

Abstract
Thiol dioxygenases are non-heme mononuclear iron enzymes that catalyze the O2-dependent oxidation of free thiols (-SH) to produce the corresponding sulfinic acid (-SO2−). Previous chemical rescue studies identified a putative FeIII-O2•− intermediate that precedes substrate oxidation in Mus musculus cysteine dioxygenase (Mm CDO). Given that a similar reactive intermediate has been identified in the extradiol dioxygenase 2, 3-HCPD, it is conceivable that these enzymes share other mechanistic features with regard to substrate oxidation. To explore this possibility, enzymatic reactions with Mm CDO (as well as the bacterial 3-mercaptopropionic acid dioxygenase, Av MDO) were performed using a substrate analogue (2-mercaptoaniline, 2ma). This aromatic thiol closely approximates the catecholic substrate of homoprotocatechuate of 2, 3-HPCD while maintaining the 2-carbon thiol-amine separation preferred by Mm CDO. Remarkably, both enzymes exhibit 2ma-gated O2-consumption; however, none of the expected products for thiol dioxygenase or intra/extradiol dioxygenase reactions were observed. Instead, benzothiazoles are produced by the condensation of 2ma with aldehydes formed by an off-pathway oxidation of primary alcohols added to aqueous reactions to solubilize the substrate. The observed oxidation of 1°-alcohols in 2ma-reactions is consistent with the formation of a high-valent intermediate similar to what has been reported for cytochrome P450 and mononuclear iron model complexes.
Keywords: Non-heme mononuclear iron enzymes, thiol dioxygenase, alcohol oxidation, benzothiazoles
Graphical Abstract
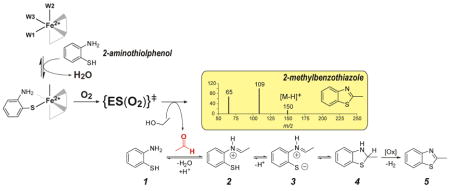
Introduction
Cysteine dioxygenase is a non-heme mononuclear iron enzyme that catalyzes the O2-dependent oxidation of L-cysteine (cys) to produce cysteine sulfinic acid (csa). Only two mammalian thiol dioxygenases have been identified to date, cysteine dioxygenase (CDO) and cysteamine (2-aminoethanethiol) dioxygenase (ADO). Among these, CDO is the best characterized [1–5]. Intracellular cys concentration is limiting in glutathione synthesis, therefore the activity of CDO directly competes with cellular redox buffering under conditions of low cys availability and oxidative stress [6]. Abnormal cys metabolism has been observed in patients suffering from a variety of neurological diseases [5, 7, 8], suggesting a correlation between impaired sulfur metabolism, oxidative stress, and neurodegenerative disease [9, 10]. Consequently, enzymes involved in sulfur-oxidation and transfer are increasingly being recognized as potential drug targets for development of antimicrobials, therapies for cancer and inflammatory disease [11–14].
Shown in Figure 1A is the crystal structure for the Rattus norvegicus CDO (pdb code 4IEV) active site [15]. The iron first coordination sphere is comprised of iron ligated along one octahedral face by the Nε-atoms of H86, H88, and H140. Prior to the addition of O2, cys coordinates to the mononuclear ferrous site of CDO in a bidentate fashion through a thiolate and neutral amine [16–18]. As illustrated in Fig. 1B, ligation of the cys thiol-group within the trigonal plane opposite H86 and H88 positions the carboxylate group of cys favorably for charge stabilization by the R60 guanidinium group.
Figure 1.

Crystal structure of the resting and substrate-bound Rattus norvegicus CDO.
Crystal structure of the Rattus norvegicus CDO active site (A, pdb code 4IEV) as compared to the substrate-bound enzyme (B, pdb code 4IEZ) [15]. Selected distances A: 1 (2.63 Å); 2 (2.73 Å); 3 (2.72 Å); B: 1′ (2.78 Å); 2′ (2.82 Å); 3′ (2.91 Å); and 4′ (3.10 Å).
It was previously demonstrated that the catalytic cycle of CDO can be ‘primed’ by 1-electron through chemical oxidation to produce CDO with ferric iron in the active site [19]. While catalytically inactive, the substrate-bound form of FeIII-CDO is more amenable to interrogation by UV-visible and EPR spectroscopy than the ‘as-isolated’ FeII-CDO enzyme. Chemical-rescue experiments were performed in which superoxide (O2•−) was introduced to the substrate-bound FeIII-CDO to produce and characterize by EPR and UV-visible spectroscopy a transient intermediate kinetically matched to csa formation. This intermediate was tentatively assigned as a substrate-bound FeIII-superoxide species similar to that spectroscopically observed for the extradiol cleaving catechol dioxygenase, homoprotocatechuate 2, 3-dioxygenase, (2, 3-HPCD) [20, 21]. While the decay of this intermediate was kinetically matched to the formation of product, the rate of csa produced was significantly slower than the steady-state turnover for Mm CDO. It is therefore not clear if this represents a catalytically relevant intermediate. More recently, stopped-flow UV-visible studies identified a transient species upon rapid mixing of the substrate-bound CDO with saturating oxygen. Supporting computational modeling of the transient UV-visible spectra suggest that this transient species is consistent with a cyclic [Fe-O-O-SCys]-structure with a quintet (S = 2) spin state. However, rapid freeze-quench Mössbauer experiments were unable to corroborate this assignment [22]. Therefore, direct spectroscopic validation of the transient intermediate produced during native catalysis is absent.
Catechol dioxygenases are key metabolic enzymes found within soil bacteria having the capacity to degrade aromatic compounds, which can be utilized as a carbon source [23, 24]. These enzymes are classified into two primary families based on the position of substrate ring cleavage and oxidation state of the catalytically essential iron within the non-heme active site. For instance, intradiol dioxygenase enzymes such as catechol 1, 2-dioxygenase (CTD) and protocatechuate 3, 4-dioxygenase (3, 4-PCD) catalyze the O2-dependent carbon-carbon bond cleavage between the substrate phenolic hydroxyl groups to yield a dicarboxylic acid muconate product. This class of enzymes stabilizes a ferric iron (FeIII) resting state, which activates the substrate for electrophilic attack by oxygen. It has been proposed that the high activation barrier for direct O2-attack on the substrate is overcome by resonance delocalization of unpaired spin-density onto the substrate from the substrate-bound high-spin ferric site to facilitate a spin-allowed reaction with triplet O2 [20, 23]. Alternatively, extradiol dioxygenases such as the aforementioned 2, 3-HCPD cleave the carbon-carbon bond adjacent to the ortho-hydroxyl groups to yield 5-carboxymethyl-2-hydroxymuconic semialdehyde. These enzymes stabilize a resting ferrous iron (FeII), or in rare instances MnII, in the active site to activate molecular oxygen for subsequent attack on the substrate [25]. For these enzymes, reversible binding of the catechol substrate precedes O2-binding at the substrate-bound ferrous active site. Oxidative addition of O2 to the ferrous iron yields a short-lived ferric-superoxo [FeIII-O2•−] species ultimately responsible for nucleophilic attack on the aromatic substrate [26].
Assuming both 2, 3-HPCD and CDO produce a FeIII-superoxo intermediate prior to substrate oxidation, it is possible that additional mechanistic parallels exist in reactions utilizing aromatic substrates. Steady-state specificity experiments with the Mus musculus CDO (Mm CDO) demonstrated that the minimal substrate for CDO must contain both amine and thiol functional groups, and that optimal activity and efficiency is observed when these groups are separated by two carbon units [27]. With this in mind, 2-mercaptoaniline (2ma) could provide an interesting comparison to the aforementioned catechol dioxygenase class of non-heme mononuclear iron enzymes. It should be noted that the bonds separating the thiol and amine functional groups in the aromatic 2ma substrate are sp2 hybridized. As these bonds cannot be rotated to the extent of aliphatic thiols, the 2ma-bound Fe-site geometry is expected to be significantly perturbed relative to the native ES-complex. Another factor to consider is the differential reactivity of aromatic versus aliphatic thiols. Potentially, these deviations may result in unique reactivity.
Neglecting potential hydrolysis, cyclization, or tautomerization, Scheme 1 illustrates the expected reaction products for analogous thiol dioxygenase, extradiol-, or intradiol cleaving activity.
Scheme 1.
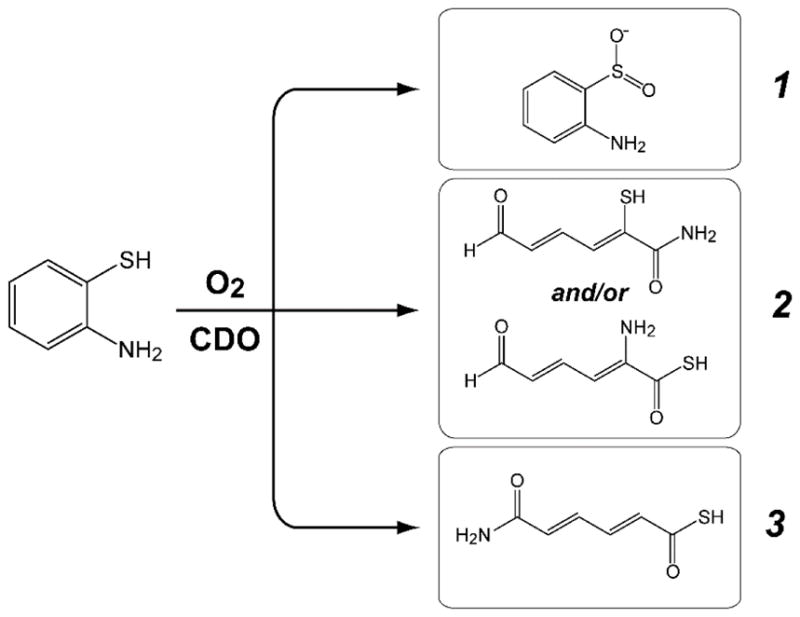
Potential reaction products for O2-dependent 2ma oxidation by Mm CDO
Potential reaction products for O2-dependent 2ma oxidation catalyzed by Mm CDO assuming (1) thiol dioxygenase [aminobenzenesulfinic acid], (2) extradiol dioxygenase- [2-mercapto-6-oxohexa-2, 4-dienamide or 2-amino-6-oxohexa-2, 4-dienethioic S-acid], or (3) intradiol dioxygenase cleaving activity [6-amino-6-oxohexa-2, 4-dienethioic S-acid].
Presented here are the results of O2-dependent 2ma reaction using two different thiol dioxygenase enzymes [Mm CDO and 3-mercaptopropionic acid dioxygenase isolated from Azotobacter vinelandii (Av MDO)]. These enzymes bind substrates bidentate [16–18] and monodentate [28, 29], respectively and thus may exhibit differential reactivity.
Materials and Methods
Enzyme purification
Recombinant Mus musculus cysteine dioxygenase (Mm CDO) and Azotobacter vinelandii 3-mercaptopropionic acid dioxygenase (Av MDO) were expressed, purified, and activity verified as described previously [29, 30]. For all batches of enzyme used in these experiments, spectrophotometric determination of ferrous and ferric iron content was measured using 2, 4, 6-tripyridyl-s-triazine (TPTZ). A detailed description of this method is presented elsewhere [18, 29]. For clarity, the concentrations reported in enzymatic assays reflect the concentration of ferrous iron within enzyme samples. The protein content in each sample was determined by Bio-Rad protein assay. Steady-state kinetic parameters observed at 20 ° C for Mm CDO and Av MDO were consistent with previously published values (Mm CDO; kcat, 0.7 ± 0.1 s−1; KM, 2.1 ± 0.3 mM; kcat/KM, 330 ± 50 M−1 s−1; Av MDO; kcat, 0.5 ± 0.1 s−1; KM, 18 ± 2 μM; kcat/KM, 28,000 ± 3000 M−1 s−1) [28, 29].
Reagents and stock solutions
2-mercaptoaniline (2ma) (99%) was purchased from Sigma-Aldrich (cat. no. 274240) and stored under argon within an anaerobic chamber to prevent disulfide formation. Stock solutions of 2ma (0.75 mM) were prepared anaerobically in ethanol (95%, ACROS) prior to subsequent dilution in aqueous buffer (10mM HEPES, 50 mM NaCl, and pH 8.5) for enzymatic assays. The benzothiazole (bt) [96%, cat. no. 101338] and 2-methylbenzothiazole (2m-bt) [99%, cat. no. 112143] standards used for spike assays and LC-MS/MS (MRM) verification of products were purchased from Sigma-Aldrich and used without further purification. However, 2-phenybenzothiazole (2p-bt) was synthesized as described below. All standards were prepared by serial dilution of a ca. 5 mM stock solution in 80:20 (buffer:ethanol). Spike assays were performed by serial dilution of the stock solution to 1–5 μM prior to HPLC analysis.
Synthesis of 2-phenybenzothiazole (2p-bt)
Synthesis of 2-phenylbenzothiazole was performed as described elsewhere [31]. Briefly, 2-mercaptoaniline (250 mg, 2 mmol), benzaldehyde (212 mg, 2 mmol) and excess acetic acid were heated for 4 hours at reflux temperature. After cooling it to room temperature, the reaction mixture was added into an ice water mixture. The solid precipitated out was collected by suction filtration. The crude solid was crystallized from ethanol/water to give 345 mg of 2p-bt in 82% yield as colorless solid.
Characterization of 2p-bt product was performed by 500 MHz 1H and 125 MHz 13C NMR experiments performed on a JOEL Eclipse Plus 500 NMR spectrometer. Chemical shifts were recorded in reference to residual solvent peaks (CDCl3 = 7.26 ppm). FT-IR spectra were recorded on a Bruker Alpha-P FT-IR Spectrometer by attenuated total reflectance on a diamond sample plate. 1H NMR (500 MHz, CDCl3) δ 8.13 – 8.09 (m, 3H), 7.89 (ddd, J = 8.0, 1.2, 0.6 Hz, 1H), 7.53 – 7.47 (m, 4H), 7.40 – 7.36 ppm (m, 1H); 13C NMR (126 MHz, CDCl3) δ 168.1, 154.2, 135.1, 133.7, 131.0, 129.1, 127.6, 126.4, 125.3, 123.3, 121.7; IR (Neat): 3064, 3018, 1477, 1432, 1312, 1224, 962, 757, 729. Melting point (113–114 °C) was obtained in capillary tubes on a Mel-Temp II apparatus, and the thermometer was uncorrected. These values are in agreement with those reported in the literature [32].
UV-visible experiments
UV-visible measurements were performed on an Agilent 8453 photo diode array spectrometer (Santa Clara, CA). Sample temperature was held constant by a 13L circulating water bath and an Agilent thermostatable cell holder (89054A) with magnetic stirrer. All measurements were made in ES Quartz cuvettes (NSG Precision Cells, Farmingdale, NY). For time dependent assays, a baseline correction at 900 nm was used. Spectra collected in kinetic mode used a 4-second interval between measurements for the first 3-minutes. Beyond this point, time intervals were increased by 5% up to 15 minutes.
In a typical UV-visible experiment, an O2-saturated buffer [10mM HEPES, pH 8.5 at 20 °C] was diluted with anaerobic buffer and ethanol to obtain the desired O2 concentration and 20% (v/v) ethanol (1.0 mL final volume). This solution was used to blank the UV-visible spectrometer. The stock 2ma solution was then added to obtain substrate concentrations ranging from ca. 30–130 μM. Reactions were initiated by injection of enzyme (final concentration, 2 μM).
Oxygen Electrode
Consumption of molecular oxygen was verified polarographically using a standard Clark electrode (Hansatech Instruments, Norfolk, England) in a jacketed 2.5 mL cell. Calibration of O2-electrode is described in detail elsewhere [30, 33]. All reactions were initiated by addition of 2.0 μM enzyme (Mm CDO or Av MDO) under identical buffer conditions as described for UV-visible experiments. Reaction temperatures were maintained at 20 ± 2 °C by circulating water bath (ThermoFlex 900, Thermo Scientific).
HPLC sample preparation
Samples were prepared for select reaction time points ranging from 1–30 minutes by removing an aliquot of the reaction (180 μL) and quenching by addition of 5 μL 2N HCl. Samples were purified by 0.22 μM cellulose acetate membrane filtration (Corning, Spin-X) prior to analysis. As a control, a zero point sample was prepared by substituting heat-denatured enzyme in the above reaction. Unless otherwise noted, the concentration of 2ma, oxygen, and alcohol in reaction buffers was fixed at 150 μM, 420 μM (35% saturated), and 20% (v/v), respectively.
HPLC instrumentation and analysis
HPLC samples were analyzed using a Dionex UltiMate 3000 HPLC equipped with a 3000 variable wavelength detector. Column, Phenomex Luna (5 μm) C18 100 Å, column [250 x 4.6 mm] with accompanying C18 [4 x 3.0 mm] guard column; mobile phase, 35:65 HPLC grade H2O:methanol; injection volume, 20 μL; isocratic flow rate, 0.5 mL/min; column temperature, 25 °C. UV-visible detection was monitored at 270 nm. Calibration curves for 2ma, bt, 2m-bt, and 2p-bt were prepared for determination of product concentration. Serial dilution of benzothiazole standards exhibited a linear response in concentrations ranging from 10–1100 μM.
LC-MS/MS (multiple reaction monitoring)
Verification of enzymatic product was performed by multiple reaction monitoring (MRM) using a triple quadrupole LC-MS/MS [Shimadzu Scientific Instruments, LCMS 8040] [34–36]. For each experiment, 10 μL of the benzothiazole standard (described above) was injected to determine the product ions and relative distribution for each ion. The molecular ion [M−H]+ for each specific benzothiazole standard [bt, 136 m/z; 2m-bt, 150 m/z; and 2p-bt, 212 m/z] was selected for secondary fragmentation in positive ion mode [37]. MRM optimization was then employed to maximize transition intensity and sensitivity for each fragment allowing for quantitation of product ions. The optimized MRM method was used to verify the formation of specific benzothiazole product ions and their relative intensities in enzymatic reactions by direct injection of assays.
GC-MS/MS sample preparation
A standard solution of benzaldehyde was prepared by dissolving 2 μL of benzaldehyde in 500 μL of dichloromethane. The same procedure was used for making standard solution of acetaldehyde. Detection of benzaldehyde in analyte samples was performed after extraction with 500 μL dichloromethane. Detection of acetaldehyde in analyte samples was performed with direct injection of the sample, without extracting with dichloromethane. Note: Acetaldehyde and benzaldehyde are highly volatile and therefore all extractions were performed in the sample vial using a Hamilton gas-tight syringe to prevent loss of volatiles.
GC-MS/MS Instrumentation and Method
GC-MS experiments were conducted in Shimadzu Scientific Instruments, GCMS-QP2010, using electron impact ionization (EI) at 150 eV and a mass selective detector. The method used for detecting acetaldehyde in the GC experiment represents 0.5 min @ 50 °C – 30 °C/min – 7.5 min @ 260 °C with injection volume of 1μL. For benzaldehyde, the GC experiment represents 1.0 min @ 50 °C – 30 °C/min – 8.0 min @ 260 °C with injection volume of 1 μL.
Results and Discussion
CDO catalyzes the O2-dependent oxidation of a variety of substrates which contain both amine and thiol functional groups separated by two carbon atoms. Understandably, the catalytic efficiency (kcat/KM) for substrates other than cys is poor, nevertheless product formation is easily observed at concentrations amenable to HPLC, NMR, and LC-MS/MS analysis [27]. Kinetic experiments were performed on an Agilent photodiode array spectrometer as formation of the 2ma-sulfinic acid product (2-aminobenzensulfinate, 2abs) is expected to be red-shifted relative to the starting substrate. Given the low solubility of 2ma, ethanol was added to the enzymatic reactions to increase the upper limit of substrate concentration in solution. As a control, equivalent Mm CDO assays with cys were carried in the same buffer to verify that the addition of ethanol did not abolish enzymatic activity. In assays performed in 20% (v/v) ethanol, only a modest loss (10–20%) of Mm CDO specific activity was observed within the time scale of the experiments (10–20 minutes).
As illustrated in Figure 2, introduction of enzyme results in rapid change in the UV-visible spectrum ranging from 250 to 450 nm. A single isosbestic point at 318 nm confirms the presence of at least 2 species in reaction mixtures. As illustrated in Fig. S1A, kinetic flooding experiments at fixed Mm CDO (2 μM) and O2 (420 μM) exhibit a linear increase in the observed rate (vobs) with increasing 2ma concentration. Similarly, Fig. S1B demonstrates that vobs also increases linearly with increasing enzyme concentration at fixed 2ma (125 μM) and O2 (420 μM) concentration. Finally, a linear increase in vobs is also observed with increasing O2-concentration (Fig. 2, inset). These experiments verify that the reaction is first-order with respect to all reactants (Mm CDO, O2, and 2ma). However, full enzymatic saturation is not observed within the 2ma (30–150 μM) or oxygen (50–900 μM) concentration range utilized in these experiments. Crucially, no change in the UV-visible data is observed upon addition of heat inactivated enzyme thus verifying that this reaction is not initiated by adventitious iron within enzyme samples.
Figure 2.
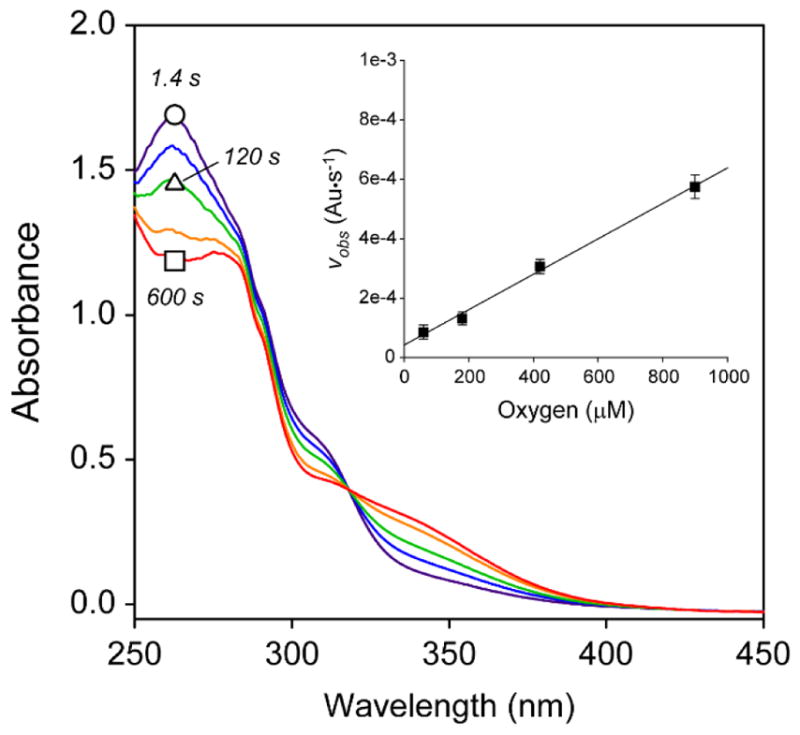
UV-visible spectra of 2ma-reactions and pseudo first order kinetic plot.
Representative UV-visible spectra illustrating the O2-dependent reaction of 2-mercaptoaniline (2ma) catalyzed by Mm CDO. Selected time points: 1.4 s (purple trace, ○), 50 s, 120 s (green, Δ), 300 s and 600 s (red, □). Assay conditions: 2 μM Mm CDO, 420 μM O2, 20% ethanol (v/v), 10 mM HEPES, pH 8.5 and 25 ± 2 °C. Inset. Pseudo first-order kinetics of initial rate (vobs) as a function of oxygen concentration. The progress of the reaction at fixed and saturating 2ma (130 μM) was monitored spectrophotometrically at 305 nm.
Consumption of oxygen was verified using a standard Clarke-type electrode. Representative O2 electrode results are shown in Supplementary Information (Figure S2). In these studies, equivalent reactions as described above were initiated by injection of enzyme. As with UV-visible experiments, the rate of O2-consumption increased linearly with increasing enzyme and 2ma concentration. As an additional control, reactions were performed using benzenethiol as a substrate. However, neither CDO nor MDO exhibited any significant consumption of oxygen upon initiation of benzenethiol-reactions (Supplemental Information, Figure S2).
A translational benefit of this reaction is that 2ma can be used to optically screen for thiol dioxygenase activity. As shown in Fig. 2, enzymatic 2ma-reactions result in the formation of absorption features ~350 nm and thus a yellow color is apparent as the reaction progresses. While spectrophotometric and O2-electrode results are useful for interrogation of the relative timing of chemical events, product analysis and assignment was performed using HPLC and LC-MS/MS as described in Materials and Methods. Figure 3 shows selected HPLC traces (spectrophotometric detection at 305 nm) for reactions of Mm CDO with 2ma in 20% (v/v) ethanol. Within this window, two peaks observed at retention times of 11.6 and 16.2 min show a clear time-dependent behavior. The 11.6 min peak decreases in intensity with time and can easily be identified as the starting 2ma by the matching retention times observed in calibration curves. LC-MS (and LC-MS/MS) was run in positive detection mode using the same mobile phase, flow rate, and column to observed the molecular ion [M−H]+ (150 m/z) of the species eluted with a retention time of 16.2 min. Since the expected mass for all dioxygenase products utilizing 2ma is 157 Da, the 150 m/z [M−H]+ species cannot be attributed to any of the reactions predicted in Scheme 1.
Figure 3.

Reverse phase HPLC chromatogram for 2ma-reactions at selected time points.
Representative HPLC chromatograms illustrating the change in analyte composition following introduction of enzyme. The initial composition of the reaction mixture is indicated by the bottom trace. The above chromatograms were quenched following 1- and 5-minutes reaction with Mm CDO. The peaks associated with 2ma (retention time, 11.6 min) and 2m-bt (retention time, 16.2 min) were verified by matching retention time with analytical standards, spike assays, and LC-MS/MS.
A fortuitous observation was made in equivalent reactions carried out where methanol was substituted for ethanol. In these reactions, the 16.2 min peak shifted to a lower retention time and the observed molecular ion [M−H]+ decreased to 136 m/z. The -14 amu shift in the molecular ion clearly suggests the absence of a (-CH2-) fragment in methanol reactions as compared to ethanol. Similarly, if benzyl alcohol is substituted for ethanol, a positive molecular ion peak of 212 m/z is observed (+76 amu shift relative to the product formed in methanol). In all cases, the size of the molecular ion [M−H]+ can be calculated by the sum of 2ma (125 amu) and primary alcohol molecular weight (methanol, 32 amu; ethanol, 46 amu; and benzyl alcohol, 108 amu) followed by subtraction of water (18 amu) and 3 hydrogen atoms. This observation indicates that the alcohol utilized in this reaction becomes directly incorporated into the oxidized product.
Based on similar reactivity reported for flavin mimics [38], cytochrome P450 [39], and mononuclear iron model complexes [40–42], we hypothesize that O2-dependent 2e− oxidation of the solvent primary alcohol results in formation of aldehydes. Rapid and near quantitative condensation of an aldehyde with the 2ma starting material is anticipated based on the well-established nucleophilic addition of amines. Following this step, it is reasonable to expect formation of a benzothiazole product as illustrated in Scheme 2.
Scheme 2.

Proposed mechanism for benzothiazole formation.
Proposed reaction mechanism for benzothiazole formation.
Many synthetic methods describe the oxidative formation of 2-substituted benzothiazoles from 2-aminothiophenol (Scheme 2, compound 1) and aldehydes under various aerobic and dehydrogenative conditions [38, 43–45]. Generally, this transformation can be envisioned from the initial, reversible condensation of an aldehyde, e.g. acetaldehyde, with the amine to form iminium intermediate 2. This species undergoes rapid intramolecular cyclization with the nucleophilic thiol. At the working pH and with the electron withdrawing iminium substituent, it is likely that the sulfur nucleophile can form a significant concentration of the thiolate intermediate, 3, further favoring the cyclization reaction. After cyclization to benzothiazoline, 4, dehydrogenative formation of the benzothiazole product (similar to NADPH oxidation) is quite facile with a variety of mild oxidants (designated [Ox] in Scheme 2).
This mechanistic model correctly predicts the resulting molecular ion size observed in the aforementioned reactions. Further corroboration for the proposed reaction can be taken tandem LC-MS/MS [multiple reaction monitoring (MRM)] results obtained for reaction mixtures. In these experiments, standard solutions (1 μM each) of benzothiazole (bt), 2-methylbenzothiazole (2m-bt), and 2-phenybenzothiazole (2p-bt) were prepared as described in Materials and Methods for direct injection. The [M+H]+ molecular ion for each standard (bt, 136 m/z; 2m-bt, 150 m/z; and 2p-bt, 212 m/z) was selected for secondary fragmentation and MRM optimization was then employed to maximize transition intensity and sensitivity for each fragment allowing for quantitative analysis of product ions. The optimized MRM method was used to compare the product of each enzymatic reaction. For simplicity, all enzymatic reactions were quenched at 5 minutes after injection of enzyme (2 μL).
Figure 4 (panel 1) shows the MRM spectra for a standard solution of 2m-bt in the same assay buffer used for kinetic experiments. In addition to the 150 m/z parent [M+H]+ ion, two additional ions are observed at 65 and 109 m/z. For comparison, the direct injection of the Mm CDO catalyzed reaction with 2ma shown in Fig. 4 (panel 2) yields an identical fragmentation pattern. Both the matching fragmentation pattern and relative intensities verify that product generated in O2-dependent assays with Mm CDO is indeed 2-methylbenzothiazole. Final corroboration was obtained by spiking known quantities of 2m-bt standards into enzymatic assays. As expected, 2m-bt coelutes exactly with the 16.2 min peak observed in HPLC chromatograms. These observations identify 2m-bt as the product of enzymatic reactions with 2ma in the presence of ethanol. For comparison, Figure 5 illustrates the same analysis used to verify formation of bt and 2p-bt in reactions utilizing 20% (v/v) methanol and benzyl alcohol, respectively.
Figure 4.
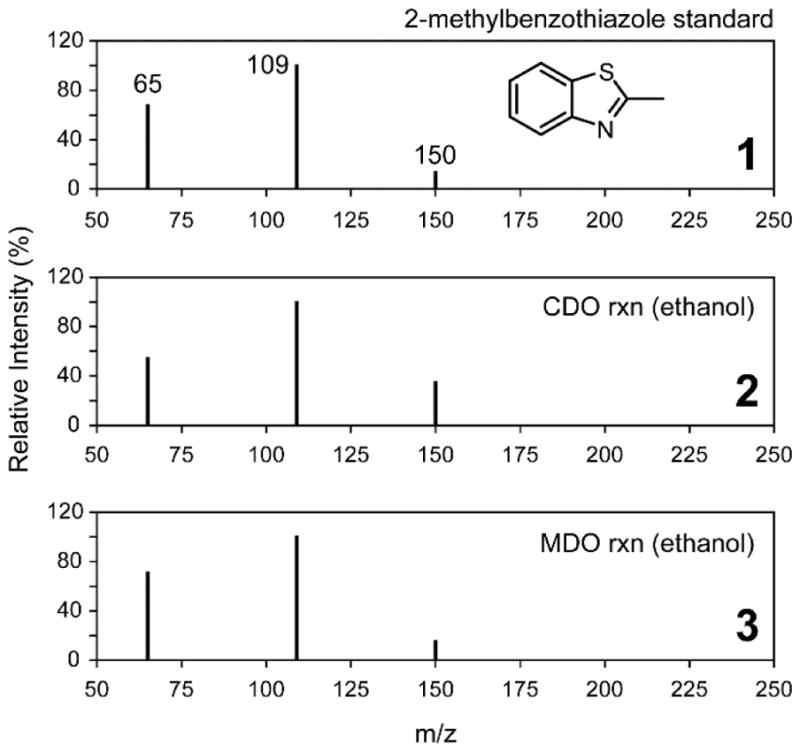
MRM spectrum of 2m-bt compared to 2ma-reactions in ethanol.
LC-MS/MS spectra of MRM [M−H]+ ions observed for the 2-methylbenzothiazole (Mol. Wt. mass, 149.21 Da) standard (panel 1). The lower panels represent direct injection of Mm CDO (2) and Av MDO (3) assays following 10 minutes reaction time. Assay conditions are identical as those described for Fig. 3.
Figure 5.

MRM spectrum of bt and 2p-bt compared to 2ma-reactions in methanol (A) and benzyl alcohol (B).
LC-MS/MS spectra of MRM [M−H]+ ions observed for the benzothiazole (A, Mol. Wt., 135.19 Da) and phenyl-benzothiazole (B, Mol. Wt., 211.28 Da) standards are shown in panel 1. Direct injection of the enzymatic assays carried out with Mm CDO in methanol (A) and benzyl alcohol (B) are shown in panel 2. Equivalent Av MDO reactions are shown in panel 3.
To this point, the evidence for aldehyde formation by enzymatic oxidation of primary alcohols has been largely inferred based on the products observed. Therefore, GC-MS analysis was performed on extracts of reactions mixtures to directly verify their presence and corroborate the proposed mechanism shown in Scheme 2. For this experiment, benzyl alcohol was selected as the co-solvent as its lower volatility (relative to formaldehyde and acetaldehyde) would decrease the possibility for loss during organic extraction. As shown in Figure 6A (trace 1), beyond the void volume peak at 1.94 min, a single peak is observed at a retention time of 2.94 minutes upon injection of a benzaldehyde standard. Enzymatic assays were performed identically to those described previously for LC-MS experiments. Reactions were initiated by addition of enzyme and allowed to turnover for 20 minutes prior to inactivation by spin-filtration (Corning, Spin-X) and extraction of organic components with dichloromethane. GC-MS chromatograms of reaction extracts exhibit a major peak at 3.33 minutes which corresponds to the benzyl alcohol solvent. While small relative to the benzyl alcohol peak, an additional peak matching the retention time of benzaldehyde (2.94 min) is readily observed in extracts of enzymatic reactions.
Figure 6.

GC-MS/MS chromatogram and MRM spectrum of benzaldehyde as compared to 2ma-reaction performed in benzyl alcohol.
GC-MS/MS chromatogram (A) and MRM [M−H]+ spectra (B) of enzymatic 2ma-reaction extracts. Panel 1 shows the secondary fragmentation pattern of the parent [M−H]+ benzaldehyde ion (107 m/z) observed in standard reactions (retention time of 2.94 min). Panel 2 shows the comparative MRM spectra for the enzymatic (Mm CDO) 2ma-reaction dichloromethane extract.
Shown in Figure 6B is the tandem mass spectrum (MRM) for the standard benzaldehyde peak compared to that observed for enzymatic reaction extracts. The molecular ion for benzaldehyde (106 m/z) was selected for secondary fragmentation and the optimized MRM analysis was run in positive detection mode. As illustrated in Fig. 6B (trace 1), beyond the molecular ion, a number of additional fragmentation ions are observed for the benzaldehyde standard (39, 51, 77, 78, 105, 106, and 107 m/z). For comparison, the mass fragmentation and relative intensities of ions observed in reaction extracts (Fig. 6B, trace 2) matches that of the benzaldehyde standard confirming the presence of benzaldehyde in reaction mixtures.
Similarly, MRM spectra obtained by direct injection of reactions carried out in 20% (v/v) ethanol verify the formation of acetaldehyde in enzymatic 2ma-reactions (Supporting Information, Figure S3). As with reactions performed in benzyl alcohol, the secondary fragmentation pattern of reaction extracts carried out in ethanol match acetaldehyde standards. Most important, control reactions performed in ethanol containing enzyme but lacking the 2ma substrate (Fig. S3, trace 3) exhibit a markedly different fragmentation pattern as either standards or enzymatic 2ma-reactions indicating that acetaldehyde is not present in the starting alcohol solvent and is only produced during enzymatic turnover with 2ma.
The scope of this chemistry was further investigated by substituting 20% (v/v) dimethyl sulfoxide (DMSO) rather than a primary alcohol as this solvent cannot form an aldehyde upon oxidation. As predicted by the proposed mechanism shown in Scheme 2, no benzothiazole products are observed in DMSO reactions. Moreover, no evidence for the formation of 3abs [157 Da] was observed by LC-MS either. Similar results were obtained in reactions with dimethylformamide (DMF) as a co-solvent and in reactions lacking any primary alcohol co-solvent. Reactions were performed utilizing the secondary alcohol 2-propanol as well but these failed to produce the expected 2, 2-dimethylbenzothiazoline product. This results of this experiment are discussed in greater detail within the Supplemental Information, Scheme 1 and Fig. S4. Collectively, these results imply that the formation of benzothiazoles is limited to the use of primary alcohols.
With the identity of reaction products bt, 2m-bt, 2p-bt confirmed by overlapping HPLC retention time and LC-MS/MS MRM, analytical determination of product yield was conducted by HPLC by comparison to primary standards. Given the proposed reaction shown in Scheme 2 it is not surprising that the product yield for these reactions is fairly low. For example, in reactions yielding 2m-bt, only 43 ± 8 μM product is observed within the first 5 minutes of the reaction. As enzyme concentration is 2 μM, this suggests 2m-bt is produced over multiple enzymatic turnovers. Beyond 10 minutes, no appreciable increase in 2m-bt concentration is observed. In fact, 2m-bt concentration appears to decrease slightly suggesting either precipitation of the benzothiazole product or further decay. Likely the rapid inactivation of the enzyme can be explained by either the release of ROS or protein denaturation precipitated by the solvent alcohol. Reactions yielding bt and 2p-bt follow a similar behavior but with attenuated product yields at 5 minutes (28 ± 6 and 3.5 ± 1.2 μM, respectively).
This reaction is first order with respect to oxygen concentration. However, from the perspective of mass balance, the absence of O-atom incorporation into the benzothiazole product (or oxidized 1°-alcohol solvents) indicates that this reaction is completely uncoupled with respect to oxygen; suggesting that all O2 consumed over the course of the reaction is released as reactive oxygen species (ROS; H2O2, O2•−, •OH). Therefore, an obvious question is whether transient Fe-oxo intermediates produced during enzymatic turnover or ROS released by uncoupled catalysis are responsible for direct oxidation of the primary alcohols in solution. However, no decrease in 2m-bt generated, nor its rate of formation was observed in reactions carried out in the presence of catalase, superoxide dismutase, or both (1 mg/mL each); in fact, 2m-bt yields are slightly increased (~20%). This suggests that, if ROS are produced in O2-dependent 2ma-reactions, they do not freely disassociate from the enzymatic active site. It is therefore more likely that a transient Fe-oxo species is responsible for the oxidation of 1°-alcohols. Potentially, ROS generated during the oxidation of the 1°-alcohols could act as an oxidant [Ox] for the dehydrogenation of benzothiazoline (4) to form the final benzothiazole product (5) shown in Scheme 2.
This uncoupled reactivity with oxygen reminiscent to what is observed for Mm CDO in reactions utilizing L-selenocysteine (sec) as a substrate. Despite modest spectroscopic perturbations from the increased polorizability of the Se-atom, both EPR and MCD spectroscopy indicate that the substrate-bound Mm CDO complex with cys and sec exhibit near equivalent coordination geometry and symmetry around the Fe-site [19, 46]. However, rapid mixing of O2 with the sec-bound enzyme produces no detectable dioxygenase products. Based on spectroscopically validated computational studies, it was proposed that the subtle electronic differences in the Fe-bound O2-adduct produced in sec-reactions differ substantially from the native reactions with cys resulting in an ‘off-pathway’ reaction coordinate [47]. The results presented here suggest that the structural constraints and/or differential reactivity of the aromatic 2ma substrate produce a similar effect on CDO and MDO.
The mononuclear non-heme iron site of Mm CDO binds substrate bidentate via neutral amine and thiolate coordination. By contrast, it has been demonstrated in both steady-state kinetic and EPR spectroscopic studies that the homologous 3-mercaptopropionic dioxygenase isolated from the soil bacteria Azotobacter vinelandii (Av MDO) binds substrate via thiolate coordination only [28, 29]. Given the change within the first coordination sphere of the enzyme-substrate complex for both thiol dioxygenase enzymes, it is possible that their reactivity with 2ma could differ. However, as shown in Fig. 4 and Fig. 5 (panel 3), identical benzothiazole products are produced in reactions utilizing Av MDO. Surprisingly, despite the monodentate substrate-coordination of MDO, no reactivity was observed toward benzenethiol (Supplemental Information, Fig. S2).
Regardless, these experiments clearly demonstrate that 2ma is capable of initiating O2-consumption for both Mm CDO and Av MDO. However, rather than the expected sulfinic acid, a benzothiazole product is generated following oxidation of 1°-alcohols in solution. Vastly decreased activities, specificity, and O2-coupling efficiencies have been reported for CDO and active site variants utilizing non-native substrates [27]. Extensive x-ray crystallographic studies have also been performed highlighting crucial outer Fe-coordination sphere interactions which facilitate efficient catalysis [48]. In some instances, relatively minor or imperceptible structural perturbations within the active site can produce significant shifts in enzymatic activity [49]. While MDO substrate specificity is significantly more relaxed as compared to CDO, both activity and O2-coupling efficiency are attenuated in reactions with cysteamine (ca) and cysteine (cys) [29, 50]. However, to our knowledge, no crystallographic studies have been reported for the substrate-bound MDO and thus outer Fe-coordination sphere interactions facilitating efficient catalysis are poorly understood.
Although the dioxygenase product (2abs) is never observed in 2ma-reactions, direct consumption of molecular oxygen is observed with pseudo-first order kinetics dependence on O2, 2ma, and enzyme concentration. It is therefore reasonable that an oxidized iron-oxo species is produced following substrate-gated O2-activaiton. Based on a similar reports for a variety of model Fe-oxo compounds [40–42] and Compound I of CytP450 [39], it is reasonable to conclude that the substrate-oxidizing transient intermediate is similarly responsible for the oxidation of the 1°-alcohol in solution in 2ma-reactions. Among similar O2-dependent oxidation of alcohols, H-atom abstraction is typically the rate limiting chemical step. This frequently results in kinetic isotope effects beyond the classical limit (kH/kD ≥ 7) [40–42, 51, 52]. By contrast, in Mm CDO O2-saturated reactions with 2ma, a kinetic isotope effect of 1.54 ± 0.13 (n = 3) was observed in equivalent reactions where the 20% (v/v) ethanol buffer was prepared using 99.5% ethanol-d6 [Sigma-Aldrich, 186414]. This suggests that H-atom abstraction from the alcohol is not the rate limiting step. However, saturation kinetics for 2ma is not observed due to its limited aqueous solubility and therefore it is likely that non-chemical events such as substrate-binding limit the rate of this reaction. Finally, while the formal oxidation state of the catalytically relevant iron-oxo intermediate [FeIII-superoxo or FeIV-oxo] remains unclear, it is expected that the structure and/or reactivity of the (ES:O2) ternary complex is different for aromatic versus aliphatic thiols. Therefore, the transient Fe-oxo intermediate responsible for oxidizing 1°-alcohols may not be relevant to native catalysis.
Concluding Remarks
Based on extensive mechanistic, spectroscopic, and crystallographic characterization a general mechanism for catalysis has been developed for non-heme mononuclear iron enzymes [53–55]. Nearly universally the monoanionic Fe-site contains a 5/6-coordinate ferrous iron with solvent molecules serving as the non-protein ligands. As isolated, the reduced Fe-site is largely unreactive toward molecular oxygen until the substrate and/or cofactor are bound within the active site [55]. The obligate-ordered binding of thiol-substrate to the Fe-site prior to O2-activation has been reported in previous Mm CDO and Av MDO studies [18, 29]. It has also been noted that both CDO and MDO can catalyze thiol dioxygenase reactions for a number of aliphatic amine/thiol substrates and that optimal activity is observed when these groups are separated by two carbon units [27]. However, the bonds separating the thiol and amine functional groups in the aromatic 2ma substrate are sp2 hybridized and thus cannot be rotated. Therefore the 2ma-bound Fe-site geometry of Mm CDO must be very different as compared to the native substrate ES-complex. Presumably, this is also true for Av MDO. Despite this obvious distinction, addition of 2ma to CDO and/or MDO triggers the immediate consumption of oxygen (Supplementary Information, Fig. S2). This suggests that the steps leading up to O2-activation are largely equivalent for reactions utilizing either aliphatic or aromatic thiols. Therefore, this ‘off-pathway’ reactivity toward 1°-alcohols may be the result of geometric distortions within the substrate/O2-bound (ES:O2) ternary complex which prevent thiol-oxidation. Another factor to consider is the differential reactivity of aromatic versus aliphatic thiols. In either case, what can be concluded is that this reactivity is inconsistent with the fixed Fe(II)-oxidation state postulated for ‘substrate-activating’ mechanisms [56]. The unusual reactivity described here further illustrates the remarkable versatility in chemical reactivity exhibited by non-heme iron oxygenase/oxidases.
Supplementary Material
Highlights.
CDO and MDO form benzothiazoles in reactions with 2-mercaptoaniline (2ma), 1°-alcohols, and O2
Aldehydes are produced by oxidation of 1°-alcohols in enzymatic 2ma-reactions
Benzothiazoles are formed by condensation of 2ma with aldehydes followed by 2-electron oxidation
Abbreviations
- CDO
cysteine dioxygenase
- MDO
3-mercaptopropionic acid dioxygenase
- ROS
reactive oxygen species
- Mm
Mus musculus
- Av
Azotobacter vinelandii
- 2ma
2-mercaptoaniline [2-aminobenzene-1-thiol]
- 3mpa
3-mercaptopropionic acid
- cys
L-cysteine
- 2abs
2-aminobenzenesulfinic acid
- 3spa
3-sulfinopropionic acid
- csa
cysteine sulfinic acid
- HPLC
high performance liquid chromatography
- LC-MS
liquid chromatography-mass spectrometry
- MRM
multiple reaction monitoring
Footnotes
This work was supported by the National Science Foundation (CHE) 1213655 (B.S.P.) and National Institute of Health (NIGMS) 1 R15 GM117511-01 (B.S.P.)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stipanuk MH. Sulfur Amino Acid Metabolism: Pathways for Production and Removal of Homocysteine and Cysteine. Annu Rev Nutr. 2004;24:539–577. doi: 10.1146/annurev.nutr.24.012003.132418. [DOI] [PubMed] [Google Scholar]
- 2.Ewetz L, Sorbo B. Characteristics of the Cysteinesulfinate-Forming Enzyme System in Rat Liver. Biochim Biophys Acta. 1966;128:296–305. doi: 10.1016/0926-6593(66)90176-7. [DOI] [PubMed] [Google Scholar]
- 3.Sorbo B, Ewetz L. The Enzymatic Oxidation of Cysteine to Cysteinesulfinate in Rat Liver. Biochem Biophys Res Commun. 1965;18:359–363. doi: 10.1016/0006-291x(65)90714-x. [DOI] [PubMed] [Google Scholar]
- 4.Lombardini JB, Singer TP, Boyer PD. Cysteine Oxygenase. J Biol Chem. 1969;244:1172–1175. [PubMed] [Google Scholar]
- 5.Dominy JE, Jr, Simmons CR, Karplus PA, Gehring AM, Stipanuk MH. Identification and Characterization of Bacterial Cysteine Dioxygenases: a New Route of Cysteine Degradation for Eubacteria. J Bacteriol. 2006;188:5561–5569. doi: 10.1128/JB.00291-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kabil O, Banerjee R. Redox Biochemistry of Hydrogen Sulfide. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon C, Emery P, Bradley H, Waring H. Abnormal sulfur oxidation in systemic lupus erythematosus. Lancett. 1992;229:25–26. doi: 10.1016/0140-6736(92)90144-r. [DOI] [PubMed] [Google Scholar]
- 8.Heafield MT, Fearn S, Steventon GB, Waring RH, Williams AC, Sturman SG. Plasma cysteine and sulfate levels in patients with motor neurone, Parkinson’s and Alzheimer’s disease. Neurosci Lett. 1990;110:216–220. doi: 10.1016/0304-3940(90)90814-p. [DOI] [PubMed] [Google Scholar]
- 9.James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, Neubrander JA. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr. 2004;80:1611–1617. doi: 10.1093/ajcn/80.6.1611. [DOI] [PubMed] [Google Scholar]
- 10.Deth R, Muratore C, Benzecry J, Power-Charnitsky V-A, Waly M. How environmental and genetic factors combine to cause autism: A redox/methylation hypothesis. NeuroToxicology. 2008;29:190–201. doi: 10.1016/j.neuro.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 11.Reddie KG, Carroll KS. Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol. 2008;12:746–754. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 12.Winyard PG, Moody CJ, Jacob C. Oxidative activation of antioxidant defence. Trends Biochem Sci. 2005;30:453–461. doi: 10.1016/j.tibs.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 14.Behave DP, Muse WB, Carroll KS. Drug Targets in Mycobacterial Sulfur Metabolism. Infect Disord Drug Targets. 2007;7:140–158. doi: 10.2174/187152607781001772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Driggers CM, Cooley RB, Sankaran B, Hirschberger LL, Stipanuk MH, Karplus PA. Cysteine Dioxygenase Structures from pH 4 to 9: Consistent Cys-Persulfenate Formation at Intermediate pH and a Cys-Bound Enzyme at Higher pH. Mol Microbiol. 2013;425:3121–3136. doi: 10.1016/j.jmb.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye S, Wu Xa, Wei L, Tang D, Sun P, Bartlam M, Rao Z. An Insight into the Mechanism of Human Cysteine Dioxygenase: Key Roles of the Thioether-Bonded Tyrosine-Cysteine Cofactor. J Biol Chem. 2007;282:3391–3402. doi: 10.1074/jbc.M609337200. [DOI] [PubMed] [Google Scholar]
- 17.McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN., Jr Structure and mechanism of mouse cysteine dioxygenase. Proc Natl Acad Sci USA. 2006;103:3084–3089. doi: 10.1073/pnas.0509262103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierce BS, Gardner JD, Bailey LJ, Brunold TC, Fox BG. Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: electronic structure of the active site and mechanistic implications. Biochemistry. 2007;46:8569–8578. doi: 10.1021/bi700662d. [DOI] [PubMed] [Google Scholar]
- 19.Crawford JA, Li W, Pierce BS. Single turnover of substrate-bound ferric cysteine dioxygenase (CDO) with superoxide anion: enzymatic reactivation, product formation, and a transient intermediate. Biochemistry. 2011;50:10241–10253. doi: 10.1021/bi2011724. [DOI] [PubMed] [Google Scholar]
- 20.Kovaleva EG, Lipscomb JD. Crystal Structures of Fe2+ Dioxygenase Superoxo, Alkylperoxo, and Bound Product Intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Münck E, Lipscomb JD. Trapping and spectroscopic characterization of an FeIII-superoxo intermediate from a nonheme mononuclear iron-containing enzyme. Proc Natl Acad Sci USA. 2010;107:16788–16793. doi: 10.1073/pnas.1010015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tchesnokov EP, Faponle AS, Davies CG, Quesne MG, Turner R, Fellner M, Souness RJ, Wilbanks SM, de Visser SP, Jameson GNL. An iron-oxygen intermediate formed during the catalytic cycle of cysteine dioxygenase. Chem Commun (Camb) 2016;52:8814–8817. doi: 10.1039/c6cc03904a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipscomb JD. Mechanism of extradiol aromatic ring-cleaving dioxygenases. Curr Opin Struct Biol. 2008;18:644–649. doi: 10.1016/j.sbi.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Broderick JB. Catechol dioxygenases. Essays Biochem. 1999;34:173–189. doi: 10.1042/bse0340173. [DOI] [PubMed] [Google Scholar]
- 25.Gunderson WA, Zatsman AI, Emerson JP, Farquhar ER, Que L, Lipscomb JD, Hendrich MP. Electron Paramagnetic Resonance Detection of Intermediates in the Enzymatic Cycle of an Extradiol Dioxygenase. J Am Chem Soc. 2008;130:14465–14467. doi: 10.1021/ja8052255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Münck E, Lipscomb JD. Trapping and spectroscopic characterization of an FeIII-superoxo intermediate from a nonheme mononuclear iron-containing enzyme. Proc Natl Acad Sci U S A. 2010;107:16788–16793. doi: 10.1073/pnas.1010015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li W, Pierce BS. Steady-state substrate specificity and O2-coupling efficiency of mouse cysteine dioxygenase. Arch Biochem Biophys. 2015;565:49–56. doi: 10.1016/j.abb.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Crowell JK, Sardar S, Hossain MS, Foss FW, Jr, Pierce BS. Non-chemical proton-dependent steps prior to O2-activation limit Azotobacter vinelandii 3-mercaptopropionic acid dioxygenase (MDO) catalysis. Arch Biochem Biophys. 2016;604:86–94. doi: 10.1016/j.abb.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pierce BS, Subedi BP, Sardar S, Crowell JK. The “Gln-Type” Thiol Dioxygenase from Azotobacter vinelandii Is a 3-Mercaptopropionic Acid Dioxygenase. Biochemistry. 2015;54:7477–7490. doi: 10.1021/acs.biochem.5b00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crowell JK, Li W, Pierce BS. Oxidative uncoupling in cysteine dioxygenase is gated by a proton-sensitive intermediate. Biochemistry. 2014;53:7541–7548. doi: 10.1021/bi501241d. [DOI] [PubMed] [Google Scholar]
- 31.Kenny RS, Mashelkar UC. Synthesis of 2-aryl and coumarin substituted benzothiazole derivatives. J Heterocycl Chem. 2006;43:1367–1369. [Google Scholar]
- 32.Guntreddi T, Vanjari R, Singh KN. Elemental Sulfur Mediated Decarboxylative Redox Cyclization Reaction of o-Chloronitroarenes and Arylacetic Acids. Organic Letters. 2015;17:976–978. doi: 10.1021/acs.orglett.5b00079. [DOI] [PubMed] [Google Scholar]
- 33.Li W, Blaesi EJ, Pecore MD, Crowell JK, Pierce BS. Second-Sphere Interactions between the C93–Y157 Cross-Link and the Substrate-Bound Fe Site Influence the O2 Coupling Efficiency in Mouse Cysteine Dioxygenase. Biochemistry. 2013;52:9104–9119. doi: 10.1021/bi4010232. [DOI] [PubMed] [Google Scholar]
- 34.Corder AL, Subedi BP, Zhang S, Dark AM, Foss FW, Pierce BS. Peroxide-Shunt Substrate-Specificity for the Salmonella typhimurium O2-Dependent tRNA Modifying Monooxygenase (MiaE) Biochemistry. 2013;52:6182–6196. doi: 10.1021/bi4000832. [DOI] [PubMed] [Google Scholar]
- 35.Subedi BP, Corder AL, Zhang S, Foss FWJ, Pierce BS. Steady-state kinetics and spectroscopic characterization of enzyme-tRNA interactions for the non-heme diiron tRNA-monooxygenase, MiaE. Biochemistry. 2014;54:363–376. doi: 10.1021/bi5012207. [DOI] [PubMed] [Google Scholar]
- 36.Yost RA, Fetterolf DD. Tandem mass spectrometry (MS/MS) instrumentation. Mass Spectrom Rev. 1983;2:1–45. [Google Scholar]
- 37.Kloepfer A, Jekel M, Reemtsma T. Determination of benzothiazoles from complex aqueous samples by liquid chromatography–mass spectrometry following solid-phase extraction. J Chromatogr A. 2004;1058:81–88. [PubMed] [Google Scholar]
- 38.Chen S, Hossain MS, Foss FW. Bioinspired Oxidative Aromatizations: One-Pot Syntheses of 2-Substituted Benzothiazoles and Pyridines by Aerobic Organocatalysis. ACS Sustain Chem Eng. 2013;1:1045–1051. [Google Scholar]
- 39.Wang Q, Sheng X, Horner JH, Newcomb M. Quantitative Production of Compound I from a Cytochrome P450 Enzyme at Low Temperatures. Kinetics, Activation Parameters, and Kinetic Isotope Effects for Oxidation of Benzyl Alcohol. J Am Chem Soc. 2009;131:10629–10636. doi: 10.1021/ja9031105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oh NY, Suh Y, Park MJ, Seo MS, Kim J, Nam W. Mechanistic Insight into Alcohol Oxidation by High-Valent Iron–Oxo Complexes of Heme and Nonheme Ligands. Angew Chem Int Ed Engl. 2005;44:4235–4239. doi: 10.1002/anie.200500623. [DOI] [PubMed] [Google Scholar]
- 41.Nam W, Lee Y-M, Fukuzumi S. Tuning Reactivity and Mechanism in Oxidation Reactions by Mononuclear Nonheme Iron(IV)-Oxo Complexes. Acc Chem Res. 2014;47:1146–1154. doi: 10.1021/ar400258p. [DOI] [PubMed] [Google Scholar]
- 42.Ghosh M, Nikhil YLK, Dhar BB, Sen Gupta S. Mechanism of Alcohol Oxidation by FeV(O) at Room Temperature. Inorganic Chemistry. 2015;54:11792–11798. doi: 10.1021/acs.inorgchem.5b01937. [DOI] [PubMed] [Google Scholar]
- 43.Yu J, Xia Y, Lu M. Iron-Catalyzed Highly Efficient Aerobic Oxidative Synthesis of Benzimidazoles, Benzoxazoles, and Benzothiazoles Directly from Aromatic Primary Amines Under Solvent-Free Conditions in the Open Air. Synth Commun. 2014;44:3019–3026. [Google Scholar]
- 44.Bahrami K, Khodaei MM, Naali F. Mild and Highly Efficient Method for the Synthesis of 2-Arylbenzimidazoles and 2-Arylbenzothiazoles. J Org Chem. 2008;73:6835–6837. doi: 10.1021/jo8010232. [DOI] [PubMed] [Google Scholar]
- 45.Gopalaiah K, Chandrudu SN. Iron(ii) bromide-catalyzed oxidative coupling of benzylamines with ortho-substituted anilines: synthesis of 1,3-benzazoles. RSC Advances. 2015;5:5015–5023. [Google Scholar]
- 46.Gardner JD, Pierce BS, Fox BG, Brunold TC. Spectroscopic and Computational Characterization of Substrate-Bound Mouse Cysteine Dioxygenase: Nature of the Ferrous and Ferric Cysteine Adducts and Mechanistic Implications. Biochemistry. 2010;49:6033–6041. doi: 10.1021/bi100189h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blaesi EJ, Gardner JD, Fox BG, Brunold TC. Spectroscopic and Computational Characterization of the NO Adduct of Substrate-Bound Fe(II) Cysteine Dioxygenase: Insights into the Mechanism of O2 Activation. Biochemistry. 2013;52:6040–6051. doi: 10.1021/bi400825c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies CG, Fellner M, Tchesnokov EP, Wilbanks SM, Jameson GNL. The Cys-Tyr Cross-Link of Cysteine Dioxygenase Changes the Optimal pH of the Reaction without a Structural Change. Biochemistry. 2014;53:7961–7968. doi: 10.1021/bi501277a. [DOI] [PubMed] [Google Scholar]
- 49.Driggers CM, Kean KM, Hirschberger LL, Cooley RB, Stipanuk MH, Karplus PA. Structure-Based Insights into the Role of the Cys–Tyr Crosslink and Inhibitor Recognition by Mammalian Cysteine Dioxygenase. J Mol Biol. 2016;428:3999–4012. doi: 10.1016/j.jmb.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fellner M, Aloi S, Tchesnokov EP, Wilbanks SM, Jameson GNL. Substrate and pH-Dependent Kinetic Profile of 3-Mercaptopropionate Dioxygenase from Pseudomonas aeruginosa. Biochemistry. 2016;55:1362–1371. doi: 10.1021/acs.biochem.5b01203. [DOI] [PubMed] [Google Scholar]
- 51.Krebs C, Galonić Fujimori D, Walsh CT, Bollinger JM. Non-Heme Fe(IV)–Oxo Intermediates. Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen K, Que L. Stereospecific Alkane Hydroxylation by Non-Heme Iron Catalysts: Mechanistic Evidence for an FeVO Active Species. J Am Chem Soc. 2001;123:6327–6337. doi: 10.1021/ja010310x. [DOI] [PubMed] [Google Scholar]
- 53.Solomon EI, Decker A, Lehnert N. Non-heme iron enzymes: contrasts to heme catalysis. Proc Natl Acad Sci USA. 2003;100:3589–3594. doi: 10.1073/pnas.0336792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 55.Costas M, Mehn MP, Jensen MP, Que LJ. Diooxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 56.Simmons CR, Krishnamoorthy K, Granett SL, Schuller DJ, Dominy JE, Begley TP, Stipanuk MH, Karplus PA. A Putative Fe2+-Bound Persulfenate Intermediate in Cysteine Dioxygenase. Biochemistry. 2008;47:11390–11392. doi: 10.1021/bi801546n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.