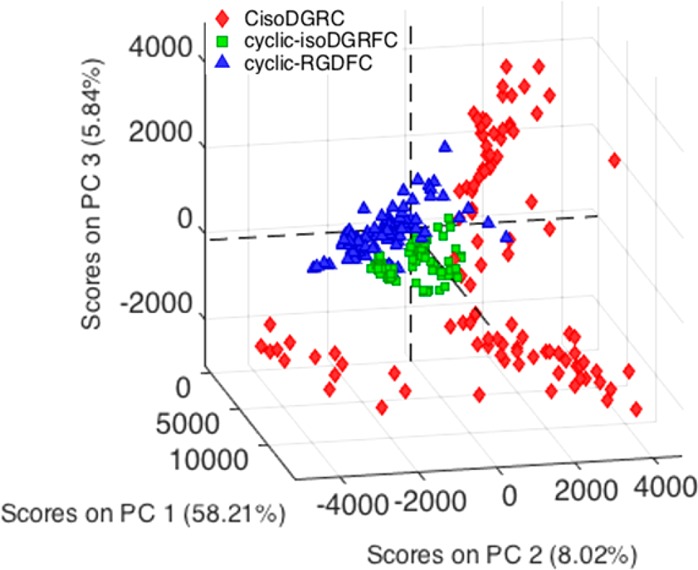
PCA score plots of SERS spectra of integrin αvβ3-bound cyclic-RGDFC-GNPs (blue), cyclic-isoDGRFC-GNPs (green), and CisoDGRC-GNPs (red).
To assess the binding dynamics, single particle tracking experiments using dark-field time-lapse microscopy were performed on the peptide-GNPs interacting with αvβ3 receptors immobilized on a glass slide. Figure 4 shows the velocity profiles for the three peptide functionalized GNPs and citrate capped GNPs. The velocity profiles of CisoDGRC-GNPs and citrate-GNPs show clear “on and off” pattern binding with the immobilized αvβ3 over the observation period, while cyclic-RGDFC-GNPs and cyclic-isoDGRFC-GNPs show reduced velocity after a single event, presumably binding to the αvβ3 integrin. It is noted that interaction between a GNP and the SiO2 surface could also result in a reduced velocity.38 The velocity profile of a cyclic-RGDFC-GNP on a bare glass slide also showed a similar “on and off” pattern, but stationary periods were transient and short-lived (Figure S4). The tracking of a stationary particle exhibited a velocity of 7 ± 1 μm s–1 (Figure S4), which is significantly different than the functionalized particle on a glass slide. This velocity for a stationary particle correlates with mean squared displacement of 0.06 ± 0.03 μm, which is the localization uncertainty in our system. The SPT results suggest a stronger binding affinity between cyclic-RGDFC-GNPs and cyclic-isoDGRFC-GNPs with the αvβ3 receptor, relative to CisoDGRC-GNPs. The apparent irreversible binding observed likely corresponds to an avidity effect associated with high ligand density on the nanoparticle in combination with ligand specificity. The ligand specificity is consistent with the SERS results.
Figure 4.
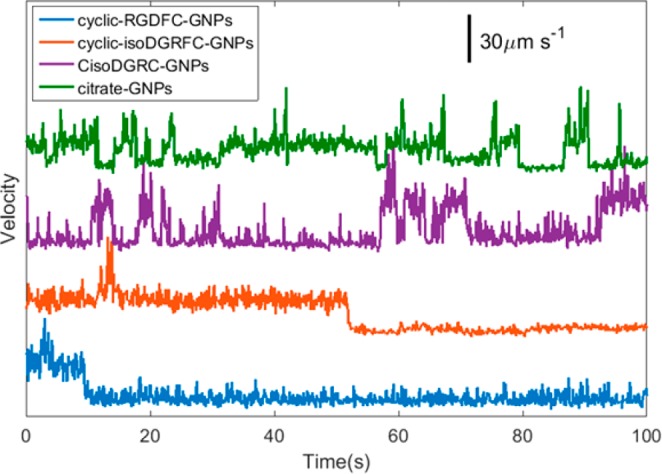
Representative velocity profiles of ligand-conjugated GNPs interacting with integrin αvβ3 immobilized on glass slides.
Investigation of Peptide-GNPs Binding with Integrin αvβ3 on the SW620 Cell Membrane
Figure 5 shows the TERS imaging results of SW620 cells incubated with cyclic-RGDFC-GNPs. As we demonstrated previously, the interaction between a TERS tip with GNPs bound to the cell membrane produces a significantly enhanced Raman signal.20,21 The TERS map in Figure 5a was generated using the intensity of the peak at 1002 cm–1, which reflects the distribution of cyclic-RGDFC-GNPs in a small area (3× 3 μm2) of the cell membrane. Due to the complexity of the cell membrane, GNPs might bind to molecules other than integrin αvβ3 (nonspecific binding) or the TERS tip may enhance other species, the MCR model constructed with SERS data (Figure 1) was applied to filter the TERS spectra and generate a MCR score map with enhanced contrast (Figure 5b). The MCR score map reduced the contrast of pixels reflecting nonspecific binding (Figure S5) but enhanced the pixels of specific TERS signal. The pixels are observed as a linear streak along the fast scanning direction, which are attributed to the lower numerical aperture (NA = 0.5) objective used in the experiments, which does not generate a pure longitudinal mode39 and produces an asymmetric tip–nanoparticle coupling.20 The observed pixels with reproducible and well-resolved TERS signals (Figure 5c) are consistent with single particles bound to receptors on flat surfaces.17,18 The observed TERS spectra are similar to the MCR component corresponding to αvβ3 receptors and contain vibrational bands associated with the amino acids in the integrin RGD-ligand binding site (Figure 4d). The small shifts of these modes observed (see Table S2 for peak assignment) are attributed to low numbers of receptors being detected compared with the ensemble averages and possibly to changes in the local electric field environment arising from the TERS tip interacting with the functionalized nanoparticle.20
Figure 5.

TERS imaging of cyclic-RGDFC-GNRs bound with sw620 cells. (a) TERS heat map (3 × 3 μm2) generated using single-peak intensity at 1002 cm–1 (step size: 93 nm). (b) MCR map generated using scores of each TERS spectra toward the MCR component corresponding to integrin αvβ3 determined from the SERS experiments. (c) TERS spectra selected from high intensity pixels in panel b. (d) Comparison between TERS spectrum and MCR component. MCR is able to filter TERS data to generate much cleaner maps.
Figure 6 shows the MCR analysis of the TERS images obtained from cyclic-isoDGRFC-GNPs and CisoDGRC-GNPs incubated with SW620. Similar to Figure 5, the MCR models were generated from the SERS spectra from each ligand interacting with the αvβ3 integrin. The obtained TERS spectra show similarity; however, fewer vibrational modes are observed in the TERS relative to the SERS spectra. This can be explained by the orientational constraint of the cell membrane on the integrin receptor. Additional TERS mapping data of SW620 cells incubated with the three peptide-GNPs can be found in the Supporting Information (Figure S6). The differences in the observed vibrational modes of the measured TERS spectra for different peptide ligands support the hypothesis that these peptide-GNPs have different binding conformations and orientations with the αvβ3 integrin.
Figure 6.

TERS detection and MCR analysis of cyclic-isoDGRFC-GNPs (upper row) and CisoDGRC-GNPs (lower row) bound with sw620 cells. Left: MCR score maps. Right: TERS spectra vs MCR components. The MCR models were generated from the SERS spectra with the purified receptor.
Figure 7 shows analysis of the TERS spectra (from triplicate experiments) from each of the peptide-GNPs bound to SW620 cells. Again the spectra were analyzed by PCA and show the same trends as were observed in the SERS data for the purified receptor. The PCA plot of TERS spectra (Figure 7a) shows that CisoDGRC-GNPs show greater heterogeneity than cyclic-RGDFC-GNPs and cyclic-isoDGRFC-GNPs, represented by the larger variance in the spectra. To further quantify these differences, the TERS spectra were further analyzed by hierarchal cluster analysis (HCA, Figure 7b). The CisoDGRC TERS spectra show significantly larger variance weighted distance between clusters than either of the other two peptides. Spectral differences could arise from differences in the plasmonic environment, such as differences in TERS tip dimension.40 To control for these plasmonic effects, the same TERS tip was used throughout the TERS experiments for each peptide to minimize tip-associated variance. Thus, the origin of differences in TERS spectra for each peptide is attributable to the interaction between the ligand functionalized nanoparticle and the integrin receptor in the cell membrane.
Figure 7.

(a) PCA and (b) HCA analysis of TERS spectra of cyclic-RGDFC-GNPs (n = 12), cyclic-isoDGRFC-GNPs (n = 11), and CisoDGRC-GNPs (n = 15) bound with SW620 cells.
To correlate the spectral variance observed in cells with ligand affinity, dark-field time-lapse microscopy was performed on cells interacting with the peptide-GNPs, allowing for the real-time tracking of single particle dynamics on the cell membrane. Figure 8a shows an example of a nanoparticle trajectory from a 2 min time-lapse video. Hyperspectral imaging was used to discriminate between nanoparticles and autofluorescent organelles (Figure S7). This allowed for the proper identification of single nanoparticles for trajectory analysis. Nanoparticle trajectories from the different functionalized GNPs in SW620 cell membrane were recorded and analyzed to determine ligand-associated changes. Multiple (7–10) trajectories were collected from GNPs conjugated with cyclic-RGDFC, cyclic-isoDGRFC, CisoDGRC, and citrate. The diffusion coefficient (D) for each particle in a two-dimensional system (membrane) was determined by the following equation:41
| 1 |
where r is the displacement and t is the time interval describing the particle movement between each frame. In this case, the mean square displacement is an average of every 10 frames in a trajectory and the time interval is 10 ms cycle time per frame. Figure S8 shows the histogram distributions of all of the calculated diffusion coefficients. The mean diffusion coefficients showed statistical differences (P < 0.001, one-way analysis of variance) for different ligand-conjugated GNPs as shown in Figure 8b. CisoDGRC-GNPs and citrate-GNPs presented higher diffusion than cyclic-RGDFC-GNPs and cyclic-isoDGRFC-GNPs in the cell membrane. The higher diffusion coefficient of the GNPs is interpreted to arise from a weaker drag force between the GNPs and the SW620 cell, presumably due to the weaker binding between the peptides and the integrin αvβ3. The SPT results on cells are consistent with the TERS observations, indicating that, by analyzing the spectral variance of TERS data, perspectives about relative binding affinities between ligand-conjugated GNPs and SW620 cells can be obtained.
Figure 8.

(a) Minimal intensity projection image (pseudo color) of a 2 min time-lapse video on a single SW620 cell incubated with GNPs. A single particle trajectory is presented in red. (b) Average diffusion coefficients of ligand-conjugated GNPs interacting with sw620 cells. **P < 0.001.
Conclusions
In this work, chemical information on specific ligand–receptor binding site of integrin αvβ3 was detected in a cancer cell membrane by TERS microscopy. Distinct Raman signals were observed using gold nanoparticles functionalized with three different peptide ligands (i.e., cyclic-RGDFC, cyclic-isoDGRFC, and CisoDGRC). Variance within the TERS spectra provides insights into chemical heterogeneity in the binding interaction that correlates with the diffusional motion of these particles in vitro. These results demonstrate the capability of TERS not only to study the binding chemistry but also to provide information about binding specificity in intact cell membranes. This capability has the potential to improve early stage drug screening.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences, part of the United States National Institutes of Health, Award R01 GM109988.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b01796.
UV–vis characterization of functionalized GNPs. SERS heatmaps from cyclic-isoDGRFC and CisoDGRC GNPs. MCR calculated Q residuals from Figure 1. Additional velocity profiles and trajectories on glass slides. TERS map of cyclic-RGDFC-GNPs bound with sw620 cells. Additional TERS mapping data of SW620 cells. Hyperspectral imaging of GNPs on cells. Histograms of diffusion coefficients from particle tracking on cells. Light scattering and zeta potential results from peptide-GNPs. Band assignments from TERS experiments. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hopkins A. L.; Groom C. R. Nat. Rev. Drug Discovery 2002, 1, 727–730. 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Patching S. G. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 43–55. 10.1016/j.bbamem.2013.04.028. [DOI] [PubMed] [Google Scholar]
- Winckler P.; Lartigue L.; Giannone G.; De Giorgi F.; Ichas F.; Sibarita J. B.; Lounis B.; Cognet L. Sci. Rep. 2013, 3, 2387. 10.1038/srep02387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin A. G.; Lounis B.; Cognet L. Biophys. J. 2014, 107, 1777–1784. 10.1016/j.bpj.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfreundschuh M.; Alsteens D.; Wieneke R.; Zhang C.; Coughlin S. R.; Tampe R.; Kobilka B. K.; Muller D. J. Nat. Commun. 2015, 6, 8857. 10.1038/ncomms9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L.; Chen Q.; Wu Y.; Qi X.; Zhou A. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1988–1995. 10.1016/j.bbamem.2015.05.007. [DOI] [PubMed] [Google Scholar]
- Aguirre C.; Cala O.; Krimm I. Curr.Protoc.Protein Sci. 2015, 81, 1–24. 10.1002/0471140864.ps1718s81. [DOI] [PubMed] [Google Scholar]
- Oxenoid K.; Chou J. J. Protein Sci. 2016, 25, 959–973. 10.1002/pro.2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet B. K.; Kobilka B. K. Trends Pharmacol. Sci. 2012, 33, 268–272. 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V.; Cherezov V.; Stevens R. C. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz Z. D.; Levin I. W. Annu. Rev. Anal. Chem. 2011, 4, 343–366. 10.1146/annurev-anchem-061010-114048. [DOI] [PubMed] [Google Scholar]
- Antonio K. A.; Schultz Z. D. Anal. Chem. 2014, 86, 30–46. 10.1021/ac403640f. [DOI] [PubMed] [Google Scholar]
- Kneipp K.; Wang Y.; Kneipp H.; Perelman L.; Itzkan I.; Dasari R.; Feld M. Phys. Rev. Lett. 1997, 78, 1667–1670. 10.1103/PhysRevLett.78.1667. [DOI] [Google Scholar]
- Xu H.; Bjerneld E.; Kall M.; Borjesson L. Phys. Rev. Lett. 1999, 83, 4357–4360. 10.1103/PhysRevLett.83.4357. [DOI] [Google Scholar]
- Boehme R.; Cialla D.; Richter M.; Roesch P.; Popp J.; Deckert V. J. Biophotonics 2010, 3, 455–461. 10.1002/jbio.201000030. [DOI] [PubMed] [Google Scholar]
- Richter M.; Hedegaard M.; Deckert-Gaudig T.; Lampen P.; Deckert V. Small 2011, 7, 209–214. 10.1002/smll.201001503. [DOI] [PubMed] [Google Scholar]
- Carrier S. L.; Kownacki C. M.; Schultz Z. D. Chem. Commun. (Cambridge, U. K.) 2011, 47, 2065–2067. 10.1039/c0cc05059h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Schultz Z. D. Analyst 2013, 138, 3150–3157. 10.1039/c3an36898j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Carrier S. L.; Park S.; Schultz Z. D. Faraday Discuss. 2015, 178, 221–235. 10.1039/C4FD00190G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander K. D.; Schultz Z. D. Anal. Chem. 2012, 84, 7408–7414. 10.1021/ac301739k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Schultz Z. D. ChemPhysChem 2014, 15, 3944–3949. 10.1002/cphc.201402466. [DOI] [PubMed] [Google Scholar]
- Xiao L.; Wang H.; Schultz Z. D. Anal. Chem. 2016, 88, 6547–6553. 10.1021/acs.analchem.6b01344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne C. K.; Jones S. A.; Chen C.; Zhuang X. Traffic 2007, 8, 389–401. 10.1111/j.1600-0854.2007.00540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan S. S.; Liu H. Y.; Vu T. Q. ACS Nano 2008, 2, 1153–1166. 10.1021/nn700399e. [DOI] [PubMed] [Google Scholar]
- Brandenburg B.; Zhuang X. Nat. Rev. Microbiol. 2007, 5, 197–208. 10.1038/nrmicro1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzo C.; Garcia-Parajo M. F.. Rep. Prog. Phys. 2015, 78, 124601. [DOI] [PubMed] [Google Scholar]
- Desgrosellier J. S.; Cheresh D. A. Nat. Rev. Cancer 2010, 10, 9–22. 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzeri S.; Zanella S.; Arosio D.; Vahdati L.; Dal Corso A.; Pignataro L.; Paolillo M.; Schinelli S.; Belvisi L.; Gennari C.; Piarulli U. Chem. - Eur. J. 2015, 21, 6265–6271. 10.1002/chem.201406567. [DOI] [PubMed] [Google Scholar]
- Mas-Moruno C.; Fraioli R.; Rechenmacher F.; Neubauer S.; Kapp T. G.; Kessler H. Angew. Chem., Int. Ed. 2016, 55, 7048–7067. 10.1002/anie.201509782. [DOI] [PubMed] [Google Scholar]
- Stupp R.; Hegi M. E.; Gorlia T.; Erridge S. C.; Perry J.; Hong Y. K.; Aldape K. D.; Lhermitte B.; Pietsch T.; Grujicic D.; Steinbach J. P.; Wick W.; Tarnawski R.; Nam D. H.; Hau P.; Weyerbrock A.; Taphoorn M. J.; Shen C. C.; Rao N.; Thurzo L.; Herrlinger U.; Gupta T.; Kortmann R. D.; Adamska K.; McBain C.; Brandes A. A.; Tonn J. C.; Schnell O.; Wiegel T.; Kim C. Y.; Nabors L. B.; Reardon D. A.; van den Bent M. J.; Hicking C.; Markivskyy A.; Picard M.; Weller M. European Organisation for Research and Treatment of Cancer (EORTC); Canadian Brain Tumor Consortium; CENTRIC study team. Lancet Oncol. 2014, 15, 1100–1108. 10.1016/S1470-2045(14)70379-1. [DOI] [PubMed] [Google Scholar]
- Bauer K. M.; Lambert P. A.; Hummon A. B. Proteomics 2012, 12, 1928–1937. 10.1002/pmic.201200041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz Z. D.; Stranick S. J.; Levin I. W. Anal. Chem. 2009, 81, 9657–9663. 10.1021/ac901789w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habuchi S.; Cotlet M.; Gronheid R.; Dirix G.; Michiels J.; Vanderleyden J.; De Schryver F. C.; Hofkens J. J. Am. Chem. Soc. 2003, 125, 8446–8447. 10.1021/ja0353311. [DOI] [PubMed] [Google Scholar]
- Taylor R. W.; Benz F.; Sigle D. O.; Bowman R. W.; Bao P.; Roth J. S.; Heath G. R.; Evans S. D.; Baumberg J. J. Sci. Rep. 2015, 4, 5940. 10.1038/srep05940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement J. E.; Leray A.; Bouhelier A.; Finot E. Phys. Chem. Chem. Phys. 2017, 19, 458–466. 10.1039/C6CP06667D. [DOI] [PubMed] [Google Scholar]
- Xiong J. P.; Stehle T.; Zhang R.; Joachimiak A.; Frech M.; Goodman S. L.; Arnaout M. A. Science 2002, 296, 151–155. 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- Artoni A.; Li J.; Mitchell B.; Ruan J.; Takagi J.; Springer T. A.; French D. L.; Coller B. S. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 13114–13120. 10.1073/pnas.0404201101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierrat S.; Hartinger E.; Faiss S.; Janshoff A.; Soennichsen C. J. Phys. Chem. C 2009, 113, 11179–11183. 10.1021/jp9030333. [DOI] [Google Scholar]
- Youngworth K.; Brown T. Opt. Express 2000, 7, 77–87. 10.1364/OE.7.000077. [DOI] [PubMed] [Google Scholar]
- Deckert-Gaudig T.; Kurouski D.; Hedegaard M. A.; Singh P.; Lednev I. K.; Deckert V. Sci. Rep. 2016, 6, 33575. 10.1038/srep33575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg H. C.Random Walks in Biology; Princeton University Press: Princeton, NJ, 1993. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.