

Abstract
In recent years, semiconducting polymer nanoparticles have attracted considerable attention because of their outstanding characteristics as fluorescent probes. These nanoparticles, which primarily consist of π-conjugated polymers and are called polymer dots (Pdots) when they exhibit small particle size and high brightness, have demonstrated utility in a wide range of applications such as fluorescence imaging and biosensing. In this review, we summarize recent findings of the photophysical properties of Pdots which speak to the merits of these entities as fluorescent labels. This review also highlights the surface functionalization and biomolecular conjugation of Pdots, and their applications in cellular labeling, in vivo imaging, single-particle tracking, biosensing, and drug delivery. We discuss the relationship between the physical properties and performance, and evaluate the merits and limitations of the Pdot probes for certain imaging tasks and fluorescence assays. We also tackle the current challenges of Pdots and share our perspective on the future directions of the field.
Keywords: fluorescence, imaging agents, polymer dots, semiconductors, sensors
On the dot
As a new class of fluorescent nanoparticles, semiconducting polymer dots, exhibit excellent characteristics such as extraordinary fluorescence brightness, fast emission rates, excellent photostability, nonblinking, and nontoxicity. This review summarizes recent advances of the polymer dots as nanoparticle labels for biological and medical applications (see figure).

1. Introduction
The development of fluorescence imaging techniques has provided effective research tools to investigate many fundamental processes in the life sciences.[1,2] Various fluorescence-based assays have also been applied to monitor a broad range of biological activities, such as conformation dynamics, interactions, and distribution of biomolecules in organelles, cells, or tissues.[3,4] More recently, advanced fluorescence imaging modalities have offered the ability to probe biomolecular interactions at the single-molecule level,[5–7] and to image subcellular structures with nanometer spatial resolution.[8–11] These advances have provided information at a level of unprecedented detail about cellular processes such as gene expression, protein transport, signal transduction, and regulatory pathways. In most cases, fluorescence detection requires exogenous fluorophores to either provide a signal readout or enhance image contrast. The potential of a fluorescence technique is significantly dependent upon the performance of the fluorophore as well as its properties, such as fluorescence brightness and photostability, which largely determine the detection limit, sensitivity, and reliability of the method.
Organic dyes appear to be the most versatile fluorophores used in biological imaging and bioassays.[12] A large variety of dye molecules, such as fluorescein, rhodamine, and cyanine, are available for different experiments, and their optical properties can be fine-tuned by various design strategies. However, the intrinsic limitations of conventional dyes, such as low absorptivity and poor photostability, have posed great difficulties for the further development of high-sensitivity imaging techniques and high-throughout assays. As a result, there has been considerable interest in brighter and more-stable fluorescent probes. For example, phycobiliproteins exhibit higher fluorescence brightness than small organic fluorophores, and genetically encoded fluorescent proteins represent a powerful toolkit for studying cellular processes.[13–15] There is also a great deal of interest in the development of brightly fluorescent nanoparticles, including semiconductor quantum dots (Qdots) and dye-loaded nanoparticles. Qdots in particular represent one of the best known nanotechnologies which has crossed into biology in the past decade.[16–18] They exhibit broad absorption bands and multi-color, narrow, and symmetric emissions with improved brightness and photostability compared to conventional fluorescent dyes. Polymer or silica nanoparticles loaded with fluorescent dyes also show higher brightness and better photostability than molecular dyes because of the large number of fluorophores per particle and the protective matrix.[19–21]
A promising strategy for generating very bright fluorescent probes is based on the development of nanoparticles consisting of highly fluorescent semiconducting polymers. There has been steady progress in creating fluorescent semiconducting polymers as the active material in polymer light-emitting devices.[22–25] Semiconducting polymers have also been extensively used as highly sensitive chemical and biological sensors.[26–35] Semiconducting polymer nanoparticles were originally developed for preparing thin films with nanoscale domains in optoelectronic devices.[36–39] These fluorescent nanomaterials have recently attracted considerable attention for both optoelectronic and biological applications.[40–44] For biological applications in particular, nanoparticles composed of semiconducting polymers can exist in many different forms having important functional implications. For example, semiconducting polymer nanoparticles can be prepared from hydrophobic semiconducting polymers,[45–80] polyelectrolytes, or hydrophilic polymers,[81–88] as self-assembled nanoparticles from amphiphilic polymers,[89–93] as hydrophobic semiconducting polymer-loaded poly(D, L-lactide-co-glycolide) (PLGA) particles,[94–97] as phospholipid-encapsulated or PEG-capped hydrophobic polymer nanoparticles,[98–102] and as semiconducting polymer-coated inorganic nanoparticles.[103–105] Among these nanoparticles, the fluorescence, colloidal stability, and functional performance are highly dependent on their particle size, composition, internal structure, and surface properties.
To make our discussions more precise, we define a polymer nanoparticle according to the description used in a previous review.[40] A polymer nanoparticle is considered to be a nanometer-sized entity (typical particle size < 100 nm), which represents a separate discontinuous phase surrounded by a continuous free-flowing medium (usually a low-molecular-weight liquid, most often water). On the basis of the classical categorization in colloid chemistry, this definition excludes true solutions of macromolecules in a low-molecular-weight solvent,[40] eventhough hyperbranched polymer molecules may adopt a three-dimensional morphology in a good solvent. Most colloidal polymer nanoparticles consist of a lyophobic polymer interior, but polyelectrolytes can also be forced to form nanoparticles. The definition of polymer nanoparticles also differentiates them from those directly grown on a substrate or dispersed in a polymeric matrix, as well as other highly disperse composite materials containing semiconducting polymers.[40]
For most biological applications, small fluorescent nanoparticles are particularly attractive as seen by the enormous efforts made in the past decade reagrding semiconductor Qdots, in which “dot” was originally used to describe their small particle size.[16–18] Since then, various fluorescent dots were developed for biological applications, including silicon dots,[106,107] carbon dots,[108,109] and dye-doped silica dots.[20] To be consistent with these small dots, Pdots specifically refer to a small subset of semiconducting polymer nanoparticles which have a particle size comparable to that of a Qdot (< 20–30 nm for encapsulated Qdots[110]). In many cases, nonfluorescent molecules may be incorporated into the Pdots for surface functionalization or multimodal imaging. However, fluorescent semiconducting polymers should be the major component of Pdots. Intuitively, therefore, Pdots should consist of semiconducting polymers present at a volume fraction or weight concentration that is at least higher than 50% and preferably higher than 80–90%. Volume fraction largely determines the fluorescence brightness for a Pdot of a given particle size. This requirement serves to distinguish Pdots from dye-loaded latex spheres or other nanoparticles containing only a small fraction of semiconducting polymers, both of which lead to a lack of the excellent optical properties inherent in Pdots. In addition, Pdots should possess a hydrophobic polymer interior because hydrophilic species can swell the particles, reduce colloidal stability, cause nonspecific labeling (through electrostatic interactions resulting from ionic groups), and affect the packing density of fluorophores as well as the fluorescence brightness.[65]
Recent research has demonstrated that Pdots exhibit excellent characteristics as fluorescent probes, including their extraordinary fluorescence brightness, fast emission rate, excellent photostability, and nonblinking and nontoxic features.[45–67] In particular, we recently carried out a systematic characterization of the optical properties and performance of Pdots as cellular labels, especially for fluorescence imaging and flow cytometry.[56,58] We found that single-particle brightness and cell-labeling brightness of Pdots are more than an order of magnitude higher than those of inorganic Qdots of comparable particle size.[56,58] We have also developed several simple yet powerful approaches for introducing functional groups and controlling the surface chemistry of Pdots. Successful functionalization is a prerequisite in employing these novel nanoprobes for cellular labeling and in vivo imaging, as well as versatile biosensors.[56–67] These developments have formed a new frontier in nanobiotechnology and nanomedicine, and represent a new direction that is orthogonal to the more established areas of conjugated polymers for optoelectronic devices and conjugated polyelectrolyte biosensors. The superior properties of Pdots over other fluorescent probes have established their potential in biology and medicine as very bright in vitro and in vivo probes.
This review is focused on recent advances and the current status of the preparation, fluorescence properties, bioconjugation, and biological applications of Pdots, which we define to be a nanoparticle composed of hydrophobic semiconducting polymers present at greater than 50% volume or weight fraction and has a diameter smaller than 20–30 nm. Other semiconducting-polymer-based nanoparticles are also considered where appropriate. In Section 2, we provide a brief history of semiconducting polymers and fluorescent polymer nanoparticles, and then discuss the fluorescence properties of Pdots and evaluate their performance in certain key areas (Section 3). As bioconjugation is one of the critical steps in converting Pdots for biological applications, Section 4 describes recent efforts to produce functionalized Pdots and bioconjugates. Section 5 provides a series of recent developments in using functionalized Pdots in cellular imaging, in vivo imaging, single-particle tracking, biosensors, and drug delivery. Finally, we speculate on key opportunities and challenges which have yet to be addressed by the scientific community in the development of Pdots. These questions should inspire future investigations in developing versatile Pdot probes and lead to new discoveries.
2. Polymer Dots as Fluorescent Probes
2.1. Semiconducting Polymers
A brief description of semiconducting polymers is necessary to understand the general properties of the corresponding nanoparticles and Pdots in particular. The majority of organic polymers are insulators. However, when they have π-conjugated structures, electrons can move along the polymer backbone through overlaps in π-electron clouds by hopping, tunneling, and related mechanisms. In general, these π-conjugated polymers in their pristine states are wide-band-gap semiconductors, the so-called semiconducting polymers. In the 1970s, organic conjugated polymers and oligomers were discovered to be metallic upon heavy doping,[111,112] a term derived from inorganic semiconductor chemistry. However, the doping in a conjugated polymer was either an oxidation or a reduction of the π-electronic system and is called p-doping and n-doping, respectively. Conjugated polymers became particularly attractive because they promised to achieve a new generation of polymeric materials which exhibited the tunable electrical and optical properties of metals or semiconductors while retaining the attractive mechanical properties and processing advantages of polymers.[113]
In general, semiconducting polymers exhibit a direct band gap, which leads to an efficient (allowed) absorption or emission at the band edge. Depending on the polymer species, a semiconducting polymer can exhibit strong fluorescence, which can be described in terms of semiconductor band theory. Upon photoexcitation, an electron is excited from the highest occupied energy band (the π band) to the lowest unoccupied energy band (the π* band), thus forming a bound state (exciton) of the excited electron and hole in the p band. The recombination of the excited electron with the hole results in a fluorescent photon. The wavelength of the absorbed light is determined by the π–π* energy gap and can be tuned by altering the molecular structure of the polymer.
Semiconducting polymers have been developed with emission colors that span the full range of the visible spectrum.[114] Important examples of fluorescent semiconducting polymers include polyfluorene (such as PDHF and PFO; Figure 1), poly(phenylene ethynylene) (such as PPE), poly(phenylene vinylene) (such as MEH-PPV and CN-PPV), fluorene-based copolymers (such as PFPV, PFBT, and PF-DBT5), and related derivatives. In many cases, photogenerated electron-hole pairs can dissociate to form free carriers which migrate through the system. The free carriers can either combine to form triplets or deactivate by other nonradiative processes (unwanted processes for fluorescence). They can also be collected to generate electric current (desirable processes for photovoltaics). The tunable electrical and optical properties, together with the versatile polymer chemistry, make semiconducting polymers a fascinating class of materials for optoelectronic devices. They have been widely demonstrated as active materials for a broad range of optoelectronic devices, including organic light-emitting diodes for flat panel displays,[22–25] photovoltaic devices for solar energy conversion,[115,116] and thin-film transistors.[117–119]
Figure 1.
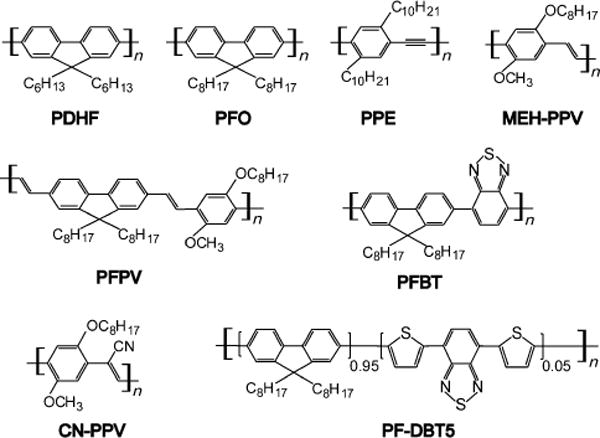
Chemical structures of highly fluorescent semiconducting polymers.
2.2. From Conjugated Polyelectrolyte Biosensors to Hydrophobic Polymer Dots
Besides their applications in light-emitting devices, fluorescent conjugated polymers have been used in a wide range of biological applications.[26–35] The applications of conjugated polymers in fluorescent biosenors have grown enormously in the past decade. In 1999, Whitten and co-workers reported efficient fluorescence quenching of a conjugated polyelectrolyte with dimethyl viologen.[26] The high sensitivity offered by quenching was attributed to a strong association between the anionic polyelectrolyte and the cationic quencher together with an efficient transport of the excitons to the quencher sites. The fluorescence quenching was ascribed to a dominant electron-transfer mechanism, while analyte-induced polymer aggregation may have also contributed to the quenching response. Since then, conjugated polyelectrolytes have dominated the use of conjugated polymers in biosensor applications. Signal amplification arising from efficient exciton migration in conjugated polymers is being exploited to produce new sensing systems of unparalleled sensitivity. The advances in the area of conjugated polyelectrolyte biosensors have been reviewed elsewhere.[31–35]
At the same time, as pointed out in the introduction, there was a clear motivation for the design of highly fluorescent nanoparticles because fluorescent dyes are not sufficiently bright and photostable for many advanced imaging tasks and high-throughout assays. Hydrophobic conjugated polymers, rather than conjugated polyelectrolytes, were selected for preparing fluorescent nanoparticles because: 1) conjugated polyelectrolytes are heavily functionalized with ionic side chains, which can cause significant nonspecific labeling by electrostatic interactions, 2) side-chain modifications with ionic groups in conjugated polymers usually reduce the fluorescence quantum yield compared to their hydrophobic counterparts, 3) synthesis of conjugated polyelectrolytes generally requires sophisticated and laborious modification of the polymer structures, whereas a large number of hydrophobic conjugated polymers with multiple emission colors are commercially available, and 4) conjugated polyelectrolytes are water soluble, thus adopting extended rodlike structures in aqueous solution. It is difficult to form densely packed nanoparticles with conjugated polyelectrolytes because of repulsive interactions and solvation effects in an aqueous environment.
The group of McNeill first demonstrated the formation of hydrophobic semiconductor Pdots which exhibit salient features as promising imaging probes.[45–55] Our recent work demonstrated the successful formation of Pdot bioconjugates and established their extraordinary brightness compared to commercially available dye-tagged antibodies and Qdots of a similar particle size.[56–67,104] Although still at the early stages of development, semiconducting polymers as fluorescent nanoparticles have attracted intense interest.[40–44] There have been significant efforts to create versatile semiconducting polymer nanoparticles (a small subset of which are Pdots), tune their properties and functions, and improve their performance for biomedical studies. The research efforts include exploring new preparation methods,[45,46,69,70,72] investigating nanoparticle formation mechanisms,[120,121] probing nanoparticle photophysics,[47,49,54,122,123] characterizing the fluorescence performance,[48,51,56,58] tuning the emission color,[47,49,71,89] engineering the particle surface,[56–58,64–66,98,102] encapsulating inorganic materials,[96,97,99,104,105] developing nanoparticle sensors,[52,59,60,62,74,77] imaging cellular structures,[56,57,63–66] and in vivo targeting in small animals.[58,72]
There are also recent studies in using conjugated polyelectrolytes for fluorescence imaging applications.[32–35] In some cases, conjugated polyelectrolytes can selectively label cellular targets in vitro or distinctively stain different protein aggregates in vivo because of the polyvalent electrostatic and hydrophobic interactions with biomolecules.[124–126] By virtue of their versatile chemistry, conjugated polyelectrolytes can be synthesized with hyperbranched structures so as to be soluble in water.[127–129] Amphiphilic conjugated polymers and oligomers can also self-assemble into spherical particles or micellar structures which exhibit tunable optical properties,[89–91] and serve as imaging probes and delivery vectors.[92,93] Moreover, traditional linear conjugated polyelectrolytes or hydrophilic semiconducting polymers, which are heavily functionalized with hydrophilic groups, can also be transformed into nanoparticles in the presence of salts, acids, or oppositely charged species.[81–88] It is important to note, however, that nanoparticle structures based on either polyelectrolytes or heavily functionalized conjugated polymers are distinct from Pdots, which consist primarily of densely packed hydrophobic polymers. Although there are a few reviews that partially cover the preparation and properties of semiconducting polymer nanoparticles,[35,40–44] a relatively comprehensive and updated review on Pdots as fluorescent probes and their biological applications is still lacking. This review places special emphasis on Pdots, which are small (< 20–30 nm) and have a high volume fraction (> 50%) of hydrophobic conjugated polymer. The small particle size, high volume fraction of semiconducting polymer, and densely packed fluorophore structure are key factors in developing the next-generation bright imaging probes and sensitive nanoparticle-based sensors.
2.3. Preparation Methods
Preparation methods of various conjugated polymer nanoparticles were described in detail in a previous review.[40] Briefly, conjugated polymer nanoparticles can be directly synthesized into nanoparticles from low-molecular-weight monomers (direct polymerization), or prepared from high-molecular-weight polymers which are already synthesized (post polymerization). The direct polymerization approach dates back to the 1980s, when several nanoparticle systems of conductive polymers were reported.[130,131] More recently, this approach has been extended to fluorescent semiconducting polymers for their nanoparticle preparations.[69–71,132] Different from conductive polymers, which can be synthesized by aqueous oxidative polymerization, fluorescent semiconducting polymers are frequently synthesized by coupling reactions catalyzed by transition metals. The reactions must be compatible with the dispersion solvents used in the direct-polymerization method. In contrast, the post-polymerization approach can use the vast library of commercially available semiconducting polymers and does not require equipment and expertise in organic and polymer synthesis. To date, fluorescent semiconducting polymer nanoparticles have been mostly prepared by this latter approach.
Typical methods of the post-polymerization approach include miniemulsion and reprecipitation techniques, in which semiconducting polymers in an organic solvent are usually used as the starting solution. Water is preferable as the final dispersion media for nanoparticles aimed at biological applications. In the miniemulsion method, semiconducting polymer nanoparticles can be formed from the emulsified droplet solution, and usually requires a water-immiscible solvent. For the reprecipitation method, the polymer is precipitated by rapidly mixing the polymer solution with water, and requires a water-miscible solvent.
By using the miniemulsion method, Landfester and coworkers first demonstrated the preparation of fluorescent semiconducting polymer nanospheres which could be used to fabricate nanoscale multiphase films in photovoltaic and light-emitting devices.[36–38] In a typical miniemulsion preparation, a solution of a semiconducting polymer in an organic solvent, such as chloroform, was emulsified in water to form stable droplets. A surfactant, such as sodium dodecyl sulphate, was also present in the water. The organic solvent was evaporated to obtain a stable dispersion of polymer nanoparticles in water. The reprecipitation method was first used by Masuhara and co-workers to prepare polythiophene particles with a diameter range of 40–400 nm,[39] and later modified by McNeill and co-workers to form Pdots.[45–54] In the reprecipitation procedure, a dilute solution of the polymer dissolved in a water-miscible organic solvent is rapidly injected into water and mixed. The sudden decrease in solubility, combined with the hydrophobic interactions between polymer chains or segments of single chains, results in a suspension of highly fluorescent Pdots.
3. Properties and Performance
3.1. Particle Size and Morphology
The miniemulsion method typically produces conjugated polymer particles, which are 40 nm to 500 nm in diameter, by varying the polymer and surfactant concentrations in the solution mixture.[38] Figure 2A shows a typical transmission electron microscopy (TEM) image of nanoparticles prepared from a ladder-type poly(p-phenylene) polymer by the miniemulsion method.[36] In a special case, nanoparticles (ca. 13 nm) of a PPV derivative were prepared by the miniemulsion technique using very dilute solutions of polymers in dichloromethane combined with a large excess of poly(ethylene glycol) (PEG) as a nonionic droplet stabilizer (semiconducting polymer/PEG ca. 0.57 wt%).[100] More recently, several types of small nanoparticles with average diameters of 2 to 5 nm were obtained using a similar preparation method and precursor solutions.[101] However, the procedure and parameters need to be further optimized because the yield of this preparation appears to be very low, and can be a significant disadvantage for a reliable preparation method.
Figure 2.

Particle sizes of semiconducting polymer nanoparticles prepared by different methods. A) TEM image of nanoparticles of a ladder-type poly(p-phenylene) polymer prepared by the miniemulsion method. Reproduced from Ref. [36] with permission. B) TEM image of Pdots from a PFBT polymer prepared by the reprecipitation method. Scale bars in (A) and (B): 100 nm. Reproduced from Ref. [57] with permission.
The miniemulsion method has also been used to prepare particles made from a semiconducting polymer. In some studies, PLGA particles loaded with semiconducting polymer were prepared by the miniemulsion technique (semiconducting polymer/PLGA ca. 1 wt%),[94,95] but the particles were large (ca. 250 nm) and had relatively low fluorophore concentration (ca. 1wt%). In a similar preparation involving a miniemulsion, semiconducting polymers were encapsulated in a phospholipid matrix, thus forming functionalized particles (ca. 100 nm diameter) with a fluorophore loading concentration of approximately 8.5wt%.[98] Furthermore, the minemulsion method can be used to generate semiconducting polymer-based composite particles (> 100 nm) consisting of inorganic materials, such as iron oxide and metal nanoparticles.[96,97,99]
The reprecipitation method has reliably generated Pdots with diameters of about 5 to 30 nm, whereas the miniemulsion approach[45–54,56–58,63–66] usually produces particles of 40 to 500 nm. Because the reprecipitation process involves a competition between interchain aggregation and intrachain collapse to form nanoparticles, the particle size can be varied by adjusting the polymer concentration in the precursor solution.[46,51] Figure 2B shows a typical TEM image of Pdots made from a PFBT polymer prepared by the reprecipitation method.[57] The observation of a spherical shape is somewhat surprising given the rigid backbone of π-conjugated polymers.[133] However, the large interfacial surface tension between the polymer and water may dominate in this size range, thus yielding a thermodynamically favored spherical shape.[134,135] Because of the strong hydrophobic interactions and large surface tension, the Pdots made of hydrophobic semiconducting polymers typically possess internal structures of densely packed chromophores, as seen by the highly efficient energy transfer inside the nanoparticles.[43,47,49,52,58–61,65,66] The phase structure of the Pdots is typically amorphous (glassy phase), and the crystalline phase can be formed partially in the glassy matrix for certain polymer species, such as PFO.[50]
Conjugated polyelectrolytes and hydrophilic semiconducting polymers can be precipitated into particles with the aid of salts, acids, or oppositely charged species.[81–85,87] The precipitation of a PPE polymer with hydrophilic amine and PEG groups from a DMSO solution into water containing salts resulted in the formation of particles with about a 500 nm diameter.[81] With similar hydrophilic polymers, smaller particles of diameters from 10 nm to 100 nm were prepared by a solvent exchange method which involved sequential ultrafiltration of acetic-acid-treated polymers.[82–85] In contrast to the hydrophobic Pdots with densely packed chromophoric structures, the polyelectrolyte-based nanoparticles consist of loosely aggregated polymer chains because of their hydrophilic nature.[32,85] Conjugated polyelectrolytes, however, can be synthesized with hyperbranched structures, thus forming a three-dimensional morphology in water.[127–129] In these structures, the particle sizes are mainly determined by segmental flexibility and generation number so uniform particle size can be obtained by polymer synthesis. In addition, amphiphilic polymers and oligmers can form spherical assemblies with particle sizes ranging from ten to hundreds of nanometers.[89,90,92,93] The assemblies can exhibit micellar structures[90] or vesicles,[136] but the internal structures of the particles remained somewhat unclear from the data reported.[89,92,93]
The physical size of a nanoparticle probe is closely related to its performance in biological applications. While there are biological applications that can tolerate large particle sizes, most imaging and bioassays require small nanoparticle labels. For example, the large size of fluorescent nanoparticles causes steric hindrance, which interferes with functions such as targeting specificity and binding affinity of the attached biomolecules. In applications involving dynamic processes, large nanoparticles can significantly impair trafficking of the attached biomolecules and restrict access to crowded cellular locations. To counter these effects, there has been extensive research efforts in developing small and compact nanoparticle probes.[137,138] In terms of per-particle fluorescence brightness, the number of fluorophores in a particle scales with the particle volume at a given chromophore packing density. The particle size combined with the fluorophore packing density determines the absorption cross section of the fluorescent polymer particles. In this regard, the hydrophobic Pdots have a significant advantage because they have densely packed chromophoric structures.
3.2. Absorption and Fluorescence
The optical properties of Pdots have been systematically characterized and evaluated for their utility as fluorescent labels.[45,46,48,51,56,58,59,65,66] Pdots prepared by reprecipitation methods usually show broadened and blue-shifted absorption spectra compared to that of the polymer in a THF solution.[46] This is consistent with an overall decrease in the conjugation length caused by bending, torsion, and kinking of the polymer backbone. Depending on the polymer, Pdots exhibit broad absorption bands ranging from λ = 350 nm to λ = 600 nm (Figure 3B), a convenient wavelength range for fluorescence microscopy and laser excitation. The light-harvesting capability of a fluorescent particle at a given wavelength is described by the absorption cross section, which can be determined from absorption spectra. An analysis of the UV/Vis absorption spectra at known Pdot concentrations indicated that the peak absorption cross section of single particles (ca. 15 nm diameter) were on the order of about 10−13 cm2, roughly ten to hundred times larger than that of CdSe Qdots in the visible and near-UV range. Their brightness was roughly three orders of magnitude greater than that of typical organic fluorescent dyes.[51,56,58]
Figure 3.
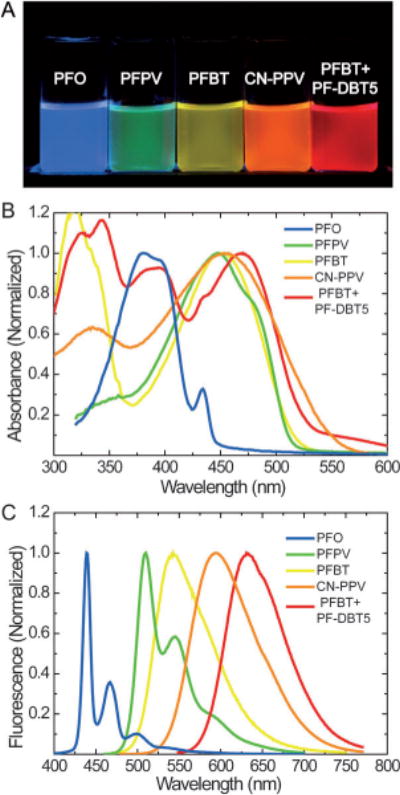
Fluorescence spectra of Pdots. A) A photograph of aqueous Pdot suspensions under UV light illumination. Absorption (B) and emission (C) spectra. The spectra of PFO, PFPV, and PFBT Pdots were reproduced from Ref. [51] with permission. The spectra of CN-PPV Pdots were reproduced from Ref. [63] with permission. The spectra of polymer blend Pdots (PFBT+PF-DBT5) were reproduced from Ref. [58] with permission.
Pdots made from different hydrophobic polymers showed multicolor emissions in the visible region (Figure 3). In most cases, the Pdots exhibited a slightly red-shifted fluorescence compared to that of the polymer in an organic solvent. This was attributed to increased interchain interactions caused by chain collapse, which produced a fraction of red-shifted aggregate species.[46] The resulting energetic disorder, combined with multiple energy transfers, generated a net red-shift in the fluorescence spectrum compared to that of the polymer in a THF solution, as has often been observed in thin films.[139] Fluorescence quantum yield is defined as the ratio of the number of photons emitted to the number of photons absorbed, thus describing the efficiency of a fluorescent probe. Depending on the polymer, the Pdot species showed moderate quantum yields from 1% to 20% in early reports.[46] Our group recently reported several new types of Pdots exhibiting high fluorescence quantum yields in the range of 50–60%,[58,63] which is comparable to those of inorganic Qdots.
To explore Pdots in two-photon fluorescence applications, McNeill and co-workers first reported that Pdots exhibited two-photon action cross sections as high as 2.0 × 105 GM, the largest reported thus far for a nanoparticle having a diameter of about 20 nm (Figure 4D).[48] The two-photon action cross-section values are three to four orders of magnitude higher than the values for conventional fluorescent dyes, and an order of magnitude higher than that for inorganic quantum dots. In a later study, Moon et al. prepared nanoparticles, having about an 8 nm diameter, by the ultrafiltration of the tartaric-acid-treated hydrophilic PPE derivative. The investigators determined the two-photon action cross sections to be 103–104 GM, and demonstrated two-photon imaging of endothelial cells in a tissue model.[84] More recently, Mecking and co-workers reported the synthesis of fluorescent nanoparticles made of polymers based on poly(arylene ethynylene) by a direct polymerization method involving Sonogashira coupling in an aqueous miniemulsion.[71] The nanoparticles exhibited sizes in the range of 60–120 nm, and the determined two-photon action cross sections ranged from 106 to 107 GM, which is in good agreement with the values of Pdots after taking particle volume into account. These studies consistently support the potential of Pdots for multiphoton fluorescence imaging applications.
Figure 4.
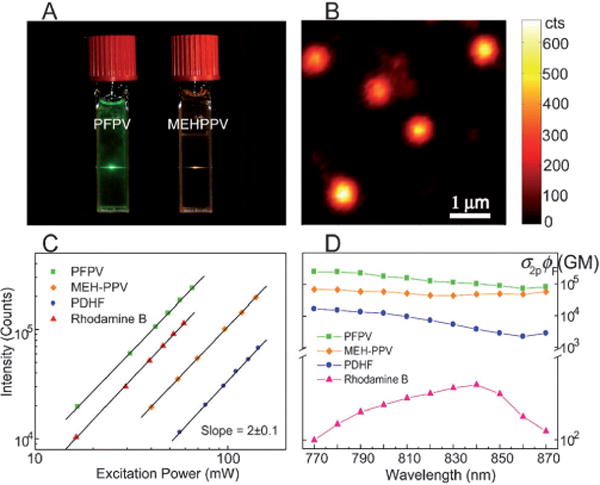
Two-photon excited fluorescence from Pdots. A) Photograph of aqueous Pdot dispersions excited with an λ= 800 nm mode-locked Ti:sapphire laser. B) Single-particle imaging of PFPV dots with two-photon excitation. C) Log-log plot of two-photon fluorescence intensities versus the excitation power. D) Semi-log plot of two-photon action cross sections (σ2pϕ) versus the excitation wavelength. Reproduced from Ref. [48] with permission.
A useful estimate of fluorescence brightness is given by the product of the peak absorption cross section and the fluorescence quantum yield. Table 1 summarizes the photophysical properties of the widely studied PFBT Pdots together with the properties of two commonly used probes purchased from Invitrogen: Qdot 565 and fluorescent IgG-Alexa 488 (ca. six dye molecules per IgG; IgG = immunoglobulin G).[56] Photophysical data indicate that under a typical laser excitation of λ = 488 nm, PFBT Pdots having an approxiamte diameter of 10 nm are about 30 times brighter than IgG-Alexa 488 and Qdot 565 probes. The absorption cross section of PFBT Pdots at λ = 488 nm is about half of its peak absorption cross section at λ = 450 nm. We performed a side-by-side brightness comparison by single-particle imaging to experimentally evaluate and compare the brightness of the three probes.[56] With a relatively low excitation power from a λ = 488 nm laser, very bright, near-diffraction-limited spots were clearly observed for individual PFBT Pdots. The IgG-Alexa 488 and Qdots exhibited much lower intensity levels and were barely detected by the camera when using the same excitation and detection conditions. The fluorescence intensity distribution of several thousands of particles indicated that PFBT Pdots were about 30 times brighter than IgG-Alexa 488 and Qdot 565 (Figure 5A), and this result is consistent with the brightness comparison based on the photophysical parameters. In a similar single-particle brightness comparison,[58] we determined that polymer-blend Pdots (PBdot) based on PFBT and a red-emissive polymer PF-DBT5 were approximately 15 times brighter than a Qdot which emitted at λ = 655 nm (Figure 5B). In a separate comparison, PFBT Pdots (ca. 15 nm) are an order of magnitude brighter than that of dye-loaded polystyrene beads (ca. 20 nm).[53] Based on these comparisons, Pdots are arguably the brightest among fluorescent nanoparticles of similar sizes.
Table 1.
Photophysical properties of PFBT Pdots, IgG-Alexa 488 and Qdot 565.[a]
| Probe (Size) | PFBT dot (ca. 10 nm) | Alexa 488[b,c] (ca. 1 nm) | Qdot 565[b] (ca. 15 nm) |
|---|---|---|---|
| Absorption/Fluorescence λmax | 460 nm/540 nm | 496 nm/519 nm | UV/565 nm |
| Extinction Coefficient at λ = 488 nm | 1.0×107 M−1 cm−1 | 5.3×104 M−1 cm−1 | 2.9×105 M−1 cm−1 |
| Quantum Yield | 0.3 | 0.9 | 0.3–0.5 |
| Fluorescence Lifetime | 0.6 ns | 4.2 ns | ca. 20 ns |
Reproduced from Ref. [56] with permission.
The data for Alexa 488 and Qdot 565 are according to the specifications provided by Invitrogen.
The parameters of Alexa 488 are for single dye molecules. An IgG-Alexa 488 probe has a hydrodynamic diameter of 12 nm and contains an average of six dye molecules, but its brightness corresponds to about 2–4 dye molecules because of self-quenching.
Figure 5.
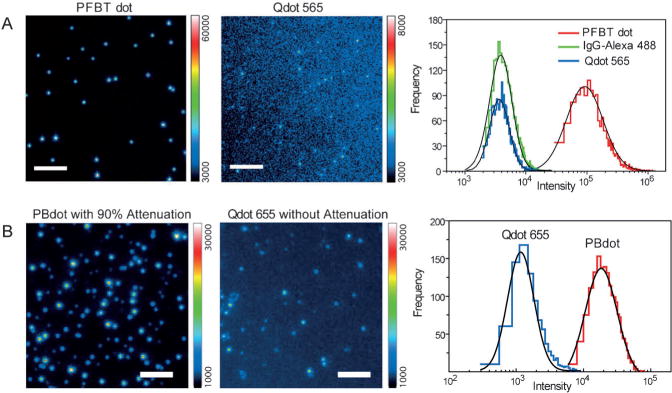
Comparison of single-particle fluorescence brightness. A) Single-particle fluorescence images of PFBT Pdots and particles of Qdot 565 obtained under identical λ = 488 nm excitation and detection conditions. Scale bar: 5 μm. The right panel shows the intensity distributions obtained from single-particle fluorescence data. Reproducedfrom Ref. [56] with permission. B) Single-particle fluorescence images of PBdot and Qdot 655. Under identical λ= 488 nm excitation, a neutral density filter with an optical density of 1 was used to obtain fluorescence images of PBdots but no attenuation was used for Qdot 655. Scale bar: 4 μm. The right panel shows the intensity distributions from single-particle fluorescence data. Reproduced from Ref. [58] with permission.
3.3. Fluorescence Lifetime, Emission Rate, and Photostability
Depending on the polymer, the fluorescence lifetime of Pdots generally varies from 100 ps to 1 ns, as determined by the time-correlated single-photon counting technique (TCSPC; Figure 6A). The fluorescence radiative rate constant, kR, and nonradiative rate constant, kNR, can be estimated by combining the quantum yield [ϕ = kR/(kR+kNR)] and the fluorescence lifetime results [τ = (kR + kNR)−1]. Pdots exhibit a fluorescence radiative rate constant in the range of 108 to 109 s−1, which is similar to or higher than that of typical fluorescent dyes (ca. 108 s−1). Single quantum dots exhibit rates of about two orders of magnitude lower.[140] In experiments, the actual emission rate is often limited by saturation. This is particularly true for experiments involving high excitation intensities, such as single-molecule imaging and particle tracking. The photon emission rate, R, is dependent on the laser excitation intensity, I, which is well described by two parameters: the maximum emission rate, Rs, and the saturation intensity, Is, using the saturation equation R = Rs(I/Is)(1+I/Is)−1.[141] Typical fluorescent dyes exhibit a saturated emission rate on the order of 106 s−1 as a result of triplet saturation.[142] This picture is complicated for Pdots with multiple densely packed chromophores. As shown in Figure 6B, the fluorescence signal of a PFBT Pdot showed power saturation behavior, from which the saturation emission rate was obtained by fitting the data with the saturation equation. Statistical analysis of several Pdots produced saturation emission rates ranging from 107 to 109 s−1.[51] The mean saturation emission rate was roughly 100 times higher than that of typical molecular dyes, and at least three orders of magnitude higher than that of colloidal semiconductor quantum dots. The fast emission rate of the Pdots is an advantage for high speed applications, such as flow cytometry and advanced dynamic imaging.
Figure 6.
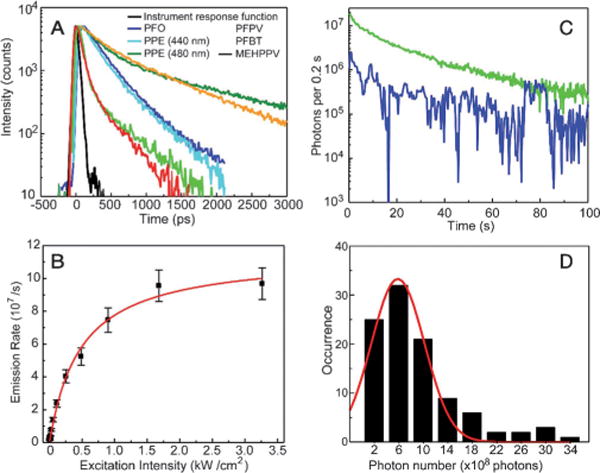
A) Fluorescence lifetimes of Pdots measured by the TCSPC technique. B) Fluorescence saturation of single PFBT Pdots with increasing excitation intensity. C) Photobleaching trajectories of single PFBT Pdots. D) Histogram of the photon numbers of individual PFBT Pdots of about a 10 nm diameter. Reproduced from Ref. [51] with permission.
The photostability of a fluorescent dye or nanoparticle can be characterized by the photobleaching quantum yield (ϕB), which is the number of molecules that have been photobleached divided by the total number of photons absorbed over a given time interval. Typical fluorescent dyes exhibit photobleaching quantum yields in the range of 10−4 to 10−6.[143] Photobleaching kinetics of aqueous Pdot suspensions were observed to vary substantially from one polymer to another.[51] Depending on the polymer, the photobleaching quantum yield was estimated to be in the range of 10−7 to 10−10 by using the rate constants obtained from the photobleaching kinetics measurements. The fluorescence quantum yield divided by photobleaching quantum yield results in the total number of photons that a nanoparticle emits prior to photobleaching (photon number). The photon number of the 15 nm diameter PFBT dots was estimated to be 109, which is three to four orders of magnitude larger than those of typical fluorescent dyes.[51,56]
Single-particle photobleaching studies provide more information about the photostability of Pdots (Figure 6C). Statistical analyses of multiple photobleaching trajectories indicated an average of about 6 × 108 photons emitted per 10 nm diameter particle, with some particles emitting more than 109 photons (Figure 6D). These numbers are more than an order of magnitude higher than those for single Qdot particles, for which the total photon number was determined to be 107 photons emitted per particle.[144] The photobleaching trajectories could be roughly categorized into two types, as exemplified by the curves shown in Figure 6C. As indicated by the green curve, most of the Pdots exhibited continuous photobleaching behavior without observable fluorescence blinking, but dimmer particles often showed some blinking (blue curve). These observations are consistent with the size-dependent blinking observed in other semiconducting polymers.[48,145] The fluorescence of the smaller (dimmer) particles fluctuates because of the small number of emitting chromophores and the reversible on/off dynamics which cause sizable fluctuations in the fraction of chromophores in the “on” state. Larger particles (> 10 nm diameter) have relatively steady fluorescence because there are contributions from a larger number of chromophores. As fluorescent probes for imaging or single-molecule experiments, the steady fluorescence of Pdots compares favorably to that of conventional dyes and quantum dots, which often exhibit pronounced blinking on time scales of microseconds to hundreds of seconds.[146]
3.4. Colloidal Stability and Chemical Stability
Three aspects describe the stability of nanoparticles for biological applications. They are whether the nanoparticles are resistant to aggregation, stable against dissociation and decomposition, and are chemically stable in the presence of biologically relevant ions and reactive species.
In regard to the first aspect, bare Pdots show excellent colloidal stability in deionized water for months without signs of aggregation or precipitation. However, the mechanism by which these intrinsically hydrophobic nanoparticles circumvent aggregation in water and exhibit good colloidal stability was not fully understood. Kee and co-workers proposed that the oxidation of semiconducting polymers during Pdot formation resulted in partially ionized defects, which in turn stabilized the nanoparticle suspension.[121] Zeta potential measurements showed that Pdots had a negatively charged surface at neutral pH, and a zeta potential of approximately −40 mV and surface charge density of (1.39–1.70) × 10−2 Cm−2. Fourier-transform infrared spectroscopy, X-ray photoelectron spectroscopy, and elemental analysis supported the presence of chemical defects that made the Pdots hydrophilic and thus well dispersed in water.
However, bare Pdots tend to aggregate in solutions which are either of moderate ionic strength or contain divalent metal ions.[64] We have shown that surface modifications can significantly improve the colloidal stability of Pdots.[56,57,64–66] For example, Pdots can be passivated with additives, such as bovine serum albumin (BSA), which maintain long-term colloidal stability and reduce nonspecific binding in labeling experiments.[56] BSA-passivated Pdots are stable for six months or longer at physiological pH values ranging from 4 to 9, and in various buffers such as HEPES, PBS, Tris, and borate buffers.[56,57,64–66] More recently, our group reported a facile method to coat hydrophobic Pdots with a surface layer of polyelectrolytes.[64] The polyelectrolyte coating results in robust and stable Pdots in solutions having a high ionic strength or containing high levels of multivalent metal ions.
Nanoparticle stability is also strongly dependent on whether the nanoparticles are easily dissociated or decomposed over time. Because Pdot formation is primarily driven by hydrophobic interactions, hydrophilic side chains can cause significant interference with the association strength among different portions of a polymer chain or between polymer chains, thus affecting nanoparticle stability. For example, loose aggregates, rather than stable and compact nanoparticles, were formed when nanoparticles were prepared using polymers with heavily functionalized hydrophilic side chains. Their aggregation nature was affected by many factors, such as polymer concentrations, ionic strength, and temperature.[85] More recently, we have systematically investigated the stability and internal structure of Pdots prepared from PFBT polymers having different densities of hydrophilic side-chain groups.[65] Our results indicate Pdot dissociation significantly increased as the density of hydrophilic side chains was increased. Polymers with low-density hydrophilic side chains formed stable and compact Pdots while those with high-density hydrophilic side chains tended to yield unstable and loose nanoparticles.[65]
The integrity of nanoparticles in biological applications also depends on their chemical reactivity towards ionic species and reactive oxygen species (ROS) in a physiological environment. For example, copper ions and ROS at physiological concentrations severely degrade Qdots, which causes a loss of luminescence and the release of toxic Cd ions.[147,148] We examined the stability of Pdots in the presence of biologically relevant ions and ROS (Figure 7). The fluorescence of Pdots was not affected by any biologically relevant ions we tested, including iron, zinc, and copper, which are among the most abundant ions encountered in biological systems. Two common and stable ROS species in the physiological environment, hypochlorous acid (HOCl) and hydrogen peroxide (H2O2), did not affect the fluorescence of Pdots. In contrast, under the same concentrations, inorganic Qdots were significantly quenched by H2O2 and iron ions, and were completely quenched by HOCl and copper ions. The stable fluorescence of Pdots can be attributed to their hydrophobic polymeric nature which tends not to have chemical interactions with metal ions and ROS. This property provides a significant advantage for using Pdots as in vivo probes.
Figure 7.
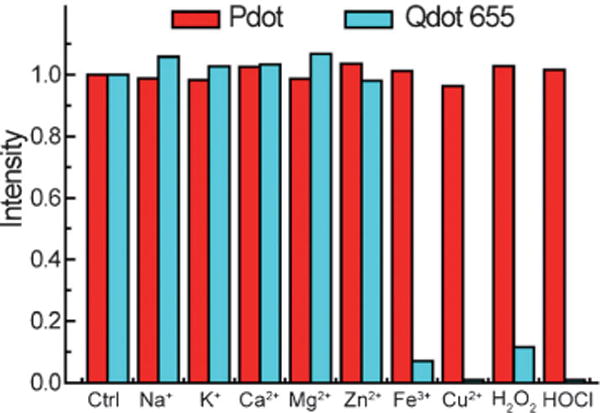
Chemical stability of Pdots in the presence of biologically relevant ions, and ROS in aqueous solutions. Qdot 655 nanocrystals were a reference. The concentration for each metal ion was 500 μM. For the ROS stability test, the H2O2 and chlorine concentration was 0.1 wt% and 0.004 wt%, respectively.
3.5. Toxicity
Biocompatibility is an important consideration for fluorescent nanoparticles aimed at biological applications. Toxicity studies of Pdots have been conducted by several groups, and all tests showed Pdots to have very low cytotoxicity. Christensen and co-workers evaluated the possible cytotoxic effects of hydrophobic Pdots having about an 18 nm diameter.[68] They assessed the viability of cells incubated with increasing amounts of PFBT Pdots using Cell Titer Blue, a dye that tracks cell viability and proliferation. The percentage of live J774A1 cells after an 18 hour incubation with Pdots was indistinguishable from the control at all concentrations tested, thus indicating that the Pdots did not have any discernible impact on cell viability and growth. In addition, images of nanoparticle-loaded cells showed a normal morphology without any disruption to the actin microfilaments. These results indicated that Pdots did not have significant cytotoxic effects on this cell line at maximal working concentrations. To explore the possibility of Pdotinduced inflammatory response, the authors monitored the expression of the proinflammatory cytokines, tumor necrosis factor α (TNF-α), and interleukin-1β (IL-1β), in Pdotloaded J774A1 cells at the mRNA level. As shown in Figure 8A, the levels of mRNA for TNF-α and IL-1β in cells that were incubated with Pdots were identical to those that were not treated with Pdots. This result suggested that Pdots did not have an inflammatory effect on this cell type.
Figure 8.
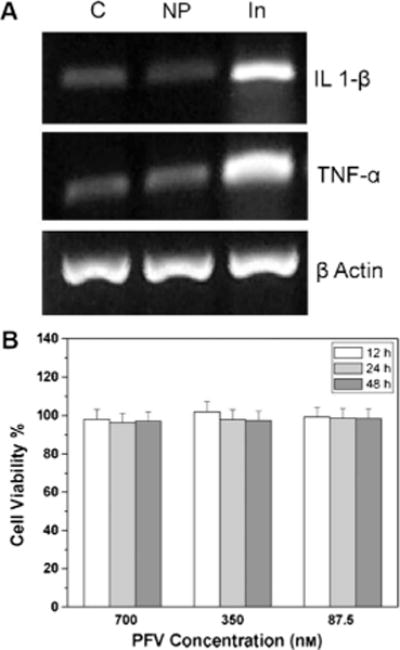
Cellular toxicity studies of Pdots. A) Monitoring of the inflammatory markers in cells treated with Pdots. J774A.1 cells were incubated with 2 nM (4 ppm) PFBT Pdots for 4 h. RNA was extracted from each sample, the total RNA was analyzed for the expression of TNF-α, IL-1β, and actin by RT-PCR, and the amplified product was separated by agarose electrophoresis. Three independent observations are represented: C: vehicle control; NP: 2 nM (4 ppm) PFBT Pdots; and In: interferon-γ (60 ng mL−1) + LPS (100 ng mL−1) as a positive control. Reproduced from Ref. [68] with permission. B) Metabolic viability of NIH/3T3 fibroblast cells after incubation with large semiconducting polymer particles at different concentrations for 12, 24, and 48 h. Reproduced from Ref. [94] with permission.
Moon and co-workers investigated the cytotoxic effects of nanoparticles (ca. 97 nm) made from a hydrophilic PPE polymer derivative.[82] BHK cells were incubated with the nanoparticles at varying amounts. Live cells were quantified at varying times using the CellTiter-Glo assay kit, which measured ATP as an indicator of metabolically active cells. Their results showed minimal inhibition of viability even at 264 μM of nanoparticles over the course of a one-week cell culture. Liu and co-workers investigated the cytotoxicity of large semiconducting polymer particles (ca. 220 nm diameter) and PLGA particles loaded with the semiconducting polymer.[94] The metabolic viability of NIH/3T3 cells did not change after incubation with these large semiconducting polymer particle suspensions, even at 700 nM for two days (Figure 8B). The nontoxic feature of semiconducting-polymer-based nanoparticles is consistent with previous studies where conjugated polymers (such as polyaniline, polypyrrole, and polythiophene derivatives) were used as electroactive biomaterials in tissue engineering applications.[149]
4. Functionalization and Bioconjugation
4.1. Encapsulation Methods
As discussed in Section 3, Pdots exhibit excellent characteristics as fluorescent probes. However, for widespread biological applications, controlling the surface chemistry and bioconjugating the Pdots are the first steps that need to be addressed. Analogous to the functionalization of Qdots, approaches based on encapsulation or embedding were initially used to produce nanoparticles having functional groups on the surface. To form nanoparticles, semiconducting polymers have been encapsulated by or embedded in materials such as silica,[46] phospholipid,[98,99] and PLGA polymers.[94,95]
Silica encapsulation is a widely used strategy to facilitate surface functionalization of nanoparticles.[16,20,150] This approach has been used to form Pdots embedded in silica with a particle size of about 10–20 nm and where a 2–3 nm thick silica shell was formed to enclose the Pdot. Besides encapsulation in silica, semiconducting polymers have also been embedded inside large PLGA particles.[94,95] The hydrodynamic diameters of the PLGA particles loaded with semiconducting polymers in water were in the range of 240 to 270 nm, as measured by dynamic light scattering (DLS). However, the fluorophore concentrations in these PLGA particles tended to be very low. As an alternative strategy, Green and co-workers encapsulated hydrophobic semiconducting polymers in phospholipid micelles.[98] The encapsulated nanoparticles showed an average diameter of 80–100 nm in water. Introducing lipid-PEG-COOH during the encapsulation process resulted in carboxylic acid functionalized particles which could be conjugated with BSA. On the basis of this strategy, the same group also encapsulated both semiconducting polymers and iron oxide nanoparticles inside phospholipid micelles.[99] Magnetic resonance imaging (MRI) was performed with the particle suspensions to demonstrate their MRI activity and prove their effectiveness as potential bimodal imaging probes.
While the above encapsulation or embedding methods showed moderate success for functionalizing the surface of the semiconducting polymer nanoparticle, they have limitations in terms of particle size and per-particle brightness. The silica embedding method can yield relatively small particle sizes, but the silica shell (2–3 nm) may suffer from hydrolysis in biological environments, and the amine functionalized silica surface also caused significant nonspecific absorption among the nanoparticles and to cellular surfaces. For the PLGA-encapsulated particles (230–270 nm) and phospholipid-encapsulated particles (80–100 nm), the particle sizes were too large for many cellular and subcellular targeting applications. In addition, the concentrations of fluorescent polymers relative to the encapsulation materials were very low in the precursor mixture at about 1% for the PLGA encapsulation,[94,95] and about 8.5% for the phospholipid encapsulation.[98] These percentages indicated relatively low fluorophore loading concentration in the final nanoparticles, and were similar to or lower than those of fluorescent-dye-loaded latex spheres. Therefore, these encapsulation strategies do not fully take advantage of the optical properties of semiconducting polymers to form nanoparticles with densely packed polymer fluorophores. As a result, the per-particle fluorescence brightness is limited because of low fluorophore loading concentrations, although in some cases, the large particle size may compensate for the brightness.
4.2. Amphiphilic Polymer Coprecipitation Methods
A reliable functionalization and conjugation method is highly desirable for preparing small, bright Pdot probes which specifically recognize and label cellular targets. Our group developed several methods that successfully addressed the challenge of Pdot bioconjugation and specific cellular targeting.[56–58,63–66] The first strategy is based on trapping heterogeneous polymer chains inside a single Pdot during nanoparticle formation. A small amount of amphiphilic polymers bearing functional groups are coprecipitated with the semiconducting polymers during Pdot preparation to modify and functionalize the nanoparticle surface. Subsequent conjugation covalently links Pdots to biomolecules which label cellular targets by specific antigen–antibody or biotin–streptavidin interactions. This functionalization and bioconjugation strategy can be easily applied to any fluorescent semiconducting polymer.
As an example, an amphiphilic comblike polystyrene polymer, PS-PEG-COOH, was used to functionalize the highly fluorescent PFBT Pdots (Figure 9A).[56] Functionalized Pdots were prepared using a precursor solution mixture with a constant PFBT concentration and PS-PEG-COOH/PFBT fractions ranging from 0 to 20 weight percent. The size and morphology of the functionalized PFBT Pdots were characterized by TEM (Figure 9B) and DLS (Figure 9C). Both measurements showed comparable particle sizes with an average diameter of about 15 nm. In terms of fluorophore density, this functionalization strategy produced good quality nanoparticle probes where more than 80 percent of the Pdots were effective fluorophores. In contrast, for Qdots and dye-loaded spheres, the effective fluorophore concentrations are limited to a few percent of the particle volume or weight because of the presence of a thick encapsulation shell (for Qdots) or self-quenching of dyes (for dye-doped spheres).
Figure 9.
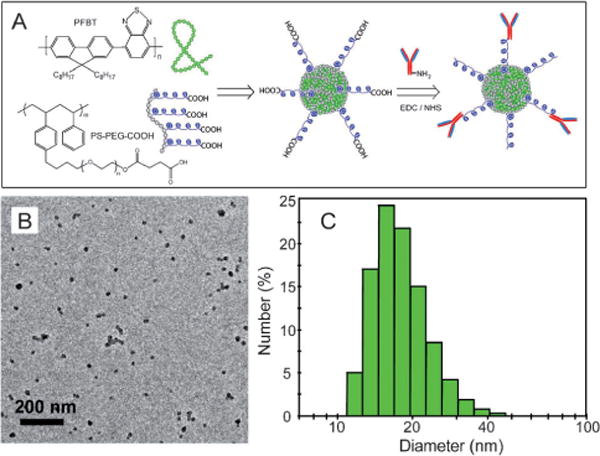
Functionalized Pdots for biomolecular conjugation. A) Surface functionalization of PFBT Pdots with the amphiphilic polymer PS-PEG-COOH. B) TEM image of functionalized PFBT Pdots. C) Hydrodynamic diameter of functionalized PFBT Pdots measured by DLS. Reproduced from Ref. [56] with permission.
In another study, the fluorescent PFBT polymer was coprecipitated with a small amount of poly(styrene-co-maleic anhydride) (PSMA) polymer and the resulting Pdots were also successfully functionalized on the surface.[57] The carboxy-functionalized Pdots were further reacted with small amine-containing molecules such as amino azides and amino alkynes to form clickable Pdots for bioorthogonal labeling using click chemistry (Figure 10A). DLS and TEM measurements showed that both the carboxy Pdots and Pdots conjugated with small molecules had comparable particle sizes, with an average diameter of about 15 nm. However, they exhibited shifted migration bands, as indicated by gel electrophoresis (Figure 10B), because of the reduced charges of the Pdot conjugates compared to the carboxy-functionalized Pdots. A fluorescence assay using an alkyne-functionalized Alexa 594 dye was further performed to verify the presence of surface azide groups, thus indicating the excellent reactivity of azide-functionalized Pdots towards a terminal alkyne group by a copper (I)-catalyzed click reaction (Figure 10C). All these results clearly indicated successful carboxy functionalization of the Pdots as well as all subsequent surface modifications.
Figure 10.
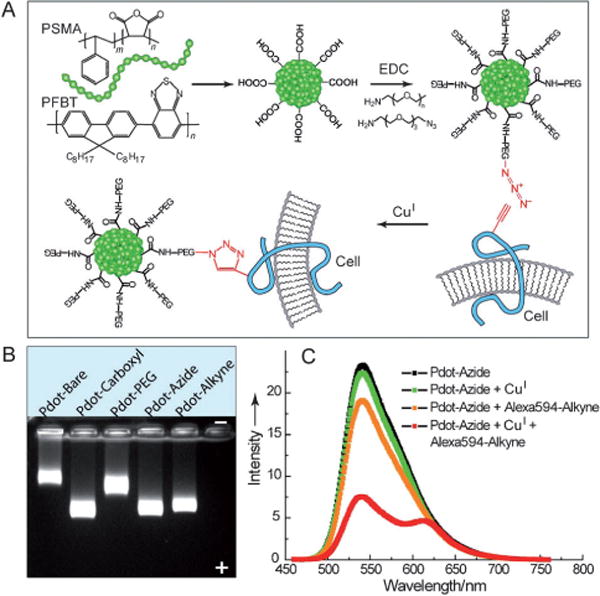
Clickable Pdots for bioorthogonal labeling. A) Pdot functionalization and subsequent bioorthogonal labeling through click chemistry. B) Gel electrophoresis of Pdots with different surface functional groups. C) A fluorescent assay using an alkyne-Alexa 594 dye to verify the presence of azide groups on the Pdot surface. Reproduced from Ref. [57] with permission.
4.3. Direct Functionalization Methods
In the encapsulation methods and the amphiphilic polymer coprecipitation scheme, bioconjugation was achieved by reacting the surface functional groups of the Pdots with biomolecules. The resulting Pdot bioconjugates were successfully used for cellular tagging,[56] bioorthogonal labeling,[57] and in vivo tumor targeting.[58] However, the functional molecules in the above schemes were attached to the Pdots primarily through noncovalent hydrophobic association. This reliance on hydrophobic association and lack of covalent attachment can be a significant drawback because the functional molecules may fall off or dissociate from the nanoparticles as a result of swelling or internal reorganization of the polymer backbone, and this in turn reduces the labeling efficiency and sensitivity in cellular assays. In addition, it is quite difficult to precisely control the density of functional groups on the surface of the Pdots by these surface modification methods.
To overcome these drawbacks, our group demonstrated a synthetic approach for covalently attaching functional groups in the initial polymer synthesis.[65] Pdots formed from these polymers can have functional groups directly available for bioconjugation, thus avoiding the need for additional surface modification procedures. We note that chemical modifications of semiconducting polymers with hydrophilic groups have been widely used to impart water solubility and functionalization to conjugated polyelectrolytes and amphiphilic conjugated polymers. Conjugated polyelectrolytes and hydrophilic conjugated polymers have been prepared in nanoparticle form with functional groups.[33,82,85] In addition, amphiphilic conjugated oligomers prefunctionalized with azide groups and biological ligands, such as mannose, can also self-assemble into nanoparticles which selectively bind proteins, bacteria, and functionalized beads.[91]
However, direct functionalization for forming hydrophobic semiconducting polymers into Pdots is distinct from these efforts. It involves very different considerations and sometimes even has opposite requirements. For example, we found that the degree of hydrophilic functionalization played a critical role in determining the stability and fluorescence performance of the final Pdots.[65] The density of hydrophilic groups must be carefully controlled at low density levels to maintain the stability and performance of Pdots for biological applications.
To investigate the effect of hydrophilic side chains on Pdot stability and performance, a series of PFBT polymers were synthesized with side-chain carboxylic acid groups at molar fractions of approximately 2%, 14%, and 50%, and were abbreviated PFBT-C2, PFBT-C14, and PFBT-C50, respectively (Figure 11). Fluorescence spectroscopy showed that the fluorescence quantum yields decreased from 30% to 17% as the density of hydrophilic functional groups increased from 2% to 50%. Single-particle fluorescence brightness comparisons revealed that the actual fluorophore packing density in PFBT-C50 Pdots was much lower than that in PFBT-C2 and PFBT-C14 Pdots, thus suggesting that the hydrophilic side chains affected the internal structures and compactness of the different Pdots. To further confirm this point, a dye-doping and leaching method was developed to examine the compactness of the fluorophore packing as well as the association strength of polymer molecules in the PFBT Pdots with different densities of hydrophilic side chains. Quantitative absorption and fluorescence analyses showed that the dye leaching and PFBT dissociation in the Pdots were strongly dependent on the density of hydrophilic side chains. For example, about 10% of PFBT and 50% of dye molecules leached out from PFBT-C50 Pdots, but no PFBTand only 6% of dye leached out from PFBT-C2 Pdots. The results, together with fluorescence resonance energy transfer (FRET) analysis, showed that PFBT-C2 Pdots had strong chain–chain associations and a densely packed internal structure through hydrophobic interactions, which is in contrast to PFBT-C50 Pdots having a high density of hydrophilic side chains. In addition, flow cytometry was performed to evaluate the role of nonspecific interactions in cellular labeling by the PFBT-C2, PFBT-C14, and PFBT-C50 Pdots. The results showed that PFBT-C50 Pdots exhibited strong nonspecific cellular labeling compared to PFBT-C2 Pdots. Based on these results, the low density of functional groups is a key factor for directly functionalizing the Pdots for biological applications. We carried out bioconjugation on the PFBT-C2 dots, and the resulting Pdot bioconjugates were specific for cellular targeting, thereby indicating that low-density functionalization was sufficient for bioconjugation and biological applications.
Figure 11.
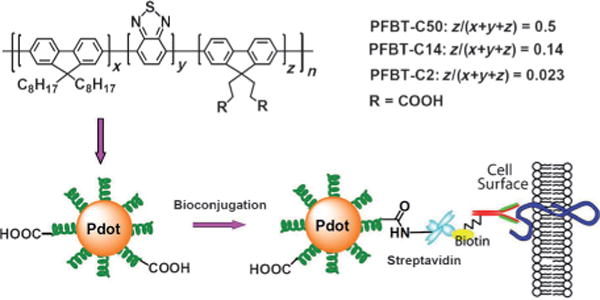
Direct functionalization of Pdots with different side-chain carboxy groups. Bioconjugation was performed on the PFBT-C2 Pdots and the resulting Pdot-bioconjugates were specific for cellular labeling. Reproduced from Ref. [65] with permission.
In a related study, our group developed a cross-linking strategy to covalently link functional polymer molecules to Pdots.[66] In this method, a PFBT polymer with side-chain amine groups was first synthesized, and a functional polymer such as poly(isobutylene-alt-maleic anhydride) (PIMA) or PSMA was used as a cross-linker. Each cross-linker molecule comprised multiple reactive units which spontaneously reacted with side-chain amine groups of the polymer to form covalent cross-links with the semiconducting polymer and simultaneously provide carboxy groups for bioconjugation. In addition to producing stable functionalized Pdots, this approach can reliably form Pdots that are 10 nm or less in particle size, which is difficult to achieve with most other methods. We successfully performed bioconjugation on these small, cross-linked Pdots, and the resulting conjugates were used to label the surface marker of breast cancer cells and the microtubules of cervical cancer cells. This application demonstrated their utility in biological imaging as well as flow cytometry experiments.
5. Biological Applications
5.1. Cellular Labeling through Endocytosis
The merits of Pdots, such as their high brightness, fast and stable emission rate, excellent photostability, and nontoxic features, are promising for experimentally demanding biological applications. In vitro cellular imaging applications of Pdots were initially conducted by a nonspecific labeling method, such as endocytosis.[51] Bare hydrophobic Pdots without any encapsulation layer appeared stable in cell culture media, and fluorescence imaging clearly indicated uptake of the bare Pdots by the cells. The high fluorescence of Pdots allowed their use at a low concentration in a cell culture. For example, Pdot fluorescence in cells could be detected with loading concentrations as low as 155 pm (270 ppb).[68] Intracellular fluorescence colocalization of bare Pdots and Texas Red dextran (TR-dex) indicated cellular uptake occurred through a common endocytic mechanism. Colocalization of bare PFBT Pdots with immunofluorescence from an anti-LAMP-1 antibody confirmed that lysosomes were the final cellular destination of bare Pdots. Inhibition of uptake by phosphoinositide 3-kinase inhibitors suggested that cellular uptake of bare Pdots occurred by constitutive macropinocytosis rather than clathrin-dependent or caveolin-dependent mechanisms.[68] Macropinocytosis is a nonspecific mechanism for sampling extracellular fluid where the contents of the vesicles are finally delivered to lysosomes. Entry of bare Pdots by this mechanism implied that the macrophage cells did not recognize the nanoparticle surface, and the bare Pdots were simply included into macropinosomes as part of the extracellular soup.
Cellular labeling by endocytosis has also been performed using nanoparticles made by encapsulating or embedding semiconducting polymers into the particle matrix. For example, PLGA particles loaded with semiconducting polymers were incubated with MCF-7 breast cancer cells, and bright fluorescence was detected in the cell cytoplasm, thus indicating efficient cellular uptake.[94] Amine-terminated folic acid was conjugated to the particles to specifically target the folate receptors which are overexpressed in cultured cancer cells. Fluorescence imaging studies showed that the internalization of folic-acid-functionalized particles in MCF-7 cancer cells was more efficient than that of the nonfunctionalized ones because of endocytosis mediated by the folate receptor.[94] In another set of experiments, the cellular uptake and cytotoxicity of several types of phospholipid-encapsulated semiconducting polymer nanoparticles, including those loaded with magnetic nanoparticles,[98,99] were studied. The data indicated the potential of Pdots as nontoxic fluorescent labels as well as multifunctional imaging probes.
In addition to nanoparticles based on hydrophobic semiconducting polymers, various conjugated polyelectrolyte nanoparticles and amphiphilic conjugated polymer nanoparticles have also been demonstrated as cellular imaging probes.[33,35,82,84,87,92,93,127,151] Cellular uptake of polyelectrolyte-based nanoparticles occured primarily through macropinocytosis or receptor-dependent endocytosis, thus the uptake was dependent upon the functional surface groups and the cell lines. It is worth noting that the molecular type (i.e. exist as solvated molecules rather than nanoparticles) of conjugated polyelectrolytes constitute a large research area within fluorescence biosensing and bioassays, where target-induced polymer aggregation through electrostatic interactions are widely used as the transduction mechanism for target detection.[32,34,35]
5.2. Immunofluorescent Labeling
Although endocytosis-based labeling showed the potential of Pdots for cellular imaging, it is intrinsically a nonspecific process and lacks the specificity of established labeling methods with organic fluorophores and Qdots, which generally involve antigen–antibody interactions. Streptavidin and IgG are widely used in bioconjugation for immunofluorescence labeling of cellular targets. Our group developed a bioconjugation method for forming Pdot-IgG and Pdot-streptavidin probes (hydrodynamic diameter ca. 10–20 nm), and tested their ability to label a specific cellular target, EpCAM, an epithelial cell-surface marker currently used to detect circulating tumor cells. Figure 12A shows that the Pdot-IgG probes successfully labeled EpCAM receptors on the surface of live MCF-7 human breast cancer cells after the cells were incubated with a monoclonal primary anti-EpCAM antibody. When the cells were incubated with just the Pdot-IgG and without the primary antibody, cell labeling was not detected (Figure 12B), thus indicating that the Pdot-IgG conjugates were highly specific for the target. Similarly, the Pdot-streptavidin probes, together with the primary anti-EpCAM antibody and biotinylated goat anti-mouse-IgG secondary antibody, also effectively labeled EpCAM on the surface of live MCF-7 cells, without detectable nonspecific binding. We further compared the labeling brightness of Pdot bioconjugates with those of commercially available Qdot-streptavidin and Alexa-IgG probes using a microfluidic flow cytometer (Figure 12C and D). Quantitative analyses of the flow cytometry data showed that the average intensity of Pdot-labeled cells was approximately 25 times brighter than that of the Qdot-labeled ones, and about 18 times brighter than that of the Alexa-IgG-labeled cells. The labeling brightness was also quantified by analyzing fluorescence images of MCF-7 cells labeled with either Pdot-streptavidin or Qdot-streptavidin: The Pdot-labeled cells were approximately 20 times brighter than the Qdot-labeled ones, and is consistent with the flow cytometry data. These results clearly established the extraordinary fluorescence brightness of Pdots and their potential for cellular imaging and biological assays.
Figure 12.
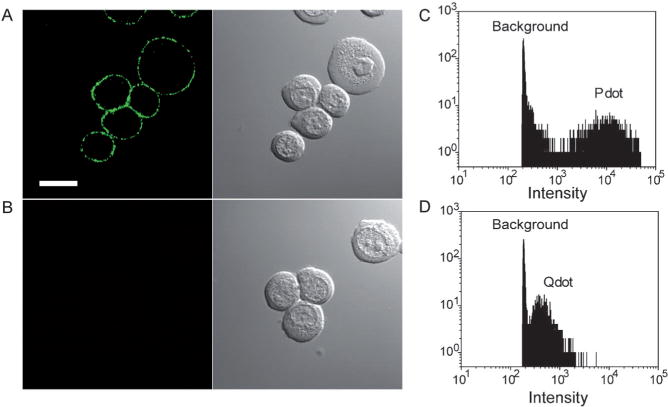
Specific cellular targeting with Pdot-bioconjugates. A) Fluorescence imaging of the cell-surface marker EpCAM on MCF-7 cells incubated sequentially with the anti-EpCAM primary antibody and Pdot-IgG conjugates. Scale bar: 10 μm. B) Fluorescence imaging of the control sample in which the cells were incubated with Pdot-IgG alone (no primary antibody). C) Fluorescence intensity distributions for Pdot-streptavidin-labeled MCF-7 cells. D) Fluorescence intensity distributions for Qdot 565-streptavidin-labeled MCF-7 cells obtained under identical experimental conditions as those used in C). Reproduced from Ref. [56] with permission.
More recently, our group reported CN-PPV Pdots which had small hydrodynamic diameters of about 10 nm and bright orange fluorescence with a quantum yield of approximately 60%.[63] Successful functionalization and bioconjugation produced CN-PPV Pdot-streptavidin probes which specifically labeled cell-surface markers and microtubule structures inside mammalian cells (Figure 13). When flow cytometry was performed to examine the specific labeling of a cell surface marker by the Pdot-streptavidin probes, the labeled cells exhibited much stronger fluorescence compared to the control samples, thus indicating the brightness and specificity of the Pdot probes. For microtubule labeling, HeLa cells were fixed, permeabilized, and then incubated with biotinylated monoclonal anti-α-tubulin antibody. The HeLa cells incubated with Pdot-streptavidin probes in the presence or absence of biotinylated antibody were imaged by confocal microcopy. The positively labeled cells showed bright fluorescence from well-resolved tubular structures, without detectable nonspecific binding in the control experiment. Under the same labeling conditions, similar high quality images of subcellular structures were not observed with the larger PFBT-streptavidin probes. The reason was attributed to the compact and small size (ca. 10 nm) of CN-PPV Pdots which exhibited very low nonspecific binding in the crowded subcellular environment when compared to the larger PFBT dots (ca. 20 nm). These results indicate compact and highly fluorescent Pdots are promising probes for subcellular imaging and bioanalytical assays.
Figure 13.
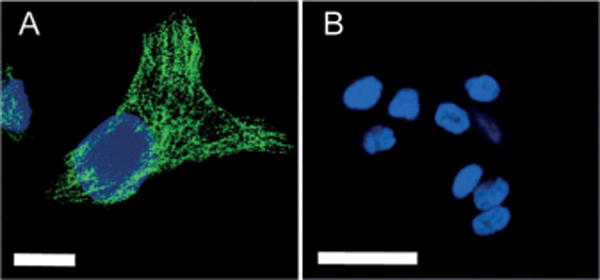
Specific subcellular imaging of Pdot-bioconjugates. A) Fluorescence imaging of microtubule structures in HeLa cells incubated sequentially with biotinylated anti-α-tubulin antibody and CN-PPV-streptavidin probes. Scale bar: 10 μm. B) Fluorescence imaging of the control sample in which the cells were incubated with Pdot-streptavidin alone (no primary antibody). Reproduced from Ref. [63] with permission.
Large particles with embedded semiconducting polymers have also been successfully used for immunofluorescence labeling of cells. For example, Liu and co-workers described PLGA-encapsulated semiconducting polymer particles (230–260 nm) which were functionalized with amine groups by varying the molar ratios of PLGA-PEG-NH2 and PLGA-OCH3.[95] Additional conjugation of the amine-functionalized particles with the antibody trastuzumab produced bioconjugated nanoparticles which could discriminate SK-BR-3 breast cancer cells from MCF-7 breast cancer cells and NIH/3T3 fibroblast cells by taking advantage of the specific binding affinity between trastuzumab and the HER2 receptor overexpressed on the SK-BR-3 breast cancer cell membrane.
5.3. Bioorthogonal Labeling by Click Chemistry
The most recognizable click reaction is the copper(I)-catalyzed azide–alkyne cycloaddition, which has been applied to research fields as diverse as materials science and chemical biology.[152–156] For biological applications, both azide and alkyne groups are considered to be bioorthogonal chemical reporters because they do not interact with any native biological functional groups. These bioorthogonal reporters can be incorporated into a target biomolecule using the biosynthetic machinery of the cell. These reporters provide chemical handles which can be subsequently tagged with exogenous probes. To demonstrate cellular labeling with Pdots and click chemistry, we visualized newly synthesized proteins which were modified by bioorthogonal noncanonical amino-acid tagging.[57] In this process, newly synthesized proteins in cells were metabolically labeled with an azide- (or alkyne-) bearing artificial amino acid, such as azidohomoalanine (AHA) or homopropargylglycine (HPG). The proteins were then amenable to tagging with fluorescent Pdots by highly selective click chemistry. Confocal imaging showed bright fluorescence for the AHA-labeled cells tagged with Pdot-alkyne by a click reaction (Figure 14). In the negative control experiment, cellular labeling was not observed, thus indicating that Pdot-alkyne tagging was highly specific for the cellular targets.
Figure 14.
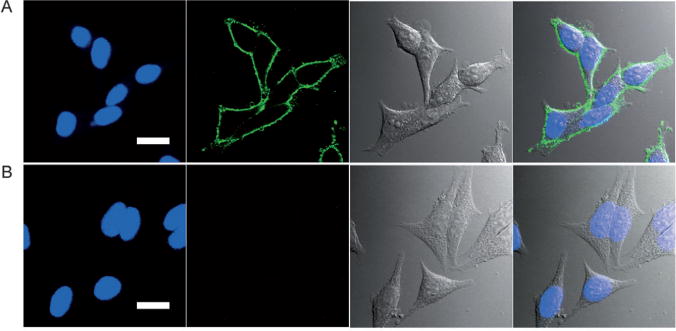
Fluorescence imaging of newly synthesized proteins in the AHA-treated MCF-7 cells tagged with Pdot-alkyne probes. A) Positive Pdot labeling in the presence of copper(I). B) Negative control for Pdot labeling carried out under identical conditions as in A) but in the absence of the reducing agent, sodium ascorbate, which generates copper(I) from copper(II). The left four panels show fluorescence images; green fluorescence was from Pdots and blue fluorescence was from the nuclear stain Hoechst 34580. The right four panels show Nomarski (DIC) and combined DIC and fluorescence images. Scale bar: 20 μm. Reproduced from Ref. [57] with permission.
The clickable Pdots were also used to selectively target glycoproteins.[57] The method involved metabolic labeling of glycans with a monosaccharide precursor which was functionalized with an azide group.[157] The azide-containing sugars were covalently tagged with imaging probes. MCF-7 cells were incubated with N-azidoacetylgalactosamine (GalNAz) for three days to enrich O-linked glycoproteins with the azido groups. The GalNAz-treated cells were tagged with Pdot-alkyne by a click reaction and subsequently viewed under a confocal microscope. Bright cellular labeling was observed for the cells positively tagged with Pdot-alkyne. Fluorescence was not observed in the negative control where cells lacked the azide groups, thus indicating that the clickable Pdots were highly specific for the glycoproteins. Notably, the very low Pdot concentrations of about 50 nM that were applied for the labeling studies were orders of magnitude less than the general concentrations used for small dye molecules (typically in the μm range). Pdot tagging using click chemistry was also highly specific for the targets, with virtually no background labeling.
5.4. In Vivo Imaging
Fluorescence imaging in vivo is mainly used for small animal studies and not generally translated into the clinic because of photon-limiting interference such as scattering, absorption, and autofluorescence, which occurs in biological media. These limitations are expected to be overcome to a great extent if a breakthrough in signal intensity is achieved by using exogenous near-infrared (NIR) fluorescent probes which are orders of magnitude brighter than the current organic dyes. In this context, semiconducting polymer nanoparticles hold great potential because of their high brightness and nontoxicity. Kim and co-workers demonstrated the successful in vivo application of semiconducting polymer nanoparticles to sentinel lymph node mapping.[72] Cyano-substituted derivatives of poly(p-phenylenevinylene) were designed to produce multicolor fluorescent nanoparticles (Figure 15), which were directly synthesized by in situ colloidal polymerization in the aqueous phase. One type of nanoparticle (NIR-cvPDs) showed polydispersed size distributions (60 ± 14 nm), a broad absorption band, and a red-to-NIR fluorescence emission peaking at λ = 693 nm with a quantum yield of 21%. The potential of NIR-cvPDs for in vivo imaging was explored for sentinel lymph node mapping in a mouse model. After the NIR-cvPDs were injected intradermally into the forepaw pad of a mouse, the nanoparticles drained rapidly from the interstitial site of injection into the lymphatic system, and finally accumulated at the regional lymph node without any sign of outflow toward the next tier nodes. The result was clear-cut imaging of sentinel lymph nodes. Biodistribution studies indicated that the lymphatically drained nanoparticles were efficiently trapped in the sentinel lymph nodes without entering the subsequent lymphatic and blood circulations.
Figure 15.
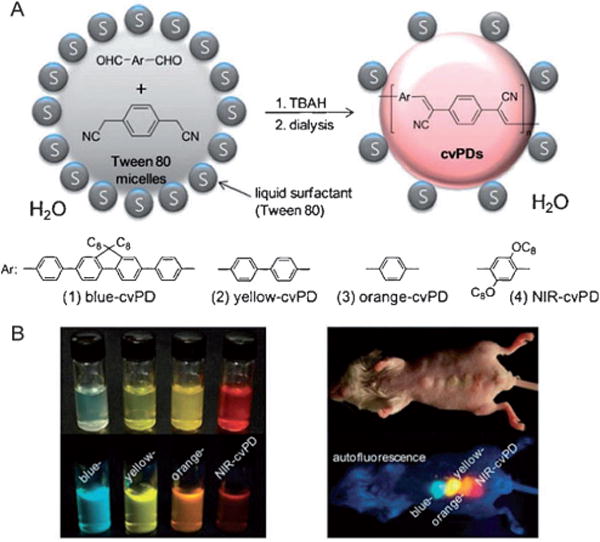
Semiconducting polymer nanoparticles for in vivo fluorescence imaging. A) Schematic diagram depicting colloidal synthesis of nanoparticles with a cyanovinylene backbone by a tetrabutylammonium hydroxide (TBAH) catalyzed Knoevenagel condensation in the hydrophobic core of solvent-free aqueous micelles. “C8” stands for n-octyl chains. B) True-color photographs of water-dispersed polymer nanoparticles (left) and a nanoparticle-injected live mouse (right) under room light (top) and UV excitation at λ = 365 nm for fluorescence (bottom). Reproduced from Ref. [72] with permission.
Delivering imaging probes to brain tumors represents one of the most challenging in vivo tasks because of the blood-brain barrier and the complex dependence on the probe size and surface properties.[158] We developed Pdot bioconjugates which consisted of optimally tailored semiconducting polymer blends for in vivo tumor targeting.[58] The Pdots of having a diameter of about 15 nm showed large absorptivity (3.0 × 107 cm−1M−1) and efficient deep-red emission (quantum yield = 0.56), thus making them aproxiamtely 15 times brighter than the Qdots emitting at λ = 655 nm. Successful Pdot functionalization produced a biocompatible surface which could be covalently conjugated to a tumor-specific peptide ligand, chlorotoxin (CTX). We evaluated the capability of the Pdot-CTX conjugate to traverse the blood-brain barrier and specifically target a tumor in a transgenic mouse model ND2:SmoA1. Pdot probes were injected into either symptomatic ND2:SmoA1 or wild-type control mice through the tail vein. The Pdot probes were either Pdot-CTX or Pdot-PEG as a control. Strong fluorescence signals were observed only in the brain tumor regions of the ND2:SmoA1 mice that received the targeting probes (Figure 16). Pdot fluorescence was not obvious in the brains of the healthy animals, thus indicating a preferential accumulation of the Pdot-CTX in ND2:SmoA1 tumors. Quantitative evaluation of Pdot accumulation and histological analysis correlated well with the biophotonic images, thus confirming the selective accumulation of the Pdot-CTX probes in the malignant brain tumors. Nanoparticle clearance and biodistribution are closely dependent on particle size.[159,160] The distribution profiles of the Pdots in the main clearance organs—liver, spleen, and kidney—were analyzed. As expected based on particle size,[160] the Pdot-CTX exhibited dominant uptake in liver, significantly lower signal in the spleen, and nearly no distribution in the kidney for both wild-type and ND2:SmoA1 mice. This distribution profile is comparable to those reported for inorganic iron oxide nanoparticles and Qdots of similar particle size.[158,160] This study represents the first demonstration of targeted delivery to diseased tissues in small animals. It suggests a great potential of Pdot-based probes for in vivo applications, such as clinical cancer diagnostics and therapeutics.
Figure 16.
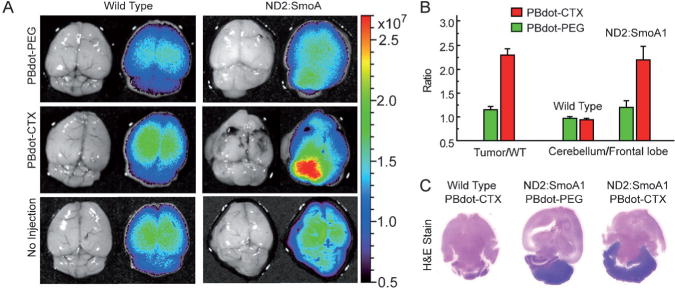
Pdot-chlorotoxin bioconjugates for in vivo tumor targeting. A) Fluorescence imaging of healthy brains in wild-type mice (left) and medulloblastoma tumors in ND2:SmoA1 mice (right). Each mouse was injected through the tail vein with 50 μL of a1 μM solution of either the nontargeting PBdot-PEG (top), or targeting PBdot-CTX (middle). As a control, some mice did not receive an injection (bottom). B) Tumor-targeting efficiency by quantifying fluorescence signals in ND2:SmoA1 versus wild-type mice and cerebellum versus frontal lobe. C) Histological examination of the mouse brains in A). The dark purple regions in the H&E-stained cerebellum of ND2:SmoA1 mice confirmed the presence of tumor. Reproduced from Ref. [58] with permission.
5.5. Single-Particle Tracking
Nanoscale two-dimensional (2D) and three-dimensional (3D) single-particle tracking methods have proven to be extraordinarily useful for investigating a wide variety of cellular processes, such as molecular transport, membrane dynamics, and the motion of motor proteins.[5,7] McNeill and co-workers evaluated the use of Pdots for nanoscale 2D and 3D tracking applications.[53] To experimentally determine the per-particle brightness for particle tracking, single Pdot particles were imaged with a CCD-equipped, inverted fluorescence microscope at an acquisition rate of 50 Hz. The detected photons per particle per image typically ranged between 1 × 106 and 1 × 105 for several hundred consecutive images. Analysis of 20 typical single-particle fluorescence intensity trajectories yielded an average of about 109 total photons emitted per particle, with some particles emitting more than 1010 photons. Based on the signal levels of isolated nanoparticles, a theoretical tracking uncertainty of better than 1 nm was estimated. Intracellular particle tracking was also demonstrated, and particle trajectories were found to be consistent with expected phenomena, such as partially confined diffusion and reversible and irreversible binding to cellular components (Figure 17). The tracking results indicate that Pdots are promising for measuring local diffusivity and nanoscale motion of individual biomolecules and subcellular structures in cells.
Figure 17.
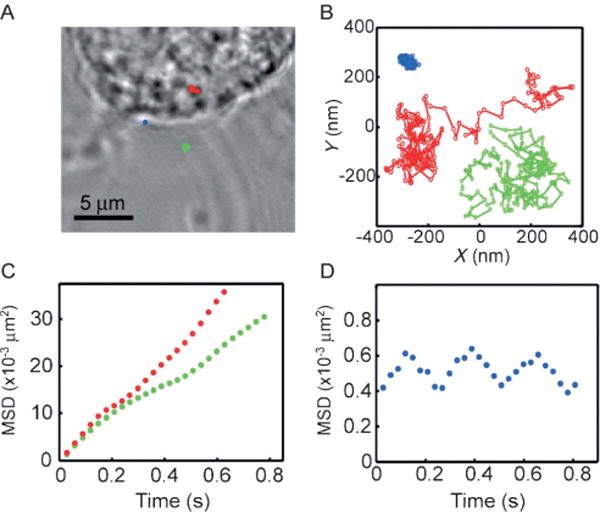
Single-particle fluorescence tracking with highly fluorescent Pdots. A) Bright-field transmission image of a fixed cell. The color marks indicate the locations of the particles. Blue corresponds to a particle bound to the membrane, green corresponds to a particle outside the cell, and red corresponds to the cell interior. B) The trajectories for the three particles obtained from fluorescence tracking. C) Mean-square displacement (MSD) of the tracked PFBT Pdots inside a cell (red circles) and outside a cell (green circles), corresponding to diffusion constants of 3.3 × 10−3 μm2s−1 and 3.6 × 10−3 μm2s−1, respectively. D) Mean-square displacement of the Pdot adhered to the membrane. Reproduced from Ref. [53] with permission.
In a related but different study, McNeill and co-workers demonstrated the tracking of individual charge carriers in Pdots by video rate fluorescence microscopy.[54] Fluctuations in the fluorescence intensity and nanometer-scale displacements in the centroid of single Pdots were observed when stationary nanoparticles were continuously imaged. The fluctuations and displacements were attributed to the hopping motion of hole polarons, which effectively quenched the fluorescence in the vicinity of the polaron. Analysis of the fluctuations in the fluorescence centroid gave a value of roughly 10−9 cm2V−1s for the zero-field hole polaron mobility. This value is one or two orders of magnitude lower than the reported values obtained by I/V analysis. However, it is consistent with the highly trapped charge carriers, because the nanoparticles would have additional conformational disorder, chemical defects, and energetic disorder compared to optimally prepared thin films. This study demonstrated that high-speed fluorescence tracking can be used to measure nanometer-scale motion and mobility of a single-charge carrier within an organic semiconductor nanostructure.
5.6. Energy-Transfer-Based Pdot Sensors
There is a growing interest in using FRET for sensing applications. FRET-based techniques have been widely demonstrated for studying biomolecular conformations and short-range interactions between molecules.[161–164] Conjugated polyelectrolytes have been extensively demonstrated as very sensitive biosensors based on an extraordinarily efficient energy transfer which is characterized as super-quenching.[27,29,32] Our discussion here focuses on a novel platform that exhibits efficient energy transfer inside the hydrophobic Pdot nanoparticles because of the densely packed structure and high-volume fraction of fluorophore polymers. The extraordinary light-gathering capability of semiconducting polymers and efficient intraparticle energy transfer has been demonstrated to enhance the per-particle fluorescence brightness, tune the emission color, and improve photostability of the Pdots. In particular, new sensing functions can be created when certain molecules with sensing functions are doped into the Pdots. The intraparticle energy transfer from the polymer donor to the dye acceptor results in emissions which are sensitive to certain analytes or responsive to environmental changes.[52,59,60,77]
Hydrophobic molecular species were initially selected for dye doping because they can be easily incorporated into the hydrophobic Pdots by hydrophobic interactions. The first example of Pdot sensor was the development of Pdots doped with the phosphorescent dye platinum octaethylporphyrin (PtOEP) for oxygen sensing.[52] The PtOEP-doped Pdots exhibited efficient intraparticle energy transfer from the polymer host to the phosphorescent dye, thus resulting in bright phosphorescence which was highly sensitive to the concentration of molecular oxygen (Figure 18A).
Figure 18.
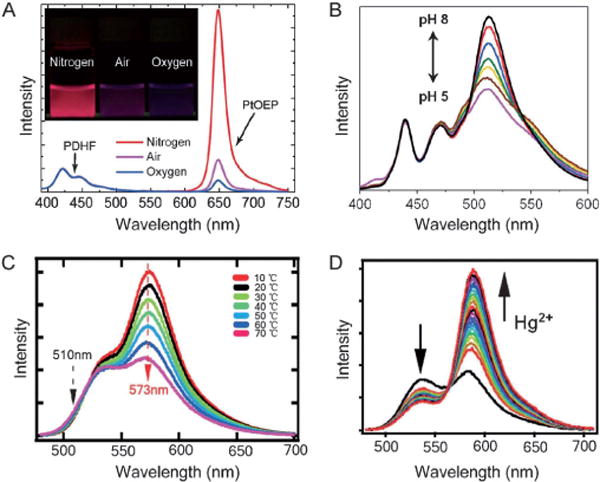
Energy-transfer-based Pdot sensors. A) Oxygen-dependent emission spectra of the PtOEP-doped PDHF Pdots. The inset shows UV illuminated photograph of the aqueous Pdot solutions saturated with nitrogen, air, and oxygen, respectively. Reproduced from Ref. [52] with permission. B) Fluorescence spectra of fluorescein-conjugated PPE Pdots at different pH values ranging from 5 to 8. Reproduced from Ref. [60] with permission. C) Fluorescence spectra of RhB-conjugated PFBT Pdots at different temperatures from 10 °C to 70 °C. Reproduced from Ref. [59] with permission. D) Fluorescence spectra of RhB-SL-doped PFBT Pdots in aqueous suspension as mercury ion concentration was increased from 0 to 0.57 μM. Reproduced from Ref. [77] with permission.
By using the successful Pdot functionalization strategies we developed, it was possible to employ a variety of fluorescent dyes for developing Pdot-based sensors.[59,60] Our group developed a pH-sensitive Pdot probe with the semiconducting polymer PPE and the pH-sensitive dye fluorescein.[60] The hydrophilic fluorescein dye was covalently coupled to the Pdot surface, and energy transfer from the polymer donor to the dye acceptor acted as a ratiometric platform for pH sensing. Intracellular pH sensing was demonstrated inside HeLa cells after the cells took up the Pdots through endocytosis. The linear pH range for the Pdot senor is between pH 5.0 and 8.0 (Figure 18B), which is suitable for most cellular studies.
In a later study, temperature-sensitive Pdots were constructed based on the semiconducting polymers PFBT and PFPV and the temperature-sensitive dye Rhodamine B (RhB).[59] To uniformly distribute the hydrophilic RhB dye into the Pdots, the small dye molecules were covalently linked to a hydrophobic polystyrene species, and then blended with the hydrophobic semiconducting polymers. Like the pH sensors, the Pdots exhibited efficient energy transfer from the polymer matrix to the dye acceptors, thus resulting in ratiometric temperature sensing. These Pdot sensors showed a linear fluorescence response in the range from 10 °C to 70 °C (Figure 18C). Efficient cellular uptake of the Pdots was observed in HeLa cells, and subsequent cellular imaging and temperature determinations were consistent with the results measured by a thermocouple, thus indicating their potential as a sensitive intracellular temperature sensor.
Nanoparticle-based ion sensors have attracted great interest because of their simplicity, selectivity, high sensitivity, and reliability.[165,166] Pdot-based ion sensors have been developed to detect biologically or environmentally relevant ions, such as Cu2+, Fe2+, and Hg2+. In a simple platform, carboxy-functionalized Pdots coordinated directly with Cu2+ or Fe2+, and thus caused fluorescence quenching because the Pdots aggregated.[62] The Cu2+-induced aggregation could be reversed by adding ethylenediaminetetraacetic acid (EDTA) to the solution, and the Fe2+-induced aggregation was not reversible. The selective fluorescence recovery allowed the sensors to differentiate between copper and iron ions and to determine their individual concentrations. In a recent study, a fluorescent Hg2+ sensor was developed on the basis of the dye-doping strategy.[77] The Pdot-based Hg2+ sensor was constructed from PFBT Pdots which were doped with a molecular dye rhodamine spirolactam (RhB-SL). The dye molecules were nonfluorescent until they were exposed to mercury ions, thus promoting an irreversible reaction and converting the nonfluorescent dye into fluorescent rhodamine. The fluorescence intensity ratio of the dye acceptor to the PFBT donor allowed ratiometric sensing of mercury ions. The light harvesting capability of the Pdots enhanced the fluorescence intensity of the rhodamine dyes by a factor of 10, thereby enabling sensitive detection of mercury ions in water (Figure 18D). Collectively, the versatile Pdot-based sensing platforms demonstrate the continued promise of Pdots for chemical sensing applications.
Very recently, Swager and co-workers demonstrated a new type of nanoparticle sensor based on energy transfer for sensing biomolecules, such as protease.[74] The nanoparticles were based on a highly fluorescent PPE polymer consisting of a red emitting perylene unit inserted randomly into the PPE backbone and a terminal unit bearing reactive succinimide groups. Taking advantage of the energy-transfer-based fluorescence amplification, the nanoparticles made from this polymer showed bright dual-peak fluorescence from both the PPE polymer and the perylene unit. Both emission peaks could be further quenched upon cross-linking the shell of nanoparticles (off state) by reaction of the succinimide groups with a short peptide. The cross-linking effectively strained the nanoparticles into a more tightly aggregated and quenched state. In response to the protease, the peptide cross-linkers were cleaved and the strain was released, thus resulting in a “turn-on” fluorescence response. Sensing assays under physiological conditions provided a 15-fold increase of the fluorescence intensity after incubation with the protease (Figure 19).[74]
Figure 19.
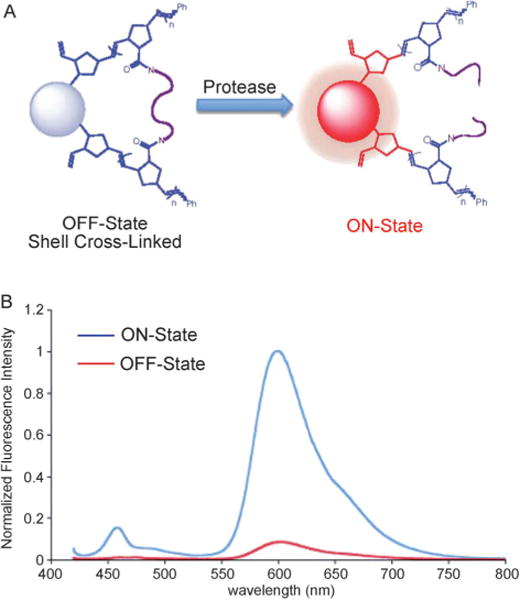
Semiconducting polymer nanoparticles for protease sensing. A) Protease-triggered “turn on” sensing scheme. The cross-linking effectively strains the nanoparticles into a more tightly aggregated and quenched state. The protease cleaves the peptide cross-linkers and releases the strain, resulting in a “turn-on” fluorescence response. B) Fluorescence spectra of shell cross-linked nanoparticle suspensions before (red) and after (blue) incubation with trypsin. Reproduced from Ref. [74] with permission.
In addition to sensing, the dye-doping strategy and energy transfer were exploited by Harbron and co-workers who developed photoswitchable Pdots doped with photochromic spirooxazine or diarylethene dyes.[75,76] In a recent study, fluorescence photoswitching of the diarylethene-doped PFBT nanoparticles was demonstrated in a cellular environment.[78] Photoswitchable probes are particularly attractive because they can potentially be used in super-resolution imaging techniques to resolve cellular structures which are much smaller than diffraction-limited spot sizes.[9,11] However, current photoswitchable Pdots generally do not exhibit the binary on/off switching needed for most schemes employed in super-resolution imaging.
5.7. Drug and Gene Delivery
Intense efforts in nanomedicine aim to use nanoparticles to specifically deliver drugs, heat, or other substances to diseased tissues.[167,168] With their flexible polymer matrix, conjugated polymer nanoparticles provide a suitable system for encapsulating drugs or other therapeutic agents. Hydrophobic Pdots as a delivery system can also overcome drug solubility issues. This advantage has significant implications because more than 40% of active substances identified through combinatorial screening programs are poorly soluble in water.[169] These drugs potentially can be doped or encapsulated inside Pdots to be delivered and released at diseased tissues. To date, there is little work in using Pdots for drug delivery. However, nanoparticles made from amphiphilic semiconducting polymers and polyelectrolytes have been explored as versatile systems for drug and gene delivery. For example, Wang and co-workers prepared conjugated polymer nanoparticles (ca. 50 nm diameter) by electrostatic assembly of the cationic conjugated fluorescent polymer PFO with anionic poly(L-glutamic acid) conjugated with the anticancer drug doxorubicin (PG-Dox).[86] This nanoparticle system delivered doxorubicin to targeted cancer cells, and because doxorubicin quenches PFO fluorescence the drug release was monitored by the fluorescence “turn-on” signal from PFO. The same group also employed a cationic poly(p-phenylene vinylene) polyelectrolyte in a cell culture study and it selectively bound and damaged bacterial cells.[170] The highly negative charge density of the bacterial cell surface made it possible for the cationic polymer to selectively bind bacteria but not mammalian cells. Upon light irradiation, the polyelectrolyte was excited, presumably generating ROS, and killed bacterial cells. Liu and co-workers also reported a drug delivery system based on conjugated polyelectrolyte nanoparticles loaded with the anticancer agent cisplatin.[88] These drug-loaded nanoparticles were demonstrated as in vivo fluorescent imaging probes which simultaneously tracked the slow and steady drug release in nude mice upon intravenous administration.
Amphiphilic and hydrophilic semiconducting polymer nanoparticles have also been explored for gene delivery.[92] In one study, a lipid-modified cationic poly(fluorenylene phenylene) polymer (PFPL) with ammonium pendant groups was synthesized and bound DNA through electrostatic interactions. The pCX-EGFP plasmid encoding the green fluorescent protein (GFP) was associated with the PFPL nanoparticles to form a nanoparticle/plasmid complex which was taken up by A549 cells through endocytosis. Green fluorescence from the GFP was observed (Figure 20A), thus indicating the nanoparticles successfully delivered plasmids into cells for further transcription and translation.[92] In another study, amine-containing hydrophilic PPEs were prepared into loosely aggregated particles.[85] These particles formed stable complexes with small interfering RNA (siRNA). The complexes were delivered to HeLa cells and a significant downregulation of a target gene was observed (Figure 20B). Although still at an early stage, these studies point to the potential of using semiconducting polymer nanoparticles as both imaging probes and delivery vehicles.
Figure 20.
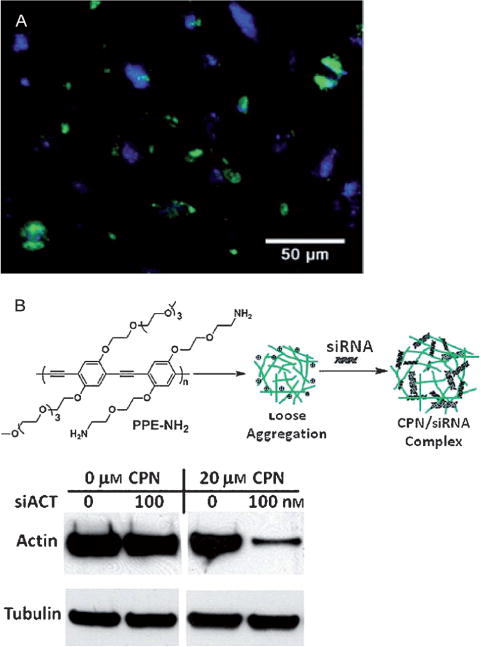
Conjugated polymer nanoparticles for gene delivery. A) Lipid-modified cationic PFPL nanoparticles for delivery of pCX-EGFP plasmids encoding the GFP. Fluorescence imaging of A549 cells after incubation with PFPL/pCX-EGFP nanoparticles showed both blue fluorescence from PFPL and green fluorescence from GFP, thus indicating the PFPL nanoparticles successfully delivered plasmids into cells. Reproduced from Ref. [92] with permission. B) Loosely aggregated PPE nanoparticles for delivery of siRNA. The top shows a schematic illustration of loosely aggregated PPE particles complexed with siRNA. The bottom shows Western blots of actin B (target) and tubulin (control). Significant reduction in the target protein was observed from the CPN/siACT transfection. Actin B expression decreased about 94% under the transfection conditions. Reproduced from Ref. [85] with permission.
Recently, there has been considerable interest in developing nanoparticle-based photosensitizing agents.[171–176] Current nanoparticle photosensitizers typically consist of photosensitizing dyes either encapsulated inside polymer/silica nanoparticles[171–174] or conjugated to a Qdot surface, and results in singlet oxygen generation by energy transfer.[175,176] These strategies were limited by low absorptivity and less efficient energy transfer. By entrapping photosensitizer drugs into Pdots, large absorptivity and highly efficient intraparticle energy transfer would yield effective nanoparticle photosensitizers. McNeill and co-workers developed a Pdot consisting of a semiconducting polymer doped with the singlet oxygen photosensitizer tetraphenylporphyrin (TPP).[49] TPP-doped Pdots have been recently shown to be promising for photodynamic therapy for both one-photon or two-photon excitations.[55,79,80] Spectroscopic studies indicated that the polymer efficiently absorbed and transferred energy to the porphyrin sensitizer, thereby resulting in highly efficient singlet oxygen generation. The quantum efficiency of singlet oxygen production was determined to be approximately 0.6–0.8, which is comparable to or higher than that of currently available photosensitizer drugs.[55] Gel electrophoresis of DNA after UV irradiation in the presence of Pdots indicated substantial DNA damage by ROS. Similar to the dye-doped Pdot sensors, the Pdot photosensitizer takes advantage of the flexible polymer matrix for encapsulation (a general feature of polymers) and the unique optical properties of Pdots (large absorptivity and efficient intra-particle energy transfer) to enhance the performance of dopant functions.
6. Current Challenges and Trends
Pdots are a new class of fluorescent nanoparticles. As is the case with all new nanoparticles, the design criteria for demanding biological applications, both in vitro and in vivo, are still being formulated. Because of the huge number of existing semiconducting polymers ranging from hydrophobic polymers to polyelectrolytes, we can envision some general design considerations for high-performance fluorescent probes and the emerging challenges. In our opinion, small and bright Pdots are extremely important for most biological imaging applications and fluorescence assays. In terms of probe performance, such as fluorescence brightness, different colored Pdots have reliably shown fluorescence quantum yields in the range of 30–60%,[56,58,63] and are comparable to those of inorganic Qdots and many organic small-molecule fluorophores. Accordingly, the absorption cross section of Pdots is the dominant factor for their fluorescence brightness. Absorption cross-section scales with the volume fraction as well as the packing density of fluorophores for a given particle size. Therefore, high fluorophore fractions and densely packed structures are highly desirable in developing bright Pdot probes. However, the preparation methods of Pdots need to be improved so that different sizes, with narrow size distributions, can be reliably produced.
For Pdot functionalization, direct functionalization seems to be a robust approach for modifying the surface of hydrophobic Pdots.[65] Hydrophilic functional groups are generally needed and can be covalently linked to the side chain or terminal units of the hydrophobic semiconducting polymers. But direct functionalization can significantly affect the per-particle brightness, internal structure, nanoparticle stability, and nonspecific labeling in biological applications. Therefore, the density of the functional groups must be carefully controlled for maximizing probe performance.[65] In addition, it is extremely difficult to control the number and geometrical distribution of chemical functional groups because of the presence of multiple reactive sites on a nanoparticle surface. In cell-based analyses, nanoparticle multivalence can cause cross-linking of cell-surface proteins involved in signal pathway activation, and thus dramatically reduce receptor binding capability. Therefore, Pdots with monofunctional groups are highly desirable for many biological applications. This challenge provides clear motivation to explore new approaches for functionalizing Pdots with a predetermined and well-controlled number of functional groups.
In terms of the spectroscopic properties of Pdots, a severe drawback is the very broad emission spectra of currently available Pdot species. For example, a widely studied Pdot species, PFBT, exhibits full width at half maximum (FWHM) at about 75 nm, which is about two times broader than those of inorganic Qdots emitting in the visible region. Most biological applications demand that multiple targets be detected simultaneously, so spectral multiplexing requires probes to possess narrow band emissions. The broad emission spectra from conventional Pdot species significantly limit their usefulness in multiplexed applications. There is an urgent need to develop new types of Pdots which can emit at different wavelengths with a narrow-band spectral width. We recently described the design and synthesis of semiconducting polymers containing boron dipyrromethene (BODIPY) units which generated multicolor, bright, and narrow-band emissive Pdots with FWHM of 40–50 nm.[67] These Pdots are promising for multiplexed applications. Most reported Pdot species exhibit emissions in the visible range, although the dye-doping method can shift the fluorescence to the nearinfrared region, such as λ = 777 nm.[61] Highly fluorescent Pdot species with both absorption and emission in the NIR region are preferable for in vivo applications because biological tissues absorb minimally at these wavelengths. These requirements provide numerous research opportunities for polymer synthetic chemists to explore new polymer species with narrow-band and/or NIR emissions.
Detailed investigations are still needed in the use of Pdots for clinical studies. In a recent paper,[160] three criteria were proposed for evaluating a nanoparticle having the potential for clinical utility: 1) a final hydrodynamic diameter (HD) of less than 5.5 nm to permit fast renal clearance and/or 2) a formulation with completely nontoxic components and/or 3) biodegradability to clearable components. More research is needed based on these three design criteria. A better method is needed to reliably obtain small Pdot size and monodispersity. Renal clearance (HD < 5.5 nm) may not be a necessity for nontoxic materials. However, the particle size does affect the biodistribution and targeting efficiency. The long-term in vivo toxicity of Pdots has to be evaluated, although Pdots appear to be biologically benign in cell culture studies. In our opinion, research in these aspects will be the key directions in developing Pdots for a variety of biological and clinical applications.
7. Summary and Outlook
Although semiconducting polymers have been investigated for over three decades, their application as fluorescent nanoparticles only prominently emerged a few years ago. Semiconductor Pdots exhibit fascinating properties as fluorescent probes, such as extraordinary brightness, fast and stable emission rates, excellent photostability, and nontoxic features. Recent investigations have revealed an underlying concept, that is, the particle size, morphology, internal structure, and functionalization methods are all critical for Pdot performance in practical biological applications. Furthermore, Pdots provide a unique platform for developing multifunctional probes which combine extraordinary fluorescence brightness and efficient intraparticle energy transfer with the flexible nontoxic polymer matrix, and allows doping with versatile sensing molecules, drugs, and other image contrast agents.
Although the advantages of Pdots over small dye molecules and inorganic Qdots are evident, it is hard to compare the three types of probes without a definite application. Small dye molecules, despite their nonoptimum brightness and photostability, are extensively used as fluorescent labels in fluorescence imaging and assays because of their well-established labeling protocols and their molecular dimensions. Pdots and Qdots behave as nanoparticles with surface properties, not as molecules, and this property complicates their interactions with biomolecules and their application in biological environments. However, the excellent properties of nanoparticle labels can address intractable problems in applications where high brightness and long-term photostability are crucial. As for Pdots and Qdots, the different subsets of fluorescent nanoparticles exhibit distinctive properties of their own. Pdots offer high brightness and non-toxicity and Qdots have narrow and symmetrical emissions. We encourage investigators to evaluate and select fluorescent probes on a case-by-case basis and avoid generalizations wherever possible.
In our opinion, the development and application of Pdot bioconjugates continues to be an active and important area in fluorescence imaging and biomedical studies. Research in the Pdot field continues to inspire the chemistry community with new discoveries and challenges. It involves cross-disciplinary collaboration between chemists, biologists, and clinicians. We envision the exploration of new Pdot species with improved performance, well-controlled surface properties, and multi-modal imaging/sensing functionalities to be the main focus of the field in the future. When fully optimized, Pdot technology is expected to have a broad and lasting impact on biomedical imaging, diagnostics, and therapeutics.
Acknowledgments
We gratefully acknowledge support from the National Institutes of Health, the National Science Foundation, and the University of Washington, USA. C.W. acknowledges financial support from “Thousand Talents Program”, the National Science Foundation of China (No. 61222508), and Jilin University.
Biographies

Changfeng Wu completed his PhD in 2008 in the group of Prof. Jason McNeill at Clemson University and then carried out postdoctoral research with Prof. Daniel T. Chiu at the University of Washington. He joined the faculty of Jilin University in 2012 and his current research is focused on the development of fluorescent nanoparticles, and spectroscopic and imaging techniques for biological applications.

Daniel T. Chiu is currently the A. Bruce Montgomery Professor of Chemistry and Professor of Bioengineering at the University of Washington. His research is focused on developing new methods based on nanotechnology and nanomaterials for probing biological processes at the single-cell and single-molecule level, and on applying these new techniques for addressing pressing biomedical problems.
Contributor Information
Prof. Changfeng Wu, State Key Laboratory on Integrated Optoelectronics, College of Electronic Science and Engineering, Jilin University, Changchun, Jilin 130012 (China)
Prof. Daniel T. Chiu, Department of Chemistry, University of Washington Seattle, WA 98195 (USA)
References
- 1.Evanko D. Nat Methods. 2005;2:901. doi: 10.1038/nmeth0905-640. This issue includes a series of review articles on fluorescence imaging techniques. [DOI] [PubMed] [Google Scholar]
- 2.Pepperkok R, Ellenberg J. Nat Rev Mol Cell Biol. 2006;7:690. doi: 10.1038/nrm1979. [DOI] [PubMed] [Google Scholar]
- 3.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3rd. Springer; 2006. [Google Scholar]
- 4.Mason WT. Fluorescent and Luminescent Probes for Biological Activity. 2nd. Academic Press; London: 1999. [Google Scholar]
- 5.Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Science. 2003;300:2061. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- 6.Xie XS, Yu J, Yang WY. Science. 2006;312:228. doi: 10.1126/science.1127566. [DOI] [PubMed] [Google Scholar]
- 7.Moerner WE. Proc Natl Acad Sci USA. 2007;104:12596. doi: 10.1073/pnas.0610081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Science. 2006;313:1642. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 9.Bates M, Huang B, Dempsey GT, Zhuang XW. Science. 2007;317:1749. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hell SW. Science. 2007;316:1153. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 11.Huang B, Wang WQ, Bates M, Zhuang XW. Science. 2008;319:810. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Nat Methods. 2008;5:763. doi: 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- 13.Tsien RY. Annu Rev Biochem. 1998;67:509. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 14.Shaner NC, Steinbach PA, Tsien RY. Nat Methods. 2005;2:905. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 15.Tsien RY. Angew Chem. 2009;121:5721. [Google Scholar]; Angew Chem Int Ed. 2009;48:5612. [Google Scholar]
- 16.Bruchez M, Jr, Moronne M, Gin P, Weiss S, Alivisatos AP. Science. 1998;281:2013. doi: 10.1126/science.281.5385.2013. [DOI] [PubMed] [Google Scholar]
- 17.Chan WCW, Nie SM. Science. 1998;281:2016. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- 18.Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Science. 2005;307:538. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao XJ, Bagwe RP, Tan WH. Adv Mater. 2004;16:173. [Google Scholar]
- 20.Ow H, Larson DR, Srivastava M, Baird BA, Webb WW, Wiesner U. Nano Lett. 2005;5:113. doi: 10.1021/nl0482478. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Wang KM, Santra S, Zhao XJ, Hilliard LR, Smith JE, Wu JR, Tan WH. Anal Chem. 2006;78:646. [Google Scholar]
- 22.Pei Q, Yu G, Zhang C, Yang Y, Heeger A. Science. 1995;269:1086. doi: 10.1126/science.269.5227.1086. [DOI] [PubMed] [Google Scholar]
- 23.Friend RH, Gymer RW, Holmes AB, Burroughes JH, Marks RN, Taliani C, Bradley DDC, Dos Santos DA, Bredas JL, Loglund M, Salaneck WR. Nature. 1999;397:121. [Google Scholar]
- 24.Müller CD, Falcou A, Reckefuss N, Rojahn M, Wiederhirn V, Rudati P, Frohne H, Nuyken O, Becker H, Meerholz K. Nature. 2003;421:829. doi: 10.1038/nature01390. [DOI] [PubMed] [Google Scholar]
- 25.Wu HB, Ying L, Yang W, Cao Y. Chem Soc Rev. 2009;38:3391. doi: 10.1039/b816352a. [DOI] [PubMed] [Google Scholar]
- 26.Chen L, McBranch DW, Wang HL, Helgeson R, Wudl F, Whitten DG. Proc Natl Acad Sci USA. 1999;96:12287. doi: 10.1073/pnas.96.22.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heeger PS, Heeger AJ. Proc Natl Acad Sci USA. 1999;96:12219. doi: 10.1073/pnas.96.22.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan CH, Wang S, Hong JW, Bazan GC, Plaxco KW, Heeger AJ. Proc Natl Acad Sci USA. 2003;100:6297. doi: 10.1073/pnas.1132025100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinto MR, Schanze KS. Proc Natl Acad Sci USA. 2004;101:7505. doi: 10.1073/pnas.0402280101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McQuade DT, Pullen AE, Swager TM. Chem Rev. 2000;100:2537. doi: 10.1021/cr9801014. [DOI] [PubMed] [Google Scholar]
- 31.Thomas SW, Joly GD, Swager TM. Chem Rev. 2007;107:1339. doi: 10.1021/cr0501339. [DOI] [PubMed] [Google Scholar]
- 32.Pu KY, Liu B. Adv Funct Mater. 2011;21:3408. [Google Scholar]
- 33.Feng XL, Liu LB, Wang S, Zhu DB. Chem Soc Rev. 2010;39:2411. doi: 10.1039/b909065g. [DOI] [PubMed] [Google Scholar]
- 34.Duarte A, Pu KY, Liu B, Bazan GC. Chem Mater. 2011;23:501. [Google Scholar]
- 35.Zhu C, Liu L, Yang Q, Lv F, Wang S. Chem Rev. 2012;112:4687. doi: 10.1021/cr200263w. [DOI] [PubMed] [Google Scholar]
- 36.Landfester K, Montenegro R, Scherf U, Guntner R, Asawapirom U, Patil S, Neher D, Kietzke T. Adv Mater. 2002;14:651. [Google Scholar]
- 37.Piok T, Gamerith S, Gadermaier C, Plank H, Wenzl FP, Patil S, Montenegro R, Kietzke T, Neher D, Scherf U, Landfester K, List EJW. Adv Mater. 2003;15:800. [Google Scholar]
- 38.Kietzke T, Neher D, Landfester K, Montenegro R, Guntner R, Scherf U. Nat Mater. 2003;2:408. doi: 10.1038/nmat889. [DOI] [PubMed] [Google Scholar]
- 39.Kurokawa N, Yoshikawa H, Hirota N, Hyodo K, Masuhara H. ChemPhysChem. 2004;5:1609. doi: 10.1002/cphc.200400117. [DOI] [PubMed] [Google Scholar]
- 40.Pecher J, Mecking S. Chem Rev. 2010;110:6260. doi: 10.1021/cr100132y. [DOI] [PubMed] [Google Scholar]
- 41.Kaeser A, Schenning APHJ. Adv Mater. 2010;22:2985. doi: 10.1002/adma.201000427. [DOI] [PubMed] [Google Scholar]
- 42.Tuncel D, Demir HV. Nanoscale. 2010;2:484. doi: 10.1039/b9nr00374f. [DOI] [PubMed] [Google Scholar]
- 43.Tian Z, Yu J, Wu C, Szymanski C, McNeill J. Nanoscale. 2010;2:1999. doi: 10.1039/c0nr00322k. [DOI] [PubMed] [Google Scholar]
- 44.Li K, Liu B. J Mater Chem. 2012;22:1257. [Google Scholar]
- 45.Szymanski C, Wu C, Hooper J, Salazar MA, Perdomo A, Dukes A, McNeill JD. J Phys Chem B. 2005;109:8543. doi: 10.1021/jp051062k. [DOI] [PubMed] [Google Scholar]
- 46.Wu C, Szymanski C, McNeill J. Langmuir. 2006;22:2956. doi: 10.1021/la060188l. [DOI] [PubMed] [Google Scholar]
- 47.Wu C, Peng H, Jiang Y, McNeill J. J Phys Chem B. 2006;110:14148. doi: 10.1021/jp0618126. [DOI] [PubMed] [Google Scholar]
- 48.Wu C, Szymanski C, Cain Z, McNeill J. J Am Chem Soc. 2007;129:12904. doi: 10.1021/ja074590d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu C, Zheng Y, Szymanski C, McNeill J. J Phys Chem C. 2008;112:1772. doi: 10.1021/jp074149+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu C, McNeill J. Langmuir. 2008;24:5855. doi: 10.1021/la8000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu C, Bull B, Szymanski C, Christensen K, McNeill J. ACS Nano. 2008;2:2415–2423. doi: 10.1021/nn800590n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu C, Bull B, Szymanski C, Christensen K, McNeill J. Angew Chem. 2009;121:2779–2783. [Google Scholar]; Angew Chem Int Ed. 2009;48:2741–2745. doi: 10.1002/anie.200805894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu J, Wu C, Sahu S, Fernando L, Szymanski C, McNeill J. J Am Chem Soc. 2009;131:18410–18414. doi: 10.1021/ja907228q. [DOI] [PubMed] [Google Scholar]
- 54.Yu J, Wu C, Tian Z, McNeill J. Nano Lett. 2012;12:1300. doi: 10.1021/nl203784m. [DOI] [PubMed] [Google Scholar]
- 55.Grimland JL, Wu CF, Ramoutar RR, Brumaghim JL, McNeill J. Nanoscale. 2011;3:1451. doi: 10.1039/c0nr00834f. [DOI] [PubMed] [Google Scholar]
- 56.Wu C, Schneider T, Zeigler M, Yu J, Schiro P, Burnham D, McNeill JD, Chiu DT. J Am Chem Soc. 2010;132:15410. doi: 10.1021/ja107196s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu C, Jin Y, Schneider T, Burnham DR, Smith PB, Chiu DT. Angew Chem. 2010;122:9626. doi: 10.1002/anie.201004260. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:9436. [Google Scholar]
- 58.Wu C, Hansen S, Hou Q, Yu J, Zeigler M, Jin Y, Burnham D, McNeill J, Olson J, Chiu DT. Angew Chem. 2011;123:3492. doi: 10.1002/anie.201007461. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:3430. [Google Scholar]
- 59.Ye F, Wu C, Jin Y, Chan Y, Zhang X, Chiu DT. J Am Chem Soc. 2011;133:8146. doi: 10.1021/ja202945g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan Y, Wu C, Ye F, Jin Y, Smith PB, Chiu DT. Anal Chem. 2011;83:1448. doi: 10.1021/ac103140x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jin Y, Ye F, Zeigler M, Wu C, Chiu DT. ACS Nano. 2011;5:1468. doi: 10.1021/nn103304m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chan Y, Jin Y, Wu C, Chiu DT. Chem Commun. 2011;47:2820. doi: 10.1039/c0cc04929h. [DOI] [PubMed] [Google Scholar]
- 63.Ye F, Wu C, Jin Y, Wang M, Chan Y, Yu J, Sun W, Hayden S, Chiu DT. Chem Commun. 2012;48:1778. doi: 10.1039/c2cc16486h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jin Y, Ye F, Wu C, Chan Y, Chiu DT. Chem Commun. 2012;48:3161. doi: 10.1039/c2cc17703j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang X, Yu J, Wu C, Jin Y, Ye F, Chiu DT. ACS Nano. 2012;6:5429. doi: 10.1021/nn301308w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu J, Wu C, Zhang X, Ye F, Gallina ME, Rong Y, Wu Y, Sun W, Chan YH, Chiu DT. Adv Mater. 2012;24:3498. doi: 10.1002/adma.201201245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rong Y, Wu C, Yu J, Zhang X, Ye F, Zeigler M, Gallina ME, Wu I, Zhang Y, Chan Y, Sun W, Uvdal K, Chiu DT. ACS Nano. 2013 doi: 10.1021/nn304376z. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fernando LP, Kandel PK, Yu JB, McNeill J, Ackroyd PC, Christensen KA. Biomacromolecules. 2010;11:2675. doi: 10.1021/bm1007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baier MC, Huber J, Mecking S. J Am Chem Soc. 2009;131:14267–14273. doi: 10.1021/ja905077c. [DOI] [PubMed] [Google Scholar]
- 70.Pecher J, Mecking S. Macromolecules. 2007;40:7733. [Google Scholar]
- 71.Pecher J, Huber J, Winterhalder M, Zumbusch A, Mecking S. Biomacromolecules. 2010;11:2776. doi: 10.1021/bm100854a. [DOI] [PubMed] [Google Scholar]
- 72.Kim S, Lim CK, Na J, Lee YD, Kim K, Choi K, Leary JF, Kwon IC. Chem Commun. 2010;46:1617. doi: 10.1039/b923309a. [DOI] [PubMed] [Google Scholar]
- 73.Lim J, Swager TM. Angew Chem. 2010;122:7648. doi: 10.1002/anie.201003111. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:7486. [Google Scholar]
- 74.Cordovilla C, Swager TM. J Am Chem Soc. 2012;134:6932. doi: 10.1021/ja301259v. [DOI] [PubMed] [Google Scholar]
- 75.Harbron EJ, Davis CM, Campbell JK, Allred RM, Kovary MT, Economou NJ. J Phys Chem C. 2009;113:13707. [Google Scholar]
- 76.Davis CM, Childress ES, Harbron EJ. J Phys Chem C. 2011;115:19065. [Google Scholar]
- 77.Childress ES, Roberts CA, Sherwood DY, LeGuyader CLM, Harbron EJ. Anal Chem. 2012;84:1235. doi: 10.1021/ac300022y. [DOI] [PubMed] [Google Scholar]
- 78.Osakada Y, Hanson L, Cui BX. Chem Commun. 2012;48:3285. doi: 10.1039/c2cc18085e. [DOI] [PubMed] [Google Scholar]
- 79.Shen XQ, He F, Wu JH, Xu GQ, Yao SQ, Xu QH. Langmuir. 2011;27:1739. doi: 10.1021/la104722q. [DOI] [PubMed] [Google Scholar]
- 80.Shen XQ, Li L, Wu H, Yao SQ, Xu QH. Nanoscale. 2011;3:5140. doi: 10.1039/c1nr11104c. [DOI] [PubMed] [Google Scholar]
- 81.Moon JH, Deans R, Krueger E, Hancock LF. Chem Commun. 2003:104. doi: 10.1039/b207186j. [DOI] [PubMed] [Google Scholar]
- 82.Moon JH, McDaniel W, MacLean P, Hancock LE. Angew Chem. 2007;119:8371. doi: 10.1002/anie.200701991. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:8223. [Google Scholar]
- 83.Moon JH, MacLean P, McDaniel W, Hancock LF. Chem Commun. 2007:4910. doi: 10.1039/b710807a. [DOI] [PubMed] [Google Scholar]
- 84.Rahim NAA, McDaniel W, Bardon K, Srinivasan S, Vickerman V, So PTC, Moon JH. Adv Mater. 2009;21:3492. [Google Scholar]
- 85.Moon JH, Mendez E, Kim Y, Kaur A. Chem Commun. 2011;47:8370. doi: 10.1039/c1cc10991j. [DOI] [PubMed] [Google Scholar]
- 86.Feng XL, Lv FT, Liu LB, Tang HW, Xing CF, Yang QO, Wang S. ACS Appl Mater Interfaces. 2010;2:2429. doi: 10.1021/am100435k. [DOI] [PubMed] [Google Scholar]
- 87.Pu KY, Li K, Liu B. Chem Mater. 2010;22:6736. [Google Scholar]
- 88.Ding D, Li K, Zhu ZS, Pu KY, Hu Y, Jiang XQ, Liu B. Nanoscale. 2011;3:1997. doi: 10.1039/c0nr00950d. [DOI] [PubMed] [Google Scholar]
- 89.Abbel R, van der Weegen R, Meijer EW, Schenning APHJ. Chem Commun. 2009:1697. doi: 10.1039/b822943k. [DOI] [PubMed] [Google Scholar]
- 90.Park SJ, Kang SG, Fryd M, Saven JG, Park SJ. J Am Chem Soc. 2010;132:9931. doi: 10.1021/ja1004569. [DOI] [PubMed] [Google Scholar]
- 91.Petkau K, Kaeser A, Fischer I, Brunsveld L, Schenning APHJ. J Am Chem Soc. 2011;133:17063. doi: 10.1021/ja2075345. [DOI] [PubMed] [Google Scholar]
- 92.Feng XL, Tang YL, Duan XR, Liu LB, Wang S. J Mater Chem. 2010;20:1312. [Google Scholar]
- 93.Pu KY, Li K, Liu B. Adv Funct Mater. 2010;20:2770. [Google Scholar]
- 94.Li K, Pan J, Feng SS, Wu AW, Pu KY, Liu YT, Liu B. Adv Funct Mater. 2009;19:3535. [Google Scholar]
- 95.Li K, Liu YT, Pu KY, Feng SS, Zhan RY, Liu B. Adv Funct Mater. 2011;21:287. [Google Scholar]
- 96.Geng J, Li K, Pu KY, Ding D, Liu B. Small. 2012;8:2421. doi: 10.1002/smll.201102353. [DOI] [PubMed] [Google Scholar]
- 97.Li K, Ding D, Huo D, Pu KY, Thao NNP, Hu Y, Li Z, Liu B. Adv Funct Mater. 2012;15:3107. [Google Scholar]
- 98.Howes P, Green M, Levitt J, Suhling K, Hughes M. J Am Chem Soc. 2010;132:3989–3996. doi: 10.1021/ja1002179. [DOI] [PubMed] [Google Scholar]
- 99.Howes P, Green M, Bowers A, Parker D, Varma G, Kallumadil M, Hughes M, Warley A, Brain A, Botnar R. J Am Chem Soc. 2010;132:9833. doi: 10.1021/ja1031634. [DOI] [PubMed] [Google Scholar]
- 100.Howes P, Thorogate R, Green M, Jickells S, Daniel B. Chem Commun. 2009:2490. doi: 10.1039/b903405f. [DOI] [PubMed] [Google Scholar]
- 101.Hashim Z, Howes P, Green M. J Mater Chem. 2011;21:1797. [Google Scholar]
- 102.Kandel PK, Fernando LP, Ackroyd PC, Christensen KA. Nanoscale. 2011;3:1037. doi: 10.1039/c0nr00746c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun B, Sun MJ, Gu Z, Shen QD, Jiang SJ, Xu Y, Wang Y. Macromolecules. 2010;43:10348. [Google Scholar]
- 104.Chan Y, Ye F, Gallina M, Zhang X, Jin Y, Wu I, Chiu DT. J Am Chem Soc. 2012;134:7309. doi: 10.1021/ja3022973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sun W, Hayden S, Jin Y, Rong Y, Yu J, Ye F, Chan Y, Zeigler M, Wu C, Chiu DT. Nanoscale. 2012;4:7246. doi: 10.1039/c2nr32055j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Warner JH, Hoshino A, Yamamoto K, Tilley RD. Angew Chem. 2005;117:4626. doi: 10.1002/anie.200501256. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:4550. [Google Scholar]
- 107.He Y, Zhong YL, Peng F, Wei XP, Su YY, Lu YM, Su S, Gu W, Liao LS, Lee ST. J Am Chem Soc. 2011;133:14192. doi: 10.1021/ja2048804. [DOI] [PubMed] [Google Scholar]
- 108.Sun YP, Zhou B, Lin Y, Wang W, Fernando KAS, Pathak P, Meziani MJ, Harruff BA, Wang X, Wang HF, Luo PJG, Yang H, Kose ME, Chen BL, Veca LM, Xie SY. J Am Chem Soc. 2006;128:7756. doi: 10.1021/ja062677d. [DOI] [PubMed] [Google Scholar]
- 109.Baker SN, Baker GA. Angew Chem. 2010;122:6876. [Google Scholar]; Angew Chem Int Ed. 2010;49:6726. [Google Scholar]
- 110.Zrazhevskiy P, Sena M, Gao XH. Chem Soc Rev. 2010;39:4326. doi: 10.1039/b915139g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shirakawa H, Louis EJ, MacDiarmid AG, Chiang CK, Heeger AJ. Chem Commun. 1977:578. [Google Scholar]
- 112.Chiang CK, Fincher CR, Jr, Park YW, Heeger AJ, Shirakawa H, Louis EJ, Gau SC, MacDiarmid AG. Phys Rev Lett. 1977;39:1098. [Google Scholar]
- 113.Heeger AJ. Angew Chem. 2001;113:2660. [Google Scholar]; Angew Chem Int Ed. 2001;40:2591. [Google Scholar]
- 114.Hide F, Diaz-Garcia MA, Schwartz BJ, Heeger AJ. Acc Chem Res. 1997;30:430. [Google Scholar]
- 115.Yu G, Gao J, Hummelen JC, Wudl F, Heeger AJ. Science. 1995;270:1789. [Google Scholar]
- 116.Günes S, Neugebauer H, Sariciftci NS. Chem Rev. 2007;107:1324. doi: 10.1021/cr050149z. [DOI] [PubMed] [Google Scholar]
- 117.Burroughes JH, Jones CA, Friend RH. Nature. 1988;335:137. [Google Scholar]
- 118.Yang Y, Heeger AJ. Nature. 1994;372:344. [Google Scholar]
- 119.Yan H, Chen ZH, Zheng Y, Newman C, Quinn JR, Dotz F, Kastler M, Facchetti A. Nature. 2009;457:679. doi: 10.1038/nature07727. [DOI] [PubMed] [Google Scholar]
- 120.Schutze F, Stempfle B, Jungst C, Woll D, Zumbusch A, Mecking S. Chem Commun. 2012;48:2104. doi: 10.1039/c2cc17066c. [DOI] [PubMed] [Google Scholar]
- 121.Clafton SN, Beattie DA, Mierczynska-Vasilev A, Acres RG, Morgan AC, Kee TW. Langmuir. 2010;26:17785. doi: 10.1021/la103063p. [DOI] [PubMed] [Google Scholar]
- 122.Collini E, Scholes GD. Science. 2009;323:369. doi: 10.1126/science.1164016. [DOI] [PubMed] [Google Scholar]
- 123.Palacios RE, Fan FRF, Grey JK, Suk J, Bard AJ, Barbara PF. Nat Mater. 2007;6:680. doi: 10.1038/nmat1959. [DOI] [PubMed] [Google Scholar]
- 124.Mcrae RL, Phillips RL, Kim IB, Bunz UHF, Fahrni CJ. J Am Chem Soc. 2008;130:7851. doi: 10.1021/ja8007402. [DOI] [PubMed] [Google Scholar]
- 125.Sigurdson CJ, Peter K, Nilsson R, Hornemann S, Manco G, Polymenidou M, Schwarz P, Leclerc M, Hammarstrom P, Wuthrich K, Aguzzi A. Nat Methods. 2007;4:1023. doi: 10.1038/nmeth1131. [DOI] [PubMed] [Google Scholar]
- 126.Åslund A, Sigurdson CJ, Klingstedt T, Grathwohl S, Bolmont T, Dickstein DL, Glimsdal E, Prokop S, Lindgren M, Konradsson P, Holtzman DM, Hof PR, Heppner FL, Gandy S, Jucker M, Aguzzi A, Hammarstrom P, Nilsson KPR. ACS Chem Biol. 2009;4:673. doi: 10.1021/cb900112v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pu KY, Li K, Shi JB, Liu B. Chem Mater. 2009;21:3816. [Google Scholar]
- 128.Pu KY, Shi JB, Cai LP, Li K, Liu B. Biomacromolecules. 2011;12:2966. doi: 10.1021/bm200563a. [DOI] [PubMed] [Google Scholar]
- 129.Li K, Pu KY, Cai LP, Liu B. Chem Mater. 2011;23:2113. [Google Scholar]
- 130.Edwards J, Fisher R, Vincent B. Makromol Chem Rapid Commun. 1983;4:393. [Google Scholar]
- 131.Armes SP, Miller JF, Vincent B. J Colloid Interface Sci. 1987;118:410. [Google Scholar]
- 132.Hittinger E, Kokil A, Weder C. Angew Chem. 2004;116:1844. doi: 10.1002/anie.200352863. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:1808. [Google Scholar]
- 133.Schwartz BJ. Annu Rev Phys Chem. 2003;54:141. doi: 10.1146/annurev.physchem.54.011002.103811. [DOI] [PubMed] [Google Scholar]
- 134.Chandler D. Nature. 2005;437:640. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 135.ten Wolde PR, Chandler D. Proc Natl Acad Sci USA. 2002;99:6539. doi: 10.1073/pnas.052153299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hoeben FJM, Shklyarevskiy IO, Pouderoijen MJ, Engelkamp H, Schenning APHJ, Christianen PCM, Maan JC, Meijer EW. Angew Chem. 2006;118:1254. doi: 10.1002/anie.200502187. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:1232. [Google Scholar]
- 137.Liu W, Howarth M, Greytak AB, Zheng Y, Nocera DG, Ting AY, Bawendi MG. J Am Chem soc. 2008;130:1274. doi: 10.1021/ja076069p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Howarth M, Liu WH, Puthenveetil S, Zheng Y, Marshall LF, Schmidt MM, Wittrup KD, Bawendi MG, Ting AY. Nat Methods. 2008;5:397. doi: 10.1038/nmeth.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nguyen TQ, Martini IB, Liu J, Schwartz BJ. J Phys Chem B. 2000;104:237. [Google Scholar]
- 140.Dahan M, Laurence T, Pinaud F, Chemla DS, Alivisatos AP, Sauer M, Weiss S. Opt Lett. 2001;26:825. doi: 10.1364/ol.26.000825. [DOI] [PubMed] [Google Scholar]
- 141.Plakhotnik T, Moerner WE, Palm V, Wild UP. Opt Commun. 1995;114:83. [Google Scholar]
- 142.Plakhotnik T, Donley EA, Wild UP. Annu Rev Phys Chem. 1997;48:181. doi: 10.1146/annurev.physchem.48.1.181. [DOI] [PubMed] [Google Scholar]
- 143.Eggeling C, Widengren J, Rigler R, Seidel CAM. Anal Chem. 1998;70:2651. doi: 10.1021/ac980027p. [DOI] [PubMed] [Google Scholar]
- 144.van Sark WGJHM, Frederix PLTM, Van den Heuvel DJ, Gerritsen HC, Bol AA, van Lingen JNJ, Donega CD, Meijerink A. J Phys Chem B. 2001;105:8281. [Google Scholar]
- 145.Grey JK, Kim DY, Norris BC, Miller WL, Barbara PF. J Phys Chem B. 2006;110:25568. doi: 10.1021/jp065990a. [DOI] [PubMed] [Google Scholar]
- 146.Yao J, Larson DR, Vishwasrao HD, Zipfel WR, Webb WW. Proc Natl Acad Sci USA. 2005;102:14284. doi: 10.1073/pnas.0506523102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mancini MC, Kairdolf BA, Smith AM, Nie SM. J Am Chem Soc. 2008;130:10836. doi: 10.1021/ja8040477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Han S, Devaraj NK, Lee J, Hilderbrand SA, Weissleder R, Bawendi MG. J Am Chem Soc. 2010;132:7838. doi: 10.1021/ja101677r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Guimard NK, Gomez N, Schmidt CE. Prog Polym Sci. 2007;32:876. [Google Scholar]
- 150.Wang L, Yang CY, Tan WH. Nano Lett. 2005;5:37. doi: 10.1021/nl048417g. [DOI] [PubMed] [Google Scholar]
- 151.Pu KY, Li K, Zhang X, Liu B. Adv Mater. 2010;22:4186. doi: 10.1002/adma.201001544. [DOI] [PubMed] [Google Scholar]
- 152.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. J Am Chem Soc. 2003;125:3192. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 153.Speers AE, Adam GC, Cravatt BF. J Am Chem Soc. 2003;125:4686. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 154.Prescher JA, Bertozzi CR. Nat Chem Biol. 2005;1:13. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 155.Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. ACS Chem Biol. 2006;1:644. doi: 10.1021/cb6003228. [DOI] [PubMed] [Google Scholar]
- 156.Sletten EM, Bertozzi CR. Angew Chem. 2009;121:7108. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:6974. [Google Scholar]
- 157.Prescher JA, Bertozzi CR. Cell. 2006;126:851. doi: 10.1016/j.cell.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 158.Veiseh O, Sun C, Fang C, Bhattarai N, Gunn J, Kievit F, Du K, Pullar B, Lee D, Ellenbogen RG, Olson J, Zhang MQ. Cancer Res. 2009;69:6200. doi: 10.1158/0008-5472.CAN-09-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Choi HS, Liu WH, Liu FB, Nasr K, Misra P, Bawendi MG, Frangioni JV. Nat Nanotechnol. 2010;5:42. doi: 10.1038/nnano.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV. Nat Biotechnol. 2007;25:1165. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ha T, Enderle T, Ogletree DF, Chemla DS, Selvin PR, Weiss S. Proc Natl Acad Sci USA. 1996;93:6264. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hohng S, Ha T. ChemPhysChem. 2005;6:956. doi: 10.1002/cphc.200400557. [DOI] [PubMed] [Google Scholar]
- 163.Clapp AR, Medintz IL, Mauro JM, Fisher BR, Bawendi MG, Mattoussi H. J Am Chem Soc. 2004;126:301. doi: 10.1021/ja037088b. [DOI] [PubMed] [Google Scholar]
- 164.Xu QH, Wang S, Korystov D, Mikhailovsky A, Bazan GC, Moses D, Heeger AJ. Proc Natl Acad Sci USA. 2005;102:530. doi: 10.1073/pnas.0408568102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kim YJ, Johnson RC, Hupp JT. Nano Lett. 2001;1:165. [Google Scholar]
- 166.Lin SY, Wu SH, Chen CH. Angew Chem. 2006;118:5070. doi: 10.1002/anie.200600771. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:4948. [Google Scholar]
- 167.Wang MD, Shin DM, Simons JW, Nie SM. Expert Rev Anticancer Ther. 2007;7:833. doi: 10.1586/14737140.7.6.833. [DOI] [PubMed] [Google Scholar]
- 168.Sinha R, Kim GJ, Nie SM, Shin DM. Mol Cancer Ther. 2006;5:1909. doi: 10.1158/1535-7163.MCT-06-0141. [DOI] [PubMed] [Google Scholar]
- 169.Nie SM, Xing Y, Kim GJ, Simons JW. Annu Rev Biomed Eng. 2007;9:257. doi: 10.1146/annurev.bioeng.9.060906.152025. [DOI] [PubMed] [Google Scholar]
- 170.Zhu CL, Yang QO, Liu LB, Lv FT, Li SY, Yang GQ, Wang S. Adv Mater. 2011;23:4805. doi: 10.1002/adma.201102850. [DOI] [PubMed] [Google Scholar]
- 171.Roy I, Ohulchanskyy TY, Pudavar HE, Bergey EJ, Oseroff AR, Morgan J, Dougherty TJ, Prasad PN. J Am Chem Soc. 2003;125:7860. doi: 10.1021/ja0343095. [DOI] [PubMed] [Google Scholar]
- 172.Kim S, Ohulchanskyy TY, Pudavar HE, Pandey RK, Prasad PN. J Am Chem Soc. 2007;129:2669. doi: 10.1021/ja0680257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.McCarthy JR, Perez JM, Bruckner C, Weissleder R. Nano Lett. 2005;5:2552. doi: 10.1021/nl0519229. [DOI] [PubMed] [Google Scholar]
- 174.Gao D, Agayan RR, Xu H, Philbert MA, Kopelman R. Nano Lett. 2006;6:2383. doi: 10.1021/nl0617179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Samia ACS, Chen XB, Burda C. J Am Chem Soc. 2003;125:15736. doi: 10.1021/ja0386905. [DOI] [PubMed] [Google Scholar]
- 176.Tsay JM, Trzoss M, Shi L, Kong X, Selke M, Jung ME, Weiss S. J Am Chem Soc. 2007;129:6865–6871. doi: 10.1021/ja070713i. [DOI] [PMC free article] [PubMed] [Google Scholar]