

Abstract
Norcantharidin, a low-toxic analog of the active anticancer compound cantharidin in Mylabris, can inhibit proliferation and induce apoptosis of multiple types of cancer cells. However, the anticancer activities of norcantharidin with respect to neuroblastoma, and its underlying mechanisms, have not been investigated. Therefore, our study was designed to determine the efficacy of norcantharidin on SK-N-SH neuroblastoma cell death and to elucidate detailed mechanisms of activity. In the present study, norcantharidin suppressed the proliferation and cloning ability of SK-N-SH cells in a dose-dependent manner, apparently by reducing the mitochondrial membrane potential and arresting SK-N-SH cells at the G2/M stage, accompanied by elevated expressions of p21 and decreased expressions of cyclin B1 and cell division control 2. Treatment by norcantharidin induced significant mitophagy and autophagy, as demonstrated by a decrease in Translocase Of Outer Mitochondrial Membrane 20 (TOM20), increased beclin1 and LC3-II protein expression, reduced protein SQSTM1/p62 expression, and accumulation of punctate LC3 in the cytoplasm of SK-N-SH cells. In addition, norcantharidin induced apoptosis through regulating the expression of B-cell lymphoma 2–associated X protein/B-cell lymphoma 2 and B-cell lymphoma 2–associated X protein/myeloid cell leukemia 1 and activating caspase-3 and caspase-9–dependent endogenous mitochondrial pathways. We also observed an increase in phosphor–AMP-activated protein kinase accompanied with a decrease in phosphor-protein kinase B and mammalian target of rapamycin expression after treatment with norcantharidin. Subsequent studies indicated that norcantharidin participates in cellular autophagy and apoptosis via activation of the c-Jun NH2-terminal kinases/c-Jun pathway. In conclusion, our results demonstrate that norcantharidin can reduce the mitochondrial membrane potential, induce mitophagy, and subsequently arouse cellular autophagy and apoptosis; the AMP-activated protein kinase, protein kinase B/mammalian target of rapamycin, and c-Jun NH2-terminal kinases/c-Jun signaling pathways are widely involved in these processes. Thus, the traditional Chinese medicine norcantharidin could be a novel therapeutic strategy for treating neuroblastoma.
Keywords: norcantharidin, neuroblastoma, SK-N-SH cells line, autophagy, apoptosis
Introduction
Neuroblastoma is the most common malignant extracranial tumor, with an incidence of 8% to 10% among pediatric malignant tumors and a mortality of 15% of all childhood cancer deaths.1 The first symptoms of neuroblastoma are usually vague (fatigue, loss of appetite, fever, and joint pain are common), making diagnosis difficult. Usually, these tumors cannot be removed completely through surgery due to metastasis at the time of diagnosis, and they have a very poor prognosis.2 Although intensive multimodality therapies have resulted in some improvement in the overall cure rate of this tumor, the therapies have considerable short- and long-term toxicities and can result in treatment-related death. Therefore, an urgent need exists for the development of more effective and less toxic anticancer drugs for neuroblastoma treatment.
The dried body of the Chinese blister beetle (Mylabris phalerata Pallas), known as Mylabris, is a traditional Chinese medicine that has been used for over 2000 years to treat abdominal masses and rabies and used as an abortifacient.3 Pharmacological studies have revealed that cantharidin (CTD), an active constituent of Mylabris, has antitumor properties and causes leukocytosis. However, its applications are limited by its gastrointestinal and urinary tracts’ side effects.4 Norcantharidin (NCTD), a demethylated derivative of CTD (Figure 1A), has been synthesized as a replacement for CTD to reduce toxic side effects while still retaining the efficacy of CTD. Nowadays, NCTD is widely used in China as an antitumor drug for inhibiting the proliferation and metastases of several kinds of carcinomas, including primary hepatoma, colorectal cancer, breast cancer, prostate cancer, and lung cancer.5-9 Unlike conventional chemotherapeutics, NCTD is preferentially toxic to cancer cells rather than normal cells,10 making this compound a promising cancer treatment agent.
Figure 1.
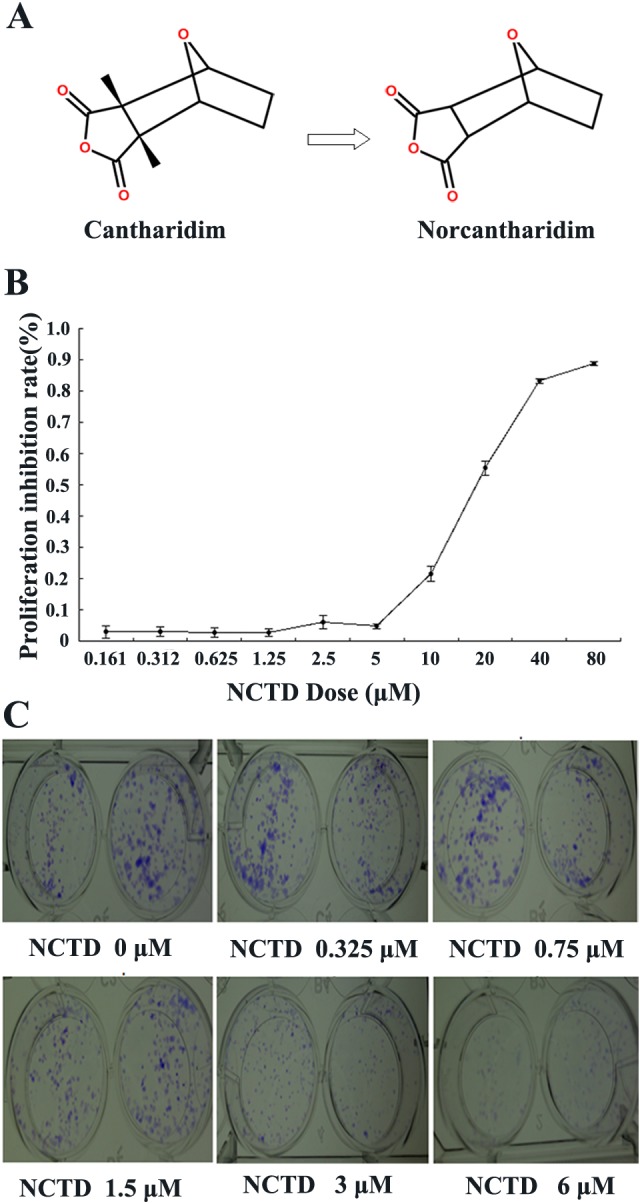
Effects of norcantharidin (NCTD) on cell viability in SK-N-SH cells. A, Molecular structure of cantharidin (CTD) and NCTD. B, Norcantharidin inhibited the proliferation of SK-N-SH cells. SK-N-SH cells were treated with the indicated concentrations of NCTD for 48 hours, and cell viability was determined by the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. C, Colony-forming test results. SK-N-SH cells were incubated with NCTD (0, 0.325, 0.75, 1.5, 3, or 6 μmol/L). Data represent one of 3 experiments yielding similar results.
The effect of NCTD in inducing the apoptosis of multiple categories of tumor cells has been frequently cited. However, the relationship between neuroblastoma and NCTD remains elusive, and whether NCTD can induce autophagy of cancer cells has not been reported yet. Previous studies have shown that NCTD is able to activate AMP-activated protein kinase (AMPK) in mammalian animal cells,8 and AMPK (a cellular protein kinase sensing energy states), if activated, can induce autophagy,11 hinting that NCTD may induce autophagy through an AMPK pathway. Autophagy is primarily a process for cell protection, playing a pivotal role in cell survival, differentiation, development, and homeostasis.12 However, unlimited autophagy tends to gradually consume intracellular components and arouse cell death.13 Certain antitumor drugs have been proven to induce autophagy,14,15 and the antitumor effects of autophagy on tumor cells have captivated increasing attention among oncologists. Accumulating evidence has demonstrated the widespread prospect of treating malignant tumors through autophagic regulation.
In the present study, we measured SK-N-SH neuroblastoma cell proliferation and induction of autophagy and apoptosis after NCTD treatment. Cell survival-related signaling proteins were also determined as further investigations. To the best of our knowledge, this is the first study to demonstrate cytotoxic activity and underlying mechanisms of NCTD with respect to SK-N-SH neuroblastoma cells in vitro.
Materials and Methods
Cell Cultures and Reagents
Analytical grade NCTD was purchased from Sigma-Aldrich (St Louis, Missouri). A stock solution of NCTD (60 μmol/L) in Dulbecco modified eagle medium (DMEM; Invitrogen, Victoria, Australia) was prepared and stored at 4°C. Other materials purchased from Sigma-Aldrich were D-Hanks’ solution, penicillin, streptomycin, fetal bovine serum (FBS), ethylenediaminetetraacetic acid, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI). Materials purchased from Cell Signaling Technology Inc (Beverly, Massachusetts, USA) were the antibodies against human cyclin A, cyclin B2, cyclin D1, cyclin D3, cyclin E2, p21, myelin transcription factor 1 (Myt1), cell division control 2 (Cdc2) proteins, phosphor-cell division control 2 (p-Cdc2), B-cell lymphoma 2 (Bcl-2), B-cell lymphoma extra large (Bcl-xl), Bcl-2–associated X protein (Bax), myeloid cell leukemia 1 (Mcl-1), procaspase-3, cleaved caspase-3, procaspase-8, procaspase-9, cleaved caspase-9, poly-ADP ribose polymerase (PARP), cleaved PARP, AMPK, phosphor-AMP-activated protein kinase (p-AMPK), protein kinase B (AKT), phosphor- protein kinase B (p-AKT), c-Jun, phosphor-c-Jun (p-c-Jun), c-Jun NH2-terminal kinases (JNK), phosphor-c-Jun NH2-terminal kinases (p-JNK), mammalian target of rapamycin (mTOR), beclin 1, microtubule-associated protein 1A/1B-light chain 3 (LC3-I), LC3-II, SQSTM1/p62, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Fluorescein isothiocyanate (FITC)-conjugated secondary antibody was obtained from Vector Laboratories (Burlingame, California).
The SK-N-SH cell line (Shanghai Institute of Cell Biology, China) was cultured in DMEM supplemented with 10% FBS, 1 mmol/L glutamine, 1% penicillin/streptomycin, 1.5 g/L sodium bicarbonate, and 1 mmol/L sodium pyruvate and maintained at 37°C in a humidified 5% CO2 atmosphere.
Cell Proliferation Assay
Cell proliferation was evaluated by the MTT assay. Cells were plated in 96-well plates at a density of 5 × 103 cells/mL. After 24 hours, NCTD was added at final concentrations of 0, 0.161, 0.312, 0.625, 1.25, 2.5, 5, 10, 20, 40, and 80 μmol/L. Cells without NCTD treatment comprised the control group. Cells were incubated with various concentrations of drugs for 48 hours, followed by addition of MTT solution (10.0 μL/well) and incubation for a further 4 hours at 37°C. At the end of the incubation period, the purple formazan crystals were dissolved in 200 μL dimethyl sulfoxide. After the crystals had dissolved, the plates were analyzed on an automated microplate spectrophotometer (Thermo Molecular Devices Co, Union City, California, USA) at 570 nm. The inhibition rate of cell proliferation was calculated for each well as follows: (A570control cells − A570treated cells)/A570control cells × 100%. The IC50 values (fifty percent inhibitory concentration values) were determined by the logit method. Experiments were performed in triplicate.
Clone Formation Assay
A clone formation assay was used to evaluate the effects of NCTD on the proliferation of SK-N-SH cells. The SK-N-SH cells were cultured in 12-well microplates (300 cells/well) in 2 mL of 100% DMEM for 24 hours. Then, cells were treated with indicated concentrations of NCTD in microplates for 7 days. Cells were stained with crystal violet (Sigma, St Louis, Missouri, USA) for 20 minutes. Images of the colonies were obtained using a digital camera (Canon EOS350D, Tokyo, Japan).
Cell Apoptosis and Death Analysis
Flow cytometry (Beckman Coulter, Fullerton, California, USA) was used to identify apoptotic cells by DNA fragmentation analysis. To determine the apoptotic rate, SK-N-SH cells were placed in 6-well plates and treated with 0, 5, 10, 20, and 40 μmol/L NCTD at a density of 2 × 104 cells/well for 24 hours and then collected. Annexin V-FITC/propidium iodide (PI) staining was performed, following the manufacturer’s protocol. Phosphatidyl serine translocation to the cell surface is an indicator of early apoptotic cells. Therefore, annexin V-positive cells were identified as apoptotic cells. The apoptotic rate was determined using CellQuest software (Becton Dickinson and company, New Jersey, USA).
Cell Cycle Analysis
The cell cycle distribution was determined by DNA staining with PI. Briefly, SK-N-SH cells were incubated with 0, 5, 10, 20, and 40 μmol/L NCTD for 24 hours. Approximately 2 × 104 cells were collected and fixed in 70% ice-cold ethanol. Cell pellets were suspended in PI and simultaneously treated with RNase at 37°C for 30 minutes. Data acquisition and analysis were performed on the flow cytometer, and the results were analyzed using CellQuest software.
Measurements of Mitochondrial Membrane Potential
The mitochondrial membrane potential (ΔΨm) was measured using JC-1 dye (Beyotime Co, Hangzhou, China), which detects mitochondrial depolarization by a change in the fluorescence intensity from red to green. The SK-N-SH cells were placed in 6-well plates and treated with 0, 5, 10, 20, and 40 μmol/L NCTD at a density of 2 × 104 cells/well for 24 hours. Cells were resuspended and then detected by flow cytometry or were harvested and smeared on slides. Images were obtained under a fluorescent microscope (DFC480; Leica Microsystems, Wetzlar, Germany).
Western Blot Assays
SK-N-SH cells were treated with 0, 5, 10, 20, and 40 μmol/L of NCTD for 24 hours. Whole-cell lysates were prepared by adding 5× sodium dodecyl sulfate (SDS) sample buffer. Equal amounts of protein were electrophoresed on 10% SDS polyacrylamide gel electrophoresis gels and transferred to polyvinylidene fluoride membranes. The membranes were then blocked for 60 minutes at room temperature with 5% nonfat dry milk/Tris buffered saline–Tween 20 (TBST) and reacted with appropriate antibodies for Cdc2, p-Cdc2, Myt1, p21, JNK, p-JNK, c-Jun, p-c-Jun, AMPK, p-AMPK, PARP, cleaved PARP, procaspase-3, procaspase-8, procaspase-9, cleaved caspase-3, cleaved caspase-8, cleaved caspase-9, Bax, Bcl-xl, Mcl-1, Bcl-2, p62, beclin-1, LC3-I, LC3-II, and GAPDH (1:1000 dilution) at 4°C overnight. Following incubation with the primary antibody, membranes were washed in TBST and incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature. Proteins were visualized by incubation with SuperSignal West Pico reagents (NCI5079; Thermo), followed by exposure to radiograph film.
Immunocytochemistry
Immunofluorescence staining was conducted using a procedure similar to that described previously.16 The SK-N-SH cells were plated on sterile coverslips and treated with 5 μmol/L NCTD and 0 μmol/L NCTD (control group) for 24 hours and fixed with 3% paraformaldehyde for 10 minutes at 37°C. After fixation, a permeabilization step was conducted with chilled methanol (100%) for 10 minutes at −20°C, and the cells were subsequently incubated in blocking solution containing 5% bovine serum albumin and 1% Triton-X 100 for 1 hour at 37°C. Cells were then incubated with LC3 antibody (MBL, Nanterre, France) or Tom20 antibody (Santa Cruz Biotechnology) for 12 hours at 4°C followed by FITC-conjugated secondary antibody for 1 hour at 37°C. The nucleus was stained with DAPI (1 mg/mL) for 10 minutes at 37°C. Fluorescence images were then captured by a confocal laser scanning microscope (LSM 700; Carl Zeiss, Oberkochen, Germany).
Statistical Analysis
All assays were performed in triplicate. Data are expressed as the mean ± standard deviation (SD). Statistical analyses were performed using an analysis of variance with SPSS 13.0 software.
Results
Norcantharidin Inhibited the Growth of SK-N-SH Cell Lines
The inhibitory effect of NCTD on SK-N-SH cells was determined by an MTT assay. Results show that NCTD significantly inhibited the viability of SK-N-SH cells in a dose-dependent manner, after cells were exposed for 48 hours to varying concentrations of NCTD (Figure 1B); the IC50 value was 15.6 μmol/L. In addition, colony formation assay showed that NCTD strongly inhibited the proliferation of SK-N-SH cells (Figure 1C).
Norcantharidin Arrested SK-N-SH Cells at the G2/M Stage and Affected the Expression of Cell Cycle Proteins
To delineate the mechanisms responsible for the inhibition of cell proliferation by NCTD, we examined the cell cycle distribution using flow cytometry. Results showed that the cycle progression was significantly inhibited at the G2/M phase, as presented in Figure 2A. To gain further information, cell cycle regulatory protein expressions were investigated by Western blot. The results showed that NCTD treatment leads to a significant dose-dependent decrease in the levels of Myt1 and Cdc2 and also the phosphorylation of Cdc2 but that p21 expression is elevated (Figure 2B). Moreover, NCTD treatment caused a pronounced decrease in the expression of cyclin D1, cyclin D3, cyclin E2, and cyclin B1, whereas cyclin A expression increased inversely (Figure 2C).
Figure 2.

Norcantharidin (NCTD) induced cell cycle arrest of SK-N-SH cells. A, SK-N-SH cells were exposed to various concentrations of NCTD (0-40 μmol/L) for 24 hours followed by analysis of cell cycle by flow cytometry. B, Western blot analysis of cell cycle-associated protein extracts obtained from SK-N-SH cells treated with different concentrations of NCTD. C, Cyclin family members were tested after NCTD treatment as described.
Norcantharidin Induced Apoptosis Dose Dependently and Reduced the Membrane Potential of Mitochondria in SK-N-SH Cells
To investigate the mechanism of NCTD inhibition of cell viability, we analyzed the effects of NCTD on cell apoptosis using flow cytometry. Annexin V(+)/PI(−) and annexin V(+)/PI(+) represent cells in early apoptosis and late apoptosis/necrosis, respectively. As shown in Figure 3A, the total apoptosis proportion in SK-N-SH cells increased as the NCTD concentration increased from 3.5% to 30.0% after treatment, suggesting that NCTD induced apoptosis in SK-N-SH cells dose dependently, especially late apoptosis.
Figure 3.

Induction of apoptosis and reduction in mitochondrial membrane potential in SK-N-SH cells by norcantharidin (NCTD). A, Norcantharidin-induced apoptosis determined by annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) staining. B, The change in mitochondrial membrane potential after 0, 5, 10, 20, or 40 μmol/L of NCTD explosion was evaluated by flow cytometry. C, Fluorescence microscopy was used to determine the mitochondrial membrane potential after different concentration (0, 20, and 40 μmol/L) of NCTD treatment. Data shown are representative of 3 different experiments.
As is well known, loss of ΔΨm is a crucial step in the process of cell death. In necrotic cells, ΔΨm and mitochondrial integrity are irreversibly compromised. Consequently, we chose the cationic dye JC-1 to examine the role of mitochondria in cell death using the flow cytometry method. We used a computer-based program to calculate the ratio of green to red fluorescence after 24 hours of treatment at varying concentrations of NCTD. As presented in Figure 3B, after incubation of SK-N-SH cells with 5 μmol/L of NCTD, no significant changes were observed in ΔΨm. However, an increase in the NCTD concentration to 10 μmol/L initiated a decrease in ΔΨm. Moreover, an increase in the NCTD concentration to 40 μmol/L produced a more than 3-fold decrease in ΔΨm, as compared with controls. Thus, NCTD induced a dose-dependent decrease in ΔΨm in SK-N-SH cells. The JC-1 aggregates showed a spectral shift in emitted light from 530 nm (ie, green-colored emissions of the monomeric JC-1 form) to 590 nm (ie, green-orange–colored emissions of the J-aggregate) upon excitation at 490 nm. Under fluorescence microscopy, the cells treated for 24 hours with 20 and 40 μmol/L NCTD both showed high green and low orange fluorescence, as compared with the control group, which showed high green and high orange fluorescence (Figure 3C).
Norcantharidin Changed Expression Levels of Bcl-2 Family Proteins and Activated Caspase-3/Caspase-9/PARP in SK-N-SH Cells
To investigate the role of Bcl-2 family in NCTD-induced apoptosis, we determined the protein expression levels by Western blot. As shown in Figure 4A, treatment with NCTD for 24 hours markedly downregulated the expression levels of the antiapoptotic proteins Bcl-2 and Mcl-1. However, NCTD treatment of the SK-N-SH cells did not obviously alter the expressions of 2 other proteins, Bax and Bcl-xl.
Figure 4.

Norcantharidin (NCTD) changed expression levels of Bcl-2 family proteins and activated caspase-3/caspase-9/PARP in SK-N-SH cells. A, Norcantharidin inhibited B-cell lymphoma 2 (Bcl-2) family members. SK-N-SH cells were treated as described previously, and the effect of NCTD on Bcl-2 family members’ expression level was determined by Western blot. B, Norcantharidin activated caspase-3 and caspase-9. SK-N-SH cells were treated with 0, 5, 10, 20, and 40 μmol/L NCTD for 24 hours. The effects of NCTD on procaspase-3, procaspase-8, procaspase-9, and PARP were evaluated by Western blot. Data represent one of 3 experiments yielding similar results.
Caspases were also activated after NCTD treatment. As observed in Figure 4B, NCTD activated caspase-3 and caspase-9 in a dose-dependent manner, although expression levels of procaspase-8 were not significantly changed. Furthermore, PARP apoptotic fragments were generated after treatments with 20 and 40 μmol/L NCTD.
Norcantharidin-Induced Autophagy and Mitophagy and Altered Expressions of Autophagy-Related Proteins
To validate NCTD-induced autophagy of SK-N-SH cells, the expression levels of Beclin-1, LC3-I, LC3-II, and p62 were measured by Western blotting after treatment of SK-N-SH cells with NCTD. As shown in Figure 5A, the treatment of SK-N-SH cells with NCTD accelerates the differentiation of LC3-I into LC3-II. Expression of p62 was downregulated, whereas the expression of beclin-1 was elevated. These results suggested that NCTD induced autophagy in SK-N-SH cells. To make further validation, the cells in each group were investigated by immunofluorescence analysis, and particles were observed under a confocal microscope for the presence of green fluorescence. As an autophagosomal membrane marker, LC3 can bind with PE and become localized on the autophagosomal inner and outer membrane. As illustrated in Figure 5B, green fluorescence was evenly expressed in the control group, whereas more green fluorescent particles were noted in the NCTD treatment group, hinting that NCTD induced the autophagy of SK-N-SH cells.
Figure 5.

Norcantharidin (NCTD) induced autophagy and mitophagy in SK-N-SH cells. A, The expression of beclin-1, LC3, and p62 were performed by Western blotting. SK-N-SH cells were treated with NCTD (0, 5, 10, 20, or 40 μmol/L) for 24 hours. B, Punctuates of LC3 proteins in NCTD-induced SK-N-SH cells. Cells were incubated with NCTD (0 or 5 μmol/L) for 24 hours and then stained with the anti-LC3 antibody. C, Norcantharidin induced mitophagy in SKHSH cells. Different concentration of NCTD (0 or 5 μmol/L) treated SK-N-SH cells for 24 hours and stained with the anti-TOM20. Cells were examined by fluorescence confocal microscopy. Green indicates fluorescein isothiocyanate (FITC)-labeled LC3 or TOM20, and blue indicates 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI)-labeled nucleus. Magnification: ×400.
The effect of mitophagy has been widely studied and is considered to be an early manifestation of cell autophagy. Mitochondrial damage compromises mitochondrial functions, thereby inducing mitophagy.17 We investigated whether mitophagy occurs after NCTD treatment to further explore the mechanism underlying NCTD-induced autophagy of SK-N-SH cells. The TOM20 protein is a mitochondrial outer membrane protein, and therefore, reductions in its expression levels can reflect mitophagy.18 Here, anti-TOM20 was detected by immunofluorescence analysis to display the distribution of mitochondrial membrane proteins and indirectly reflect the status of mitophagy in response to treatment of SK-N-SH cells with NCTD. The green fluorescence shown in Figure 5C represents the distribution of TOM20. As compared with cells in the control group, TOM20 levels were significantly diminished, and perinuclear aggregation of TOM20 was noted in the NCTD treatment group, suggesting that NCTD induced mitophagy in SK-N-SH cells.
Effect of NCTD on the Expression of Intracellular Signaling Proteins
Some antitumor medications reportedly activate AMPK and inhibit AKT/mTOR signaling pathways and induce autophagy and apoptosis of cancer cells.19,20 Hence, expression of mTOR and phosphorylated levels AKT and AMPK in SK-N-SH cells treated with NCTD were analyzed by Western blot. As Figure 6A showed that expression of mTOR did not significantly change after treatment with a low concentration of NCTD, the levels declined considerably after treatment with 40 μmol/L NCTD. Overall, the expressions of AKT and AMPK before versus after NCTD treatment were not significantly different, but the expression of p-AKT was downregulated, whereas those of p-AMPK were upregulated with increasing concentrations of NCTD. Moreover, previous studies have demonstrated that NCTD-induced apoptosis of tumor cells is accompanied by the activation of the JNK signaling pathway.21,22 Western blot revealed that JNK expression did not significantly change in SK-N-SH cells after treatments, whereas expression levels of p-JNK, c-Jun, and p-c-Jun were elevated in a dose-dependent manner (Figure 6B), suggesting that NCTD participates in the apoptosis of SK-N-SH cells via the JNK/c-Jun signaling pathway.
Figure 6.

Effects of norcantharidin (NCTD) on intracellular signaling protein expression levels in SK-N-SH cells. A, Norcantharidin activates the AMP-activated protein kinase (AMPK) and protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway in SKNCH cell. SK-N-SH cells were treated with 0, 5, 10, 20, and 40 μmol/L NCTD for 24 hours. The effects of NCTD on AMPK, phosphor–AMP-activated protein (p-AMPK), AKT, phosphor-protein kinase B (p-AKT), and mTOR expression levels were evaluated by Western blotting. B, Norcantharidin activates the c-Jun NH2-terminal kinases (JNK)/c-Jun pathway in SK-N-SH cells. After treatment, SK-N-SH cells were subjected to Western blotting for JNK, phosphor-c-Jun NH2-terminal kinases (p-JNK), c-Jun, and phosphor-c-Jun (p-c-Jun). Both experiments were individually performed 3 times and present representative data.
Discussion
Previous studies have demonstrated that NCTD, the demethylated analog of the antitumor compound CTD found in the Chinese traditional medicine Mylabris, induces an inhibitory effect upon the growth and proliferation of multiple tumor cells in a dose-dependent manner. However, until now, no previous studies have examined the treatment of neuroblastoma with NCTD. In this study, SK-N-SH neuroblastoma cells were treated with varying concentrations of NCTD to investigate the molecular mechanisms underlying the inhibitory effect of NCTD on the growth of tumor cells. An MTT assay revealed that NCTD can significantly suppress the growth of SK-N-SH cells in a dose-dependent manner. A clone formation assay revealed that the number of cell clones formed decreased with increasing NCTD concentration, suggesting that NCTD exerts an inhibitory effect on cell growth (Figure 1). We ascribe the inhibitory effect to the promotion of cell apoptosis and autophagy by NCTD.
Apoptosis, also known as type I programmed cell death, has been widely studied in relation to the treatment of malignant tumors. In accordance with these studies, our flow cytometry results showed that NCTD can effectively induce apoptosis of SK-N-SH cells in a dose-dependent manner (Figure 3A). B-cell lymphoma 2 and caspase family members play crucial roles in cell apoptosis. Levels of Bcl-2, Bcl-xl, and Mcl-1 on the mitochondrial membrane decreased, leading to an increase in Bax/Bcl-2, Bax/Bcl-xl, and Bax/Mcl-1. Thus, the permeability of the outer mitochondrial membrane is enhanced and the ΔΨm is decreased, releasing apoptosis-promoting substances, such as cytochrome C, from the mitochondria to initiate the activation of caspase-9, which eventually activated downstream effector caspase-3 leading to cell death.23,24 In this study, expressions of Bcl-2 and Mcl-1 decreased with increasing concentrations of NCTD. However, Bax and Bcl-xl were insensitive to NCTD, resulting in an increase in Bax/Bcl-2 and Bax/Mcl-1 in the presence of NCTD, thereby cleaved active caspase-9 and caspase-3 were demonstrated. The activated caspase-3 cleaves PARP and causes morphological and biological apoptotic changes,25 which eventually inhibit proliferation and accelerate apoptosis of SK-N-SH cells. As no obvious changes were observed in caspase-8 levels, we speculate that SK-N-SH cell apoptosis induced by NCTD does not involve exogenous apoptosis pathways (Figure 4).
Autophagy, also known as type II programmed cell death, is an independent cell death process different from apoptosis.26 Excessive autophagy is likely to accelerate apoptosis promoting cell death.27 Previous studies have demonstrated that NCTD is capable of activating AMPK in mammalian animal cells.8 AMP-activated protein kinase, a protein kinase that senses energy status, is vital to the regulation of metabolism, autophagy, and apoptosis.11 As a signaling pathway molecule, AMPK is able to induce autophagy directly through the ULK1 protein or indirectly by inhibiting the mTOR signaling pathway.20 In this study, NCTD activated AMPK through upregulation of p-AMPK and suppressed the expression of mTOR, indicating that NCTD is associated with autophagy (Figure 6A). Subsequently, we demonstrated that treatment of SK-N-SH cells with NCTD upregulates the expression of beclin-1, accelerates differentiation from LC3-I to LC3-II, downregulates the expression of p62, and also relocates LC3 from the cytoplasm to the caryotheca, indicating that NCTD induces autophagy in SK-N-SH cells (Figure 5A and 5B).
Mitophagy, a selective form of macroautophagy leading to cell autophagy, refers to the scavenging of injured mitochondria by an autophagosome and then fusing of the autophagosome with a lysosome and its subsequent degradation, which completes the degradation of the injured mitochondria and thus maintains the stability of the intracellular environment.28 Persistent mitophagy eventually leads to cell autophagy. The depolarization of mitochondria is a vital early event in mitophagy.29 In this study, NCTD significantly reduced the ΔΨm of SK-N-SH cells (Figure 3B and 3C), successfully induced mitophagy (Figure 5C), and subsequently accelerated the incidence of cell autophagy.
Autophagy is considered to maintain cell metabolism, whereas excessive autophagy accelerates cell autophagic apoptosis.30 Both apoptosis and autophagy can induce programmed cell death. However, the relationship between apoptosis and autophagy remains elusive. Under certain circumstances, the induction of autophagy can accelerate the incidence of apoptosis.31 In this study, after treatment with 5 μmol/L NCTD for 24 hours, fluorescent confocal microscopy detected the apparent aggregation of LC3 proteins in SK-N-SH cells (Figure 5B). However, an insignificant increase in the level of cleaved PARP was shown in this concentration of NCTD treatment (Figure 4B), and the flow cytometry revealed late apoptosis in SK-N-SH cells (Figure 3A), indicating that autophagy was followed by apoptosis in NCTC-treated SK-N-SH cells.
To gain further insights into the mechanism of NCTD-induced SK-N-SH cell death, we examined the effects of NCTD treatment on cell signaling pathways involved in autophagy and apoptosis. Mitogen-activated protein kinase (MAPK) is a type of serine/threonine protein kinase that is able to amplify and direct the cascade of external signals and cater to environmental changes by regulating molecular and cellular physiological reactions. The c-Jun amino-terminal kinase, which is a vital member of the MAPK family, is known to be a stress-activated protein kinase, as it can regulate intracellular and extracellular stress responses. Recent studies have demonstrated that the incidence of multiple diseases could involve activation of JNK/c-Jun, reduced expressions of Bcl-2 and Mcl-1, and accelerated release of mitochondrial cytochrome C into cytoplasm, thereby leading to caspase activation and cell apoptosis.32,33 Additional studies have revealed that phosphorylated activation of JNK allows for beclin-1 activation,34 upregulation of LC3-II expression, downregulation of p62 expression, and thus acceleration of tumor cell autophagy.35,36 Anand and Babu37 demonstrated that activation of the JNK signaling pathway in nerve cells is accompanied by increased expression of p-JNK and p-c-Jun and that p-JNK plays a role in mediating the death of nerve cells. In this study, we found that NCTD significantly enhanced the expression of p-JNK and p-c-Jun in SK-N-SH cells in a dose-dependent manner. Consequently, we confirmed that NCTD participates in the autophagy and apoptosis of SK-N-SH cells by activating the JNK/c-Jun signaling pathway (Figure 6B). Beclin-1 is a key regulator of autophagy and apoptosis.38 Under normal conditions, beclin-1 constantly binds with Bcl-2 family proteins, including Bcl-2, Bcl-xl, and Mcl-1,39,40 and suppresses the incidence of autophagy. Medication-activated JNK could downregulate Bcl-2 expression. The separation of beclin-1 from beclin-1/Bcl-2 complex resulted in increased levels of free beclin-1, thus enhancing autophagy.41
Previous research has demonstrated that the AKT/mTOR signaling pathway is critical to cell proliferation and that activation of this pathway is correlated with the incidence and progression of varying malignant tumors.42,43 Some antitumor drugs can induce early autophagy and late apoptosis of tumor cells through the AKT/mTOR signaling pathway.44,45 Activated AKT can activate Bcl-2 or Bcl-xl, suppress the release of cytochrome C, and inhibit caspase proteinase, thereby suppressing the incidence of apoptosis. Activated AKT is also capable of transmitting survival signals by acting directly upon the cell cycle, inhibiting the expression of p21 and upregulating the expression of cyclin D. In this study, the expression of p-AKT was decreased at elevated concentrations of NCTD in SK-N-SH cells, corresponding to the expression level of mTOR, indicating that NCTD mediates autophagy and apoptosis of SK-N-SH cells via the inhibition of AKT/mTOR signaling pathway (Figure 6A).
As is well known, almost all types of malignant tumors are characterized by abnormal cell proliferation caused by cell cycle disorders.46 Several lines of evidence from this study have suggested that NCTD-induced apoptosis in human tumor cells might be associated with arrest of the cell cycle at the G2/M phase via changes in the expression of cell cycle-related genes, including cyclins, cyclin-dependent kinases (CDKs), and cyclin-dependent kinase inhibitors (such as p21).47,48 In the present study, Western blot analysis revealed that the expressions of cyclin D1, cyclin D3, cyclin E2, and cyclin B1 were downregulated, whereas the expression of cyclin A was upregulated (Figure 2C), indicating that the number of cells at the G2/M stage gradually increased with increasing NCTD concentrations, which is consistent with the findings of flow cytometry (Figure 2A), suggesting that NCTD is able to inhibit the proliferation of SK-N-SH cells probably by interfering with cellular mitosis. Among Cip/Kip family members, p21 and p27 can interact with CDK and cyclin compounds to arrest cells at the G1 or G2 stages. The transcription factor Myt1 can result in phosphorylation of Cdc2 at Tyr15 and Thr14, thereby arresting cells at the G2 stage. It has been reported that AMPK can suppress the cell cycle and affect cell proliferation by upregulating the expression of p21 protein.49 Our results indicate that NCTD treatment leads to a significantly dose-dependent decrease in levels of Myt1, Cdc2, and p-Cdc2 but that expression of p21 is elevated (Figure 2B), hinting that NCTD activates AMPK, inhibits AKT, upregulates p21 expression, and suppresses the activity of cyclin B1 and Cdc2, thus arresting cells at the G2/M stage.
Taken together, as we can see in Figure 7, NCTD treatment firstly reduces the ΔΨm and then initiates a program of mitochondrial autophagy, which causes the incidence of cell autophagy and cell apoptosis via mechanisms that involve activation of the AMPK signaling pathway and inhibition of mTOR expression. In addition, AMPK is able to accelerate the expression of the cell cycle-related protein p21, inhibit the activity of cyclin B1/Cdc2 via formation of p21/cyclin B1/Cdc2, downregulate the expression of cyclin D1, and arrest SK-N-SH cells at the G2/M stage of the cell cycle. Inhibition of p- AKT may also upregulate the expression of p21 and inhibit cell proliferation. On one hand, suppression of AKT phosphorylation accelerates the incidence of autophagy by downregulating the expression of mTOR, on the other hand, downregulating the expression of Bcl-2 and Mcl-1, causing a loss of ΔΨm, resulting in the activation of caspase-9 and caspase-3, leading to cleavage of PARP and eventually causing apoptosis of SK-N-SH cells. Negative regulation of Bcl-2, Mcl-1, and activation of beclin-1 equally participates in autophagy and apoptosis of SK-N-SH cells. In addition, activation of the JNK/c-Jun signaling pathway is also involved in NCTD-induced autophagy and apoptosis of SK-N-SH cells, as activation of this pathway not only upregulates the expressions of beclin-1 and LC3-II, downregulates the expression of p62, and participates in the incidence of autophagy but also downregulates the expressions of Bcl-2 and Mcl-1 and induces cell death via a mitochondrial apoptosis pathway. Consequently, NCTD is able to induce autophagy and apoptosis of neuroblastoma SK-N-SH cells via multiple signaling pathways. We conclude that NCTD may provide avenue for the development of novel therapeutics.
Figure 7.

Schematic diagrams showing the mechanisms underlying norcantharidin (NCTD)-induced SK-N-SH cell death.
Abbreviations
- AKT
protein kinase B
- AMPK
AMP-activated protein kinase
- Bax
B-cell lymphoma 2-associated X protein
- Bcl-2
B-cell lymphoma 2
- Bcl-xl
B-cell lymphoma-extra large
- Cdc2
cell division control 2
- CDK
cyclin-dependent kinase
- CTD
cantharidin
- DAPI
4′,6-diamidino-2-phenylindole dihydrochloride
- DMEM
Dulbecco modified eagle medium
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- JNK
c-Jun NH2-terminal kinases
- LC3-I
microtubule-associated protein 1A/1B-light chain 3
- MAPK
mitogen-activated protein kinase
- Mcl-1
myeloid cell leukemia 1
- mTOR
mammalian target of rapamycin
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- Myt1
myelin transcription factor 1
- NCTD
norcantharidin
- p-AKT
phosphor-protein kinase B
- p-AMPK
phosphor–AMP-activated protein
- PARP
poly-ADP ribose polymerase
- p-Cdc2
phosphor-cell division control 2
- p-c-Jun
phosphor-c-Jun
- p-JNK
phosphor-c-Jun NH2-terminal kinases
- PI
propidium iodide
- SDS
sodium dodecyl sulfate
- TBST
Tris buffered saline–Tween 20
- ΔΨm
mitochondrial membrane potential
- TOM20
Translocase Of Outer Mitochondrial Membrane 20
Footnotes
Authors’ Note: Zeping Han and Baoxia Li contributed equally to this study.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the the Fundamental Research Funds for the Central Universities (21615424) and the Natural Science Foundation of Guangdong Province in China (2014A030313356).
References
- 1. Schulte JH, Schulte S, Heukamp LC, et al. Targeted therapy for neuroblastoma: ALK inhibitors. Klin Padiatr. 2013;225(6):303–308. [DOI] [PubMed] [Google Scholar]
- 2. Castel V, Segura V, Berlanga P. Emerging drugs for neuroblastoma. Expert Opin Emerg Drugs. 2013;18(2):155–171. [DOI] [PubMed] [Google Scholar]
- 3. Moed L, Shwayder TA, Chang MW. Cantharidin revisited: a blistering defense of an ancient medicine. Arch Dermatol. 2001;137(10):1357–1360. [DOI] [PubMed] [Google Scholar]
- 4. Kadioglu O, Kermani NS, Kelter G, et al. Pharmacogenomics of cantharidin in tumor cells. Biochem Pharmacol. 2014;87(3):399–409. [DOI] [PubMed] [Google Scholar]
- 5. Yeh CH, Yang YY, Huang YF, Chow KC, Chen MF. Induction of apoptosis in human Hep3B hepatoma cells by norcantharidin through a p53 independent pathway via TRAIL/DR5 signal transduction. Chin J Integr Med. 2012;18(9):676–682. [DOI] [PubMed] [Google Scholar]
- 6. Zhang L, Ji Q, Liu X, et al. Norcantharidin inhibits tumor angiogenesis via blocking VEGFR2/MEK/ERK signaling pathways. Cancer Sci. 2013;104(5):604–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen YJ, Kuo CD, Chen SH, et al. Small-molecule synthetic compound norcantharidin reverses multi-drug resistance by regulating Sonic hedgehog signaling in human breast cancer cells. PLoS One. 2012;7(5):e37006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shen B, He PJ, Shao CL. Norcantharidin Induced DU145 cell apoptosis through ROS-mediated mitochondrial dysfunction and energy depletion. PLoS One. 2013;8(12):e84610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xie J, Zhang Y, Hu X, et al. Norcantharidin inhibits Wnt signal pathway via promoter demethylation of WIF-1 in human non-small cell lung cancer. Med Oncol. 2015;32(5):145. [DOI] [PubMed] [Google Scholar]
- 10. Liao HF, Su SL, Chen YJ, Chou CH, Kuo CD. Norcantharidin preferentially induces apoptosis in human leukemic Jurkat cells without affecting viability of normal blood mononuclear cells. Food Chem Toxicol. 2007;45(9):1678–1687. [DOI] [PubMed] [Google Scholar]
- 11. Rehman G, Shehzad A, Khan AL, Hamayun M. Role of AMP-activated protein kinase in cancer therapy. Arch Pharm (Weinheim). 2014;347(7):457–468. [DOI] [PubMed] [Google Scholar]
- 12. Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tormo D, Checińska A, Alonso-Curbelo D, et al. Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell. 2009;16(2):103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Friedhuber AM, Chandolu V, Manchun S, Donkor O, Sriamornsak P, Dass CR. Nucleotropic doxorubicin nanoparticles decrease cancer cell viability, destroy mitochondria, induce autophagy and enhance tumour necrosis. J Pharm Pharmacol. 2015;67(1):68–77. [DOI] [PubMed] [Google Scholar]
- 15. Jiao G, Ren T, Guo W, Ren C, Yang K. Arsenic trioxide inhibits growth of human chondrosarcoma cells through G2/M arrest and apoptosis as well as autophagy. Tumour Biol. 2015;36(5):3969–3977. [DOI] [PubMed] [Google Scholar]
- 16. Lee JW, Kim KS, An HK, Kim CH, Moon HI, Lee YC. Dendropanoxide induces autophagy through ERK1/2 activation in MG-63 human osteosarcoma cells and autophagy inhibition enhances dendropanoxide-induced apoptosis. PLoS One. 2013;8(12):e83611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Melser S, Lavie J, Bénard G. Mitochondrial degradation and energy metabolism. Biochim Biophys Acta. 2015;1853(10 pt B):2812–2821. [DOI] [PubMed] [Google Scholar]
- 18. Chan NC, Salazar AM, Pham AH, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20(9):1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kannaiyan R, Manu KA, Chen L, et al. Celastrol inhibits tumor cell proliferation and promotes apotosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/AKT signaling pathways. Apoptosis. 2011;16(10):1028–1024. [DOI] [PubMed] [Google Scholar]
- 20. Zhao P, Dou Y, Chen L, et al. SC-III3, a novel scopoletin derivative, induces autophagy of human hepatoma HepG2 cells through AMPK/mTOR signaling pathway by acting on mitochondria. Fitoterapia. 2015;104:31–40. [DOI] [PubMed] [Google Scholar]
- 21. Chen YJ, Kuo CD, Tsai YM, Yu CC, Wang GS, Liao HF. Norcantharidin induces anoikis through Jun-N-terminal kinase activation in CT26 colorectal cancer cells. Anticancer Drugs. 2008;19(1):55–64. [DOI] [PubMed] [Google Scholar]
- 22. An WW, Gong XF, Wang MW, Tashiro S, Onodera S, Ikejima T. Norcantharidin induces apoptosis in HeLa cells through caspase, MAPK, and mitochondrial pathways. Acta Pharmacol Sin. 2004;25(11):1502–1508. [PubMed] [Google Scholar]
- 23. Vera Y, Erkkilä K, Wang C, et al. Involvement of p38 mitogen-activated protein kinase and inducible nitric oxide synthase in apoptotic signaling of murine and human male germ cells after hormone deprivation. Mol Endocrinol. 2006;20(7):1597–1609. [DOI] [PubMed] [Google Scholar]
- 24. Edlich F, Banerjee S, Suzuki M, et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell. 2011;145(1):104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu X, Kim CN, Pohl J, Wang X. Purification and characterization of an interleukin-1beta-converting enzyme family protease that activates cysteine protease P32(CPP32). J Biol Chem. 1996;271(23):13371–13376. [PubMed] [Google Scholar]
- 26. Lalaoui N, Lindgvist LM, Sandow JJ, Ekert PG. The molecular relationships between apoptosis, autophagy and necroptosis. Semin Cell Dev Biol. 2015;39:63–69. [DOI] [PubMed] [Google Scholar]
- 27. Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16(7):966–975. [DOI] [PubMed] [Google Scholar]
- 28. Hirota Y, Kang D, Kanki T. The physiological role of mitophagy: new insights into phosphorylation events. Int J Cell Biol. 2012;2012:354914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27(2):433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liang C. Negative regulation of autophagy. Cell Death Differ. 2010;17:1707–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang YX, Xu SQ, Chen XH, Liu RS, Liang ZQ. Autophagy Involvement in olanzapine-mediated cytotoxic effects in human glioma cells. Asian Pac J Cancer Prev. 2014;15(19):8107–8113. [DOI] [PubMed] [Google Scholar]
- 32. Ju J, Qi Z, Cai X, Liu N, Wang S, Chen Y. Toosendanin induces apotosis through suppression of JNK signaling pathway in HL-60 cells. Toxicol In Vitro. 2013;27(1):232–238. [DOI] [PubMed] [Google Scholar]
- 33. Morel C, Carlson SM, White FM, Davis RJ. Mcl-1 integrates the opposing actions of signaling pathways that mediate survival and apoptosis. Mol Cell Biol. 2009;29(14):3845–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang XY, Wu XQ, Deng R, Sun T, Feng GK, Zhu XF. Upregulation of sestrin 2 expression via JNK pathway activation contributes to autophagy induction in cancer cells. Cell Signal. 2013;25(1):150–158. [DOI] [PubMed] [Google Scholar]
- 35. Xie CM, Chan WY, Yu S, Zhao J, Cheng CH. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic Biol Med. 2011;51(7):1365–1375. [DOI] [PubMed] [Google Scholar]
- 36. Kim A, Yim NH, Ma JY. Samsoeum, a traditional herbal medicine, elicits apoptotic and autophagic cell death by inhibiting Akt/mTOR and activating the JNK pathway in cancer cells. BMC Complement Altern Med. 2013;13:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anand SS, Babu PP. c-Jun N terminal kinases (JNK) are activated in the brain during the pathology of experimental cerebral malaria. Neurosci Lett. 2011;488(2):118–122. [DOI] [PubMed] [Google Scholar]
- 38. Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin1. Oncogene. 2010;29(12):1717–1719. [DOI] [PubMed] [Google Scholar]
- 39. Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. [DOI] [PubMed] [Google Scholar]
- 40. Maiuri MC, Le Toumelin G, Criollo A, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26(10):2527–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beck JT, Ismail A, Tolomeo C. Targeting the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway: an emerging treatment strategy for squamous cell lung carcinoma. Cancer Treat Rev. 2014;40(8):980–989. [DOI] [PubMed] [Google Scholar]
- 43. Guo Y, Li Y, Shan Q, He G, Lin J, Gong Y. Curcumin potentiates the anti-leukemia effects of imatinib by downregulation of the AKT/mTOR pathway and BCR/ABL gene expression in Ph+ acute lymphoblastic leukemia. Int J Biochem Cell Biol. 2015;65:1–11. [DOI] [PubMed] [Google Scholar]
- 44. Kumar D, Shankar S, Srivastava RK. Rottlerin-induced autophagy leads to the apoptosis in breast cancer stem cells: molecular mechanisms. Mol Cancer. 2013;12(1):171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. [DOI] [PubMed] [Google Scholar]
- 47. Fan YZ, Zhao ZM, Fu JY, Chen CQ, Sun W. Norcantharidin inhibits growth of human gallbladder carcinoma xenografted tumors in nude mice by inducing apoptosis and blocking the cell cycle in vivo. Hepatobiliary Pancreat Dis Int. 2010;9(4):414–422. [PubMed] [Google Scholar]
- 48. Chen YN, Chen JC, Yin SC, et al. Effector mechanisms of norcantharidin- induced mitotic arrest and apoptosis in human hepatoma cells. Int J Cancer. 2002;100(2):158–165. [DOI] [PubMed] [Google Scholar]
- 49. Zhuang Y, Miskimins WK. Cell cycle arrest in metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27kip or p21Cip. J Mol Signal. 2008;3:18. [DOI] [PMC free article] [PubMed] [Google Scholar]