

Summary
Dengue, caused by four dengue virus serotypes (DENV-1 to DENV-4), is a highly prevalent mosquito-borne viral disease in humans. Yet, selection pressures driving DENV microevolution within human hosts (intrahost) remain unknown. We employed a whole-genome segmented amplification approach coupled with deep sequencing to profile DENV-3 intrahost diversity in peripheral blood mononuclear cell (PBMC) and plasma samples from 77 dengue patients. DENV-3 intrahost diversity appears to be driven by immune pressures as well as replicative success in PBMCs and potentially other replication sites. Hotspots for intrahost variation were detected in 59%–78% of patients in the viral Envelope and pre-Membrane/Membrane proteins, which together form the virion surface. Dominant variants at the hotspots arose via convergent microevolution, appear to be immune-escape variants, and were evolutionarily constrained at the macro level due to viral replication defects. Dengue is thus an example of an acute infection in which selection pressures within infected individuals drive rapid intrahost virus microevolution.
Keywords: dengue virus, intrahost, quasispecies, next-generation sequencing, whole genome, human infection, selection pressure, microevolution, fitness, convergent evolution
Graphical Abstract
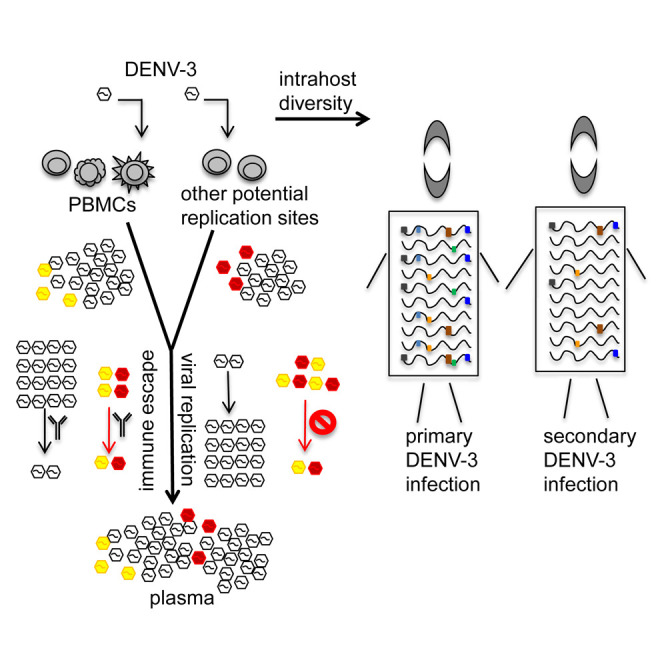
Highlights
-
•
DENV-3 intrahost diversity was analyzed in PBMCs and/or plasma from 77 dengue patients
-
•
Virus microevolution is shaped by immune pressure and replication sites including PBMCs
-
•
Hotspots arose via convergent microevolution and are likely immune-escape variants
-
•
Hotspot variants were evolutionarily constrained by replication defects
Dengue, caused by DENV-1 to -4, is a highly prevalent viral disease. Parameswaran et al. profile DENV-3 intrahost diversity in 77 patients, showing that intrahost virus microevolution occurs in PBMCs and potentially other replication sites. They find that intrahost variants, likely immune-escape hotspots, arise via convergent microevolution, yet are evolutionarily constrained by replication defects.
Introduction
Dengue virus (DENV) is a mosquito-borne Flavivirus with a single-stranded RNA genome that causes an estimated 390 million infections and up to 96 million dengue cases worldwide every year (Bhatt et al., 2013). There are four closely related serotypes of DENV (DENV-1 to DENV-4), each of which encodes three structural proteins (Capsid [C], pre-Membrane/Membrane [prM/M], and Envelope [E]) and seven non-structural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5). Infections with DENV can result in a spectrum of clinical manifestations, ranging from asymptomatic infection to the debilitating acute febrile illness, dengue fever (DF), to the life-threatening dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS) (WHO, 1997). Major determinants of dengue pathogenesis include virulence of the infecting DENV strain, host genetic factors, and pre-existing host immune responses from prior infection(s) with a different DENV serotype (Halstead and Yamarat, 1965, Messer et al., 2003, Nguyen et al., 2008, OhAinle et al., 2011, Rico-Hesse et al., 1997). In sequential infections, disease severity appears to be determined by a complex interplay of protective and enhancing components from pre-existing immunity to viral antigens (Halstead, 2009, Peiris and Porterfield, 1979, Rothman and Ennis, 1999). Such intricacies of the human immune response to DENV infection have made it difficult not only to identify the precise mechanisms that trigger progression to severe disease but also to engineer vaccines and therapeutics for combating the disease.
Replication of DENV within each host produces a population of genetically related but distinct genomes (referred to as intrahost diversity) due to the error-prone nature of the viral replicase, the RNA-dependent RNA polymerase (RdRP) (Domingo and Holland, 1997). These intrahost variants are thought to serve as templates on which evolutionary mechanisms act to shape variation at the consensus level between hosts (i.e., interhost diversity), leading to the emergence of genetically distinct strains and genotypes of DENV. Genetic variations in intrahost populations have been proposed to influence disease outcome and pathogenesis in chronic human infections with RNA viruses such as HIV and hepatitis C virus (HCV) (Farci et al., 2002, Joos et al., 2005, Lee et al., 2008, Moreau et al., 2008, Sullivan et al., 2007), which provide considerable time frames (months to years) for discernible virus evolution in response to intrahost selection pressures. Similar observations have also been reported in chronic infections with influenza virus and norovirus, viruses that are usually associated with acute infections (Bull et al., 2012, Debbink et al., 2014, Rogers et al., 2015, Valkenburg et al., 2013). These studies have characterized the emergence of individual variants associated with immune evasion or drug resistance. Unlike chronic infections, only a handful of studies have reported on the evolutionary mechanisms and viral genetics driving virus evolution in acute human infections such as Ebola (Gire et al., 2014, Ni et al., 2016), chikungunya (Stapleford et al., 2016), influenza A (Sobel Leonard et al., 2016), Middle East respiratory syndrome (Park et al., 2016), and dengue (Parameswaran et al., 2012, Rodriguez-Roche et al., 2016, Sessions et al., 2015, Sim et al., 2015, Thai et al., 2012), where virus evolution is severely constrained by time (days to weeks). Consequently, limited information exists about the fitness and pathogenesis profiles of individual intrahost DENV variants that emerge in acute human infections. Information on intrahost viral diversity can provide new perspectives in assessing infection outcome, disease pathogenesis, and vaccine or therapeutic efficacy in individuals with dengue or other closely related viral infections such as Zika.
In this study, we employ a whole-genome segmented amplification approach coupled with high-throughput sequencing to profile intrahost viral diversity across the entire coding region of the DENV-3 genome with considerable depth of coverage. Using snapshots of DENV-3 intrahost diversity in 31 plasma and 68 peripheral blood mononuclear cell (PBMC) samples from 77 individuals (including 22 with paired plasma and PBMC samples) enrolled in a prospective pediatric hospital-based study in Nicaragua, we demonstrate that DENV-3 diversification in human acute dengue (microevolution) is shaped by convergent selection pressures, including pre-existing immunity, and possibly by replication in sites other than PBMCs, while it is constrained by severe defects in replicative ability.
Results and Discussion
Samples and Methods
All PBMC and plasma samples used in this study were collected on the first day of presentation from individuals aged 6 months to 14 years with primary (1°; 53 samples) or secondary (2°; 46 samples) acute, symptomatic DENV-3 infection during the 2009–2010 epidemic season in Managua, Nicaragua (Tables 1 and S1) (Narvaez et al., 2011). The majority of PBMC (82%) and plasma (91%) samples were from individuals who presented with DF. The rest of the PBMC (18%) and plasma (9%) samples were from individuals who presented with DHF/DSS. Supporting clinical information is provided in Table S1.
Table 1.
Summary of Samples Used in This Study
| Sample Typea | Year of Collection | Disease Severity | Immune Status | No. of Samplesb | No. of Paired Samplesb |
|---|---|---|---|---|---|
| PBMCs | 2009–2010 | DF | primary | 32 | 11 |
| secondary | 23 | 9 | |||
| DHF/DSS | primary | 5 | 0 | ||
| secondary | 8 | 2 | |||
| Plasma | 2009–2010 | DF | primary | 16 | 11 |
| secondary | 13 | 9 | |||
| DHF/DSS | primary | 0 | 0 | ||
| secondary | 2 | 2 |
PBMC and plasma samples (including 53 primary and 46 secondary samples) were obtained from individuals aged 6 months to 14 years who were enrolled in the hospital-based pediatric dengue study and tested positive for DENV-3.
Only nucleotide loci with coverage of ≥1,000 reads and locus-specific Sanger quality scores of ≥30 were considered for calculating genome coverage. Samples with ≥50% genome coverage are reported.
The DENV-3 genome was amplified via reverse transcription and PCR using sequence-specific primer pairs to generate 12 overlapping amplicons per sample (Figure S1A). Multiplexed libraries for high-throughput sequencing were constructed from pooled amplicon mixes and sequenced on an Illumina HiSeq 2000 platform to yield 150-nucleotide (nt) paired-end reads in forward (read 1) and reverse (read 2) orientations. Reference genomes were reconstructed for each sample using Bowtie2 (Langmead and Salzberg, 2012) and SAMtools (Li et al., 2009), and in-house python scripts were used to calculate variation at the nucleotide, codon, and amino acid levels for each locus in the DENV-3 coding region (see STAR Methods). After filtering for coverage (≥1,000 reads) and Sanger quality scores (≥30), median coverage per codon locus was 22,975 and 20,024 for read 1 and read 2 datasets, respectively. Only samples with ≥50% coverage of the DENV-3 genome were retained for downstream analyses; 68 PBMC and 31 plasma samples from 77 individuals, including paired PBMC-plasma datasets from 22 individuals, passed the threshold for genome coverage. Coding-region coverage in these samples varied between 50.7% and 99.9% (median of 98.9%) for both read 1 and read 2 datasets combined (Table S1).
Percent variant diversity at nucleotide, codon, and amino acid loci was calculated as the percent of reads spanning each coordinate that was different from consensus. Nucleotide- and codon-level diversities provide aggregate measures of both non-synonymous and synonymous mutations (i.e., mutations that do and do not change amino acid encoding, respectively), while amino acid diversity only captures non-synonymous mutations. In our study, variant abundances of ≥1.0% were reproducible across replicates (see STAR Methods) and were considered variants of high confidence. Unless otherwise specified, we used high-confidence variants for investigating diversity patterns. Samples from both sexes were included in all intrahost diversity comparisons; no significant differences between sexes were observed for any of the calculated DENV-3 intrahost diversity metrics (data not shown).
Intrahost Immune Pressures Shape DENV-3 Diversity in Acute Human Infections
We evaluated snapshots of intrahost diversity in samples from distinct subsets of individuals with 1° (53 samples) or 2° (46 samples) dengue to assess the effects of immune-driven selection pressures on intrahost DENV evolution. We compared the percent loci per protein that are variant in any sample between 1° and 2° dengue and found significantly fewer unique variant loci in 2° dengue cases, both in PBMC (Figure 1 A; left panel, p < 0.001) and plasma (Figure 1A; right panel, p < 0.01) samples. These observations were not due to systematic disparities in the yields of viral RNA, since measures of genome equivalents (GE) per milliliter of extracted viral RNA were not significantly different between 1° and 2° cases for both PBMC and plasma samples (Figure 1B; left and right panels). Thus, DENV-3 appears to be evolving at fewer distinct loci genome-wide in 2° dengue compared with 1° dengue, suggesting that virus evolution is constrained by pre-existing immune pressures in 2° dengue. In addition, we examined the locations of unique variant loci on the exposed and membrane-associated surfaces of the E protein, which is a prominent target for antibody-mediated immunity in human dengue. We detected fewer unique variant loci across all samples in 2° dengue cases compared with 1° cases (Figure 1C; top and bottom panels) on both the exposed and the membrane-associated surfaces of E; much of this difference was contributed by PBMC-specific variant loci. Thus, consistent with genome-wide differences between 1° and 2° dengue, the breadth of the viral variant repertoire in E appears to be reduced in 2° dengue compared with 1° dengue. Such global differences in viral composition between naive individuals and individuals with pre-existing immunity to DENV indicate that the extent of diversity in the intrahost DENV-3 population is directed by the immune repertoire during acute dengue in humans. Further studies investigating particular epitopes will help resolve the specificities of pre-existing immune pressures contributing to differential rates of viral microevolution in 1° and 2° dengue.
Figure 1.

DENV-3 Intrahost Diversity Is Shaped by Selection Pressures
(A) Percent unique loci per protein with amino acid diversity, calculated as the percent of amino acids in a protein that show ≥1% variation, in PBMC (left panel) and plasma (right panel) samples from 53 individuals with 1° and 46 individuals with 2° dengue. ∗p ≤ 0.01, ∗∗p ≤ 0.001 (Wilcoxon signed-rank test). Each line represents one protein.
(B) Comparisons of genome equivalents per milliliter (GE/mL) of extracted RNA in 53 1° and 46 2° cases from PBMC (left panel) and plasma (right panel) samples, median ± SD, boxes represent 25th and 75th percentiles and whiskers are 10th and 90th percentiles. n.s., non-significant (Wilcoxon test for unpaired samples).
(C) All loci with ≥1% variation at the amino acid level, superimposed on the exposed surface (left panel) or the membrane-associated surface (right panel) of the DENV-3 E protein homodimer (PDB: 1UZG). Amino acids are colored according to whether they exhibit diversity in PBMC samples only (PBMCs, red), in plasma samples only (Plasma, blue), or in both PBMCs and plasma (Both, green). Data are shown separately for 53 1° (top panel) and 46 2° (bottom panel) dengue cases. EDIa/b, EDIIa/b, EDIIIa/b represent E domains I, II, and III from monomer chains a and b, respectively.
(D) Percent amino acid variants for loci that exhibit diversity in PBMCs only (PBMCs), compared with loci that exhibit diversity in both PBMCs and plasma (Both), median ± SD, boxes represent 25th and 75th percentiles and whiskers are 10th and 90th percentiles. In the left panel, the y axis represents the percent abundance of non-synonymous variants in PBMCs. In the right panel, the y axis represents the percent abundance of synonymous variants in PBMCs. ∗∗p ≤ 0.0001 (Wilcoxon test), n = 22 paired PBMC and plasma samples.
(E) Protein-specific comparisons of percent amino acid variants in 22 paired PBMC and plasma samples, colored by whether individual loci display ≥1% amino acid diversity in PBMC samples only (yellow), in plasma samples only (red), or in both PBMC and plasma samples (green). Data are shown for the two proteins (prM/M and NS3) that exhibit significant differences in % amino acid (AA) variants between PBMCs and plasma. All other proteins (including E, which is shown as an example) exhibit no significant differences in % AA variants between PBMCs and plasma. ∗∗p ≤ 0.0001, n.s., non-significant (Wilcoxon signed-rank test).
(F) Comparisons of genome equivalents per milliliter (GE/mL) of extracted RNA in 12 paired PBMC and plasma samples. n.s., non-significant (Wilcoxon signed-rank test for paired samples).
Selection Pressures Shape DENV-3 Microevolution in Plasma
We used patterns of DENV-3 intrahost diversity in paired PBMC and plasma samples from a subset of 22 individuals to assess the role of selection pressures mediated by human immune pathways or virion intracellular replicative success in shaping differences in DENV variants at the PBMC-plasma (i.e., intracellular-extracellular) interface and to explore the relative contributions from PBMCs and other potential viral replication sites to circulating virion populations in plasma.
We examined the percent abundance of non-synonymous variants in PBMCs and plasma, binning loci by whether variants were found at levels ≥1% in PBMCs only (PBMCs), or in both PBMC and plasma samples (Both). The median percent abundance of variant DENV-3 genomes that were found in PBMCs only was significantly lower than the median percent abundance of variant genomes found in both PBMCs and plasma, for both non-synonymous and synonymous variants (Figure 1D, left and right panels, p < 0.0001). Thus, variants that appear in both PBMCs and plasma are more abundant than variants that are restricted to PBMCs, suggesting that DENV variants found in plasma have been selected based on their intracellular replicative success in PBMCs.
We also found evidence for immune-driven selection pressures shaping differences between plasma and PBMC DENV populations. In particular, significantly higher abundances for non-synonymous variants were observed in plasma compared with PBMCs for the prM/M and NS3 proteins (Figure 1E, p < 0.0001), which are major targets of the human B cell and T cell immune responses, respectively (de Alwis et al., 2014, Dejnirattisai et al., 2010, Rivino et al., 2013, Simmons et al., 2005, Weiskopf et al., 2013).
Furthermore, we observed many dissimilarities between PBMCs and plasma DENV populations that could not be attributed to replication defects or immune pressure mechanisms. For instance, minimal intersection was detected in the locations of variant loci on the highly antigenic E protein (Figure 1C, green loci), as well as in other proteins (Figures 1E and S2A), inferred from the presence of fewer green lines representing loci with ≥1% amino acid variants in both PBMC and plasma samples, compared with orange and red lines representing loci with ≥1% amino acid variants in PBMC or plasma samples, respectively. These findings were not due to differences in the depth of sequencing coverage (data not shown), or in the number of viral copies used as input for generating sequencing libraries (Figure S2B), because these parameters were comparable between paired PBMC and plasma samples. These observations of large-scale differences between plasma and PBMC DENV variant populations, when taken together with prior knowledge from human autopsy studies about the existence of significant non-PBMC compartments for viral replication in phagocytes, lymph node, spleen, alveolar macrophages in lung, and perivascular cells in brain (Aye et al., 2014, Balsitis et al., 2009, Jessie et al., 2004), suggest contribution to plasma DENV populations from replication compartments other than PBMCs.
Hotspots for DENV-3 Intrahost Diversity in Acute Human Dengue
We next sought to identify hotspots for intrahost viral diversity, defined as loci with detectable intrahost diversity in multiple hosts, in PBMC and plasma samples from individuals with DENV-3 infection. Hotspots may arise de novo within each host due to convergent selection pressures, or may be attributed to the co-transmission of wild-type (WT) and variant viruses within a host population. The probability of detecting diversity hotspots due to selection pressures has been assumed to be low in acute human dengue (Descloux et al., 2009, Lin et al., 2004, Parameswaran et al., 2012, Thai et al., 2012, Wang et al., 2002a, Wang et al., 2002b), primarily because of the time constraint (8–14 days) on virus evolution within hosts.
We analyzed percent variant abundance together with percent prevalence of variants at each codon locus (depicted by the color and size of each data point) across the entire DENV-3 coding region and found that very few codon loci were hotspots for intrahost diversity in PBMC and plasma samples (Figure 2 A, top and bottom panels, respectively). However, prominent hotspots for diversity were identified in the majority (59%–78%) of samples at three codon coordinates: (1, 2) 214 and 215, corresponding to amino acids 100 and 101 in the membrane portion of prM/M (hereafter prM100 and prM101), and (3) 595, corresponding to amino acid 315 in the highly conserved AB-loop region in E Domain III (E315) (Figures 2A and 2B). To confirm the presence of the more abundant E315 variant, we designed primers that specifically targeted and amplified either WT or E315 DENV RNA and validated the specificity of these primers using RNA from WT or E315 variant viruses (Figure S3A). We tested a subset of seven serum samples used in this study and were able to directly detect the E315 hotspot variant at various levels in all seven clinical samples (Figure S3B).
Figure 2.

Hotspots for Intrahost Diversity in the DENV-3 Genome
(A) Percent intrahost codon diversity (y axis) across the coding region of DENV-3 (x axis) in 68 PBMC (top panel) and 31 plasma (bottom panel) samples; each data point is colored according to the number of samples in which ≥1% intrahost diversity is detected at that locus.
(B) Major variants at the hotspot loci in prM/M (top) and E (bottom). Red asterisk, furin cleavage site.
(C) Prevalence of major hotspot loci (i.e., loci with ≥1% variant abundance in ≥50% of samples) in 68 plasma (right panel) and 31 PBMC (left panel) samples in both read orientations (read 1 in yellow and read 2 in red).
(D) Percent samples with diversity at each hotspot locus, shown for increasing cutoffs for percent intrahost codon diversity, in the 68 PBMC (left panel) and 31 plasma (right panel) datasets.
(E) Average percent intrahost (yellow) and percent interhost (red) diversity in codon (left panel) or amino acid (right panel) coordinates for each hotspot locus. Interhost diversity was calculated across all global DENV-3 isolates for which full-genome sequences were available (n = 690).
The dominant variants at these hotspots were identical across samples (Figure S2C) and exhibited altered amino acid sequence compared with consensus: (1) glycine to serine at prM100, (2) methionine to leucine at prM101, and (3) histidine to leucine at E315 (Figure 2B). prM100 and prM101 variants were significantly more prevalent in plasma (59.3% and 62.9%, respectively), compared with PBMC samples (2.9% and 1.4%, respectively) (p < 0.0001, Fisher's exact test), while the E315 variant was highly prevalent in both PBMC (77.6%) and plasma (69.7%) samples (Figure 2C, average of read 1 and read 2 values). Hotspots detected were not PCR artifacts from the amplicon and library preparation (data not shown). Even at lower thresholds of ≥0.5% variant abundance, the prM100 and prM101 variants were only detected in a small number of PBMC samples (8.8%–13.2% of samples; Figure 2D). The E315 variants thus appear to originate in PBMCs (in addition to other cell types), whereas it is conceivable that the prM100 and prM101 variants arise in viral replication sites distinct from PBMCs, although we have not ruled out the possibility that these variants arose in PBMCs and are simply enriched in the extracellular environment. The prM100,101 and E315 variants constitute 2.2% and 1.9%, respectively, of an estimated 1.2 × 107 GE/mL of circulating viral populations in plasma (extrapolated from Figure 1F; median yield of 1.2 × 106 GE in 50 μL of RNA extracted from 100 μL of plasma) from individuals with detectable levels (≥1.0%) of these variants (Figure S2C, median abundances). We also analyzed interhost diversity in 690 full-length DENV-3 consensus genomes (GenBank) and found that even though DENV-3 exhibited codon-level diversity at prM100 and E315 (Figure 2E, left panel, red bars), virus populations were invariant in the interhost amino acid sequence at all three hotspots (Figure 2E, right panel, red bars, and Table S2), suggestive of macro-evolutionary (i.e., consensus-level) constraints at these loci. In addition to major hotspots, we detected several minor hotspots, defined as loci with 1.0% or higher variant abundance in ≥10% of samples, in both PBMC and plasma samples (Figure S2D, all minor hotspots marked with ˆ). Of these minor hotspots, only one (codon 1071 in polyprotein) appeared to be shared between PBMC and plasma samples at a diversity threshold of 1.0% (Figure S2D). All minor hotspots (loci marked with ˆ) exhibited interhost diversity at the codon level (Figure S2E, left panel, red line, and Table S2), but interhost amino acid diversity was only observed at five out of nine minor hotspots (Figure S2E, right panel, red line, and Table S2). Overall, our findings demonstrate that hotspots for intrahost viral diversity can be detected in acute human dengue. Furthermore, the presence of hotspot variants at macroevolutionarily conserved loci highlight the importance of host-specific selection pressures in DENV-3 intrahost evolution.
Convergent Evolution at Major Hotspot Loci
We next explored linkage patterns between polymorphisms in the sequences of reconstructed haplotypes spanning the prM100,101 and E315 major hotspot loci to identify whether (1) variants at the prM100 and prM101 hotspot loci were present on the same viral genome, and (2) viruses with variation at the prM100,101 and E315 hotspots arose de novo in different samples due to convergent selection pressures, or were present in multiple hosts due to co-transmission with consensus viral genomes. In samples with detectable levels of both prM variants at the population level, we observed that all haplotypes with the prM100 variant also included the prM101 variant (and vice versa), demonstrating that the prM variants were linked, i.e., present on the same variant viral genome (Figure 3 A, red arrowheads). Moreover, we identified haplotypes with variants at only one prM hotspot (Figure S2F), which implied that DENV-3 was evolving independently at each of the prM100 and prM101 hotspots. The prM100,101 variant thus appears to be the result of sequential evolution at prM100 and prM101 on the same viral genome. Our haplotype analysis also revealed significant interhost diversity at three loci proximal to the prM100,101 and E315 hotspots (Figures 3A and 3B; black arrowheads). Four different haplotypes with a wide range of frequencies were detected among the consensus genomes in our samples, each with unique combinations of nucleotide diversity at these loci (Figures 3A and 3B, and Table S3). Nucleotides at all three polymorphic loci were 100% concordant between the dominant WT consensus genome and the prM/M or E variant genome for all samples (Figures 3A and 3B and Table S3; variant genomes marked with ∗). This was true even for samples with low-frequency consensus haplotypes (C-A-T and A-G-G haplotypes for prM/M and E, respectively), and for samples in which mixed infections with multiple WT haplotypes were detected (Figures 3A and 3B; genomes marked with #). The genetic concordance between WT and variant haplotypes in our samples strongly support the independent emergence and/or expansion of these hotspot variants within each individual, although we cannot completely exclude contributions from co-circulating variants at the prM100,101 and E315 loci. Taken together, our observations indicate that variants at the prM100,101 and E315 hotspot loci arose by convergent evolution during acute dengue, in at least four independent events, corresponding to the presence of the variants in four distinct haplotypes.
Figure 3.

Sequence Diversity in Reconstructed Haplotypes Spanning the Intrahost Diversity Hotspots in prM/M and E
Color-coded nucleotide diversity in regions flanking intrahost diversity hotspots in (A) prM/M and (B) E; each row represents a unique reconstructed haplotype with the hotspot variant in 8 PBMC/plasma samples for the prM/M hotspot and 39 PBMC/plasma samples for the E hotspot (samples delineated by thin white lines). Consensus haplotypes and their observed frequencies across all Nicaraguan samples are shown on the left, separated by thick white lines. Haplotypes with the prM100,101 and E315 variants are denoted with asterisks (∗), WT variant haplotypes distinct from the consensus haplotype are marked with #, and underlined loci in B represent the AB-loop region containing the E hotspot. Red asterisk, furin cleavage site. Red arrowheads, prM and E hotspot variants. Black arrowheads, other variants present at >1% abundance.
Replication Phenotypes and Origins of Hotspot Variants
The four-plasmid reverse genetics approach (Messer et al., 2012) was used to engineer chimeric infectious clones containing non-structural genes from a 1989 Sri Lankan DENV-3 isolate (GenBank: JQ411814.1) and either (1) Nicaraguan consensus sequence for the structural genes (WT), (2) WT Nicaraguan consensus with the Gly100Ser and Met101Leu mutations in the membrane portion of prM/M (prM100,101 mutant), or (3) WT Nicaraguan consensus with the His315Leu mutation in the AB loop in E (E315 mutant). We evaluated the replication phenotypes of WT and mutant viruses in C6/36 mosquito cells by directly competing each mutant virus with WT virus in co-infection experiments (Quiner et al., 2014). Viral RNA mixtures from the input and from cellular supernatants collected 3, 4, and 5 days post infection (d.p.i.) were sequenced by the Sanger method to capture ratios of WT and mutant nucleotides at the prM100,101 and E315 hotspots using the polySNP program. The replicative index, calculated as the log2 value of relative levels of WT and mutant viruses in the supernatant compared with relative levels in the input, was negative for prM100,101 or E315 mutant viruses at 3–5 d.p.i., indicating that both mutants were replication defective in C6/36 cells compared with WT (Figure 4 A). In addition, we assessed the replication phenotypes of WT and mutant viruses in a human monocytic U937 cell line that expresses DC-SIGN, a co-receptor for DENV entry (Tassaneetrithep et al., 2003) (U937-DC-SIGN). Single infections were performed in U937-DC-SIGN cells with 625 GE/cell each of WT, prM100,101, and E315 viruses. All three viruses were able to establish infection in U937-DC-SIGN cells; however, the percent of infected cells was considerably reduced in infections with prM100,101 and E315 variants compared with WT virus at 1, 2, and 3 d.p.i., as determined with both anti-E and anti-NS3 antibodies (Figure 4B, left and right panels, respectively). No revertants were detected at 3 d.p.i. with either variant (data not shown). The E315 variant also exhibited a temperature-sensitive (32°C versus 37°C) plaquing phenotype on baby hamster kidney (BHK-21) cells with better plaquing at 32°C, whereas the prM100,101 variant plaqued relatively well at 37°C (Figure S4). Our results thus demonstrate that the prM100,101 and E315 mutant viruses establish infections in C6/36 mosquito cells, U937-DC-SIGN human cells, and in BHK-21 mammalian cells with considerably reduced efficiencies due to the deleterious effects of these mutations on viral replicative ability.
Figure 4.

Phenotypic Differences between Wild-Type, prM100,101, and E315 Viruses
(A) C6/36 cells were infected with a mixture containing WT virus and the prM100,101 (yellow) or E315 (red) mutant viruses. The replication index was measured as the log2 value of relative levels of mutant and WT genomes in the supernatant at 3–5 d.p.i., compared with the corresponding ratios in the input. A negative index indicates that the replicative ability of mutant virus is lower than WT virus.
(B) U937-DC-SIGN cells were mock-infected (mock, blue) or infected with WT (aqua), prM100,101 (yellow), and E315 (red) viruses, fixed, and stained with anti-E (left panel) and anti-NS3 (right panel) antibodies at various time points post infection. All data points for (A and B) represent the average of two independent experiments, mean ± SD.
The prM100,101 and E315 mutant viruses do not exhibit any defects in entry and behave similar to WT virus, as demonstrated by directly comparing entry capabilities of the three viruses using U937-DC-SIGN cells at 37°C (Figure S5), suggesting that the replication defect for both prM100,101 and E315 mutant viruses is downstream of virus entry. We do not anticipate that these mutations have a direct impact on viral RNA replication, as none of the DENV structural proteins, except RNA elements in the Capsid gene (Byk and Gamarnik, 2016, Clyde et al., 2008, de Borba et al., 2015, Friebe et al., 2011, Selisko et al., 2014), have been implicated in a direct role in RNA replication. In studies by Zheng et al. (2014), mutations at prM98 in DENV-1 were shown to result in impaired prM processing and decreased viral infectivity. prM100 and prM101 are situated only 2–3 amino acids away from prM98, and given the absence of entry and replication defects, we hypothesize that the prM100,101 hotspot mutations likely impair prM processing and DENV infectivity, by a mechanism similar to prM98 (Zheng et al., 2014). Even though prM antibodies have been identified by several groups (Beltramello et al., 2010, Dejnirattisai et al., 2010), the immune pressures driving evolution at the prM/M locus are poorly understood, and the origin of the prM100,101 variant viruses remains elusive. In contrast, significantly more is known about the functionalities and antigenicity of loci in the E protein, and specifically the E315 locus. Previous studies have shown that E315 plays a pH-sensing role in mediating viral fusion with the endosome at the post-entry stage (Nelson et al., 2009), suggesting that viruses with the E315 hotspot mutation are likely impaired at the stage of viral membrane fusion with the endosomal membrane. Interestingly, the process of viral membrane fusion appears to be a significant target of anti-DENV antibodies during infection in humans, with fusion loop antibodies making up a significant proportion of the population of broadly reactive antibodies that are elicited during infection with DENV (Goncalvez et al., 2004, Lai et al., 2013). Given the convergent nature of the immune response and the prevalence of the E315 variant, it is conceivable that the E315 variant arose as an immune-escape variant to fusion loop antibodies. Indeed, Goncalvez et al. (2004) demonstrated that after 11 cycles of passage in the presence of anti-fusion loop mAb 1A5, an escape mutation was isolated at E317 in DENV-2, further supporting our claim that immune pressures exerted by fusion loop antibodies played a key role in convergent selection for variants at the analogous E315 hotspot locus in DENV-3. Taken together, we conclude that the E315 hotspot variant, despite possibly possessing an immune-escape advantage, is constrained to the intrahost evolutionary system because of the severe defects associated with viral membrane fusion post entry.
Concluding Remarks
We have thus demonstrated that intrahost DENV-3 populations can rapidly and convergently evolve in acute human infections, with virus microevolution possibly shaped by viral replication compartments distinct from PBMCs, driven in part by immune selection pressures and constrained by replication defects at the macro-evolutionary scale. In particular, we identified a highly prevalent DENV variant (E315) that likely arose as an immune-escape variant in response to pressures exerted by fusion loop antibodies, yet was constrained by severe replicative defects likely incurred due to a deficiency in viral membrane fusion with the host endosome post entry. The identification of such convergent immune pressures that appear to result in the selection of prevalent immune-escape variants, especially ones that are constrained by replication defects or fitness costs, could be harnessed for intelligent selection of new candidates and targets for vaccine/drug design. Such snapshots of intrahost DENV diversity help to identify mechanisms driving virus microevolution in human dengue and provide new perspectives in assessing infection outcome, disease pathogenesis, and vaccine/therapeutic efficacy in individuals.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-E (5D4) | Hybridoma from ATCC | 5D4-11 (ATCC® HB-49™), RRID: CVCL_J928; Henchal et al. (1982) |
| anti-NS3 (E1D8) | Hybridoma generated in Harris Laboratory | E1D8, Balsitis et al. (2009) |
| Bacterial and Virus Strains | ||
| E. coli TOP10 competent cells | Invitrogen | C4040-10 |
| Biological Samples | ||
| PBMC and plasma samples | This paper: Table S1 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Kanamycin Sulfate | Life Technologies | 11815-024 |
| Chloramphenicol >=98% (TLC) | Sigma-Aldrich | C0378-25G |
| Critical Commercial Assays | ||
| RNeasy Mini kit | Qiagen | Cat No./ID: 74104 |
| QIAamp Viral RNA Mini Kit | Qiagen | Cat No./ID: 52904 |
| QIAquick Gel Extraction Kit | Qiagen | Cat No./ID: 28704 |
| Nextera XT DNA Sample Preparation Kit | Illumina | FC-131-1096 |
| SuperScript III One-Step RT-PCR System | Invitrogen | 12574-035 |
| mMessage mMachine T7 Kit | Life Technology | AM1344M |
| MinElute Gel Extraction Kit (50) (70bp to 4kb) | Qiagen | Qia 28604 |
| Deposited Data | ||
| Fastq files for all these samples have been deposited into | NCBI's Sequence Read Archive | BioProject ID PRJNA394021 |
| Experimental Models: Cell Lines | ||
| Baby hamster kidney cells (BHK-21 clone 15) | Gift from B. Childs (Center for Vector-born Diseases, University of California, Davis, CA) | N/A |
| C6/36 cells, 37°C in 5% CO2. | Gift from Dr. Ralph Baric, University of North Carolina, Chapel Hill | N/A |
| U937-DC-SIGN cells | Gift of Dr. Aravinda de Silva, University of North Carolina, Chapel Hill | N/A |
| Oligonucleotides | ||
| Sequences of primers used for amplifying the DENV-3 genome | Table S4 of this paper | N/A |
| DV3Ewt_r: CGACCTTAATGAGTATTGTCCCAT | This paper | N/A |
| DV3E∗_r: CGACCTTAATGAGTATTGTCCCAa | This paper | N/A |
| PP49_DENV3_32_F (GACTCGGAAGCTTGCTTAAC) | This paper | N/A |
| primers (5’- GACTCGGAAGCTTGCTTAAC-3’ and 5’-TATTGACAGGCTCCTCCTTCTTAG-3’) designed to capture the prM100,101 and E315 hotspot loci | This paper | N/A |
| Sequencing primer for prM100,101: 5’-TCAATATGCTGAAACGCGTG-3’ | This paper | N/A |
| Sequencing primer for E315: 5’-CGGACAGGTTTGGATTTCAATG-3’ | This paper | N/A |
| Recombinant DNA | ||
| plasmid A-D | Gift of Dr. Ralph Baric | Messer et al., 2012 |
| plasmid A Nicaraguan WT | This paper | N/A |
| plasmid A Nicaraguan prM100,101 | This paper | N/A |
| plasmid A Nicaraguan E315 | This paper | N/A |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| ShoRAH | Zagordi et al., 2011 | http://www.cbg.ethz.ch/software/shorah |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Eva Harris (eharris@berkeley.edu).
Experimental Model and Subject Details
Ethics Statement
All studies were approved by the Institutional Review Boards (IRBs) of the University of California, Berkeley, and of the Nicaraguan Ministry of Health. Written consent was obtained from the parents/legal guardians of all pediatric subjects. Assent was also obtained from subjects six years of age or older.
Study Population
PBMC and plasma samples were obtained from subjects in an ongoing prospective hospital-based study of pediatric dengue in Nicaragua established in 1998 (Hammond et al., 2005), which enrolls patients who present with suspected dengue to the National Pediatric Reference Hospital in Managua, Nicaragua. Plasma and PBMCs are collected from patients for three sequential days during the acute phase of illness, at convalescence (days 14-28) and longitudinally for 18 months. Criteria for identifying dengue-positive cases and for classifying by immune status are as described (Gutierrez et al., 2013, Narvaez et al., 2011). Dengue cases are classified as DF, DHF, or DSS according to the 1997 WHO Dengue Guidelines (Narvaez et al., 2011, WHO, 1997). Thirty-one plasma and 68 peripheral blood mononuclear cell (PBMC) samples from 77 individuals (including 22 with paired plasma and PBMC samples) were analyzed in this study. The gender identity as well as the age of the patients is listed in Table S1. Extensive epidemiological and clinical data are available for these samples. The sample size for this study was determined by availability of pediatric patient samples for primary and secondary dengue cases caused by DENV-3 within a single endemic year (2009-2010). Subgroups within the 77 patients in the study were selected based on sample type (plasma versus PBMCs), clinical classification (for example, primary versus secondary dengue, Dengue Fever versus Dengue Hemorrhagic Fever and Dengue Shock Syndrome), or gene origin of variation (based on DENV-3 gene annotation).
Cell Culture
Baby hamster kidney cells (BHK-21 clone 15) were maintained in αMEM (Gibco) supplemented with 5% heat-inactivated Fetal Bovine Serum (FBS; Gibco), 10 mM HEPES (Gibco), and 100 U/ml penicillin and 100 μg/ml streptomycin (Pen/Strep; Gibco) at 37°C in 5% CO2. C6/36 cells (gift of Dr. Ralph Baric) were maintained in MEM with HEPES supplemented with 10% heat-inactivated FBS, 1X non-essential amino acids (NEAA; Gibco), 2 mM L-glutamine (Gibco) and Pen/Strep at 32°C in 5% CO2. U937 cells expressing DC-SIGN (U937-DC-SIGN, gift of Dr. Aravinda de Silva) were maintained in RPMI 1640 medium (Gibco) supplemented with 5% FBS, 1% Pen/Strep and 1% HEPES at 37°C in 5% CO2.
Method Details
RNA Extraction and DENV-3 Genome Amplification
A hybrid TRIzol/RNeasy protocol was developed for viral RNA recovery from PBMCs. Briefly, PBMCs were lysed in TRIzol (Life Technologies), and aqueous phase RNA was extracted after addition of chloroform. Next, 3.5 volumes of Buffer RLT (RNeasy Mini kit; Qiagen) and 2.5 volumes of 100% ethanol were added to the RNA solution, and the mixture was applied to an RNeasy column. All washes and RNA elutions were performed as described (RNeasy kit protocol). Viral RNA from plasma samples was extracted using the QIAamp Viral RNA Mini Kit (Qiagen) as described in the manufacturer’s protocol. RNA was eluted in 50 μl of RNase-free water containing linear polyacrylamide (1 μl; Life Technologies), carrier RNA (0.1 μl; Qiagen), and SUPERase-In (1 μl; Life Technologies). Twelve RT/PCR amplifications were set up for each sample, with various combinations of forward and reverse primers (Table S4) and reagents from the SuperScript III One-Step RT-PCR System (Invitrogen). For each reaction, we used 5 ul of denatured RNA, 17.5 ul of a 2X reaction mix, 0.25 ul each of forward and reverse primers (100 uM), 0.7ul of the enzyme mix, and 11.3 ul of RNase-free water. DENV-3 RNA was reverse-transcribed (50°C for 60 minutes) and cDNA was amplified as follows: 1 cycle of denaturation (94°C for 2 minutes), 35 cycles of denaturation (94°C for 15 seconds), annealing (55°C for 30 seconds), and extension (68°C for 3 minutes), and a final extension step (68°C for 10 minutes)}. The conditions were identical for all amplicons except amplicon 4, for which the extension time was shortened to 1 minute. DENV-3 amplicons were purified on a 1% agarose gel, and DNA was recovered using the QIAquick Gel Extraction Kit (Qiagen). For each sample, PCR amplicons were pooled in equimolar ratios, and indexed libraries were prepared for high-throughput sequencing using the Nextera XT DNA Sample Preparation Kit (Illumina). Nextera XT libraries were pooled and sequenced for 150 cycles in two lanes on Illumina’s HiSeq 2000 platform to generate paired-end datasets.
Consensus Genome Imputation and Variant Analysis
Reads were segregated based on their forward (read 1) and reverse (read 2) indices. The paired-end datasets for reads 1 and 2 were analyzed separately for all samples.
Filtering for Read Quality
The Fastx toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) was used to (a) trim reads with low quality (<Q30) at the 3’ end, (b) remove reads less than 50 nucleotides in length, and (c) only retain reads of high quality sequence (≥Q30) at ≥90% of read length.
Read Alignments and Variant Identification
We generated a DENV-3 consensus genome based on all sequenced Nicaraguan DENV-3 isolates from the 2009-2010 epidemic year (n = 108; NCBI Virus Variation Database). Read 1 and read 2 datasets for each sample were aligned to the Nicaraguan DENV-3 consensus genome using Bowtie2 (Langmead and Salzberg, 2012). SAMtools (Li et al., 2009) was used to create a pileup file from the Bowtie2 output, and pileup files were filtered based on a minimum read quality score of 30 and minimum coverage of 5 reads. Consensus genomes were created for each dataset using in-house scripts, and gaps were filled with Nicaraguan DENV-3 consensus sequences. All datasets were re-aligned to their corresponding consensus genomes using Bowtie2, and pileup files generated by SAMtools were filtered based on quality scores (≥30). Fastq files for all the sequences generated in this study have been deposited into NCBI's Sequence Read Archive, and can be accessed using BioProject ID PRJNA394021. In-house scripts were used to calculate percent variant abundance in nucleotide, codon and amino acid coordinates at all loci with a minimum coverage of 5 reads.
Threshold for Variant Abundance
Replicate amplicon libraries were analyzed in a pairwise manner to set abundance thresholds for high-confidence variant detection. In replicate amplicon libraries from four PBMC samples, all variants that were detected at an abundance of ≥0.5% in replicate A were also detected in replicate B (Figure S1B, left panels) and vice versa (data not shown). However, the threshold for variant detection in both replicates was considerably higher for replicate libraries from the three serum samples (≥1.0%; Figure S1B, right panels). Additionally, frequency distribution of loci by percent variant abundance in replicate B, for various thresholds of percent variant abundances in replicate A, revealed that concordance of 50% or higher between replicates was achieved at loci with ≥1.0% variant abundance in both PBMC and serum samples (Figure S1C, left and right panels, respectively). For identifying variants, we implemented the following thresholds: (a) loci should have a minimum coverage of 1000 reads, (b) variant abundance must be ≥1%, and (c) variant cannot be in a primer-binding region.
Reconstructing Haplotypes
The software package ShoRAH (Zagordi et al., 2011) was used to reconstruct 150 nucleotide-haplotypes spanning the prM100,101 and E315 hotspot loci. Only haplotypes with a reconstruction quality score of ≥0.9 were considered for diversity analysis.
Generation of WT, prM100,101 and E315 DENV-3 Viruses
A four-plasmid infectious clone system (plasmids A-D; gift of Dr. Ralph Baric (Messer et al., 2012)) based on a Sri Lankan DENV-3 template was used to derive Nicaraguan WT DENV-3 and DENV-3 with mutations at the prM100,101 or E315 hotspot loci. We introduced four amino acid changes in the C, prM and E sequences in plasmid A (P27S, I286V, V361T and A609V in the DENV-3 polyprotein) to generate infectious clones that were identical in structural protein sequence to the Nicaraguan DENV-3 consensus genome (“WT”). The prM100,101 mutations were inserted by site-directed mutagenesis in plasmid A using the QuikChange site-directed mutagenesis kit (Stratagene). The E315 mutation was synthesized in the context of a plasmid A fragment spanning nucleotides 1-2034 of the DENV-3 genome (Bio Basic, Inc.). Infectious clones for WT, prM100,101 and E315 viruses were derived as described (Messer et al., 2012), with some modifications. All plasmids were individually propagated in E. coli TOP10 competent cells (Invitrogen) and grown on LB plates with selective antibiotics (plasmid A encoding for WT, prM100,101 and E315 was selected with ampicillin, plasmids B and C with kanamycin, and plasmid D with chloramphenicol) at 30°C for 24 hours. Sequences of colony-purified plasmids were confirmed by Sanger sequencing, and restriction digests were set up as follows: plasmid A with SpeI, AhdI and BsmBI yielding a 2.0-kb fragment; plasmid B with BglI and BsmBI yielding a 1.1-kb fragment; plasmid C with BglI yielding a 3.9-kb fragment; and plasmid D with BglI and BsmbI yielding a 3.0-kb fragment. Fragments were gel-purified using a MinElute Gel Extraction Kit (Qiagen), mixed in equimolar ratios, and ligated with T4 ligase (NEB) for 2 hours at 16°C. Full-length transcripts of DENV-3 cDNA constructs were generated in vitro as described using the mMessage mMachine T7 Kit (Ambion) with the following modifications: (a) 20 μl reaction mixtures were supplemented with 2 μl of a 30 mM GTP stock, resulting in a 1.3:1 ratio of cap analog to GTP, and (b) the reaction was incubated at 37°C for 3 hours. For electroporation, DENV-3 transcripts were added to 400 μl of BHK cell suspension (1 x 107 cells/ml in RNase-free PBS) in an electroporation cuvette, and pulsed once with 270 V (975 μF) in a Bio-Rad Gene Pulser II electroporator. The transfected BHK cells were grown in a 75 cm2 flask and incubated at 37°C for 5 days. For virus amplification, 2 ml of viral supernatant from BHK cells was used to infect C6/36 cells in a 75 cm2 flask, and 18 ml of C6/36 media (1X MEM + 2% FBS + 1% Pen/Strep + 1% NEAA + 1% L-Glutamine) was added to the cells. Supernatants were harvested five days post-infection, supplemented to 1X SPG (sucrose-phosphate-glutamate stabilizer, prepared in-house), clarified by centrifugation, and frozen at −80°C or passaged serially in C6/36 cells.
Virus Titration
For plaque assays, ten-fold serial dilutions of virus culture supernatant (undiluted, and dilutions of 10−1 to 10−5) were added in duplicate to cultured BHK-21 cells in 24-well plates and incubated for 1 h at 37°C. After discarding the inoculum, the cells were overlaid with 1 ml of 0.8% carboxymethyl cellulose in MEM supplemented with 2% FBS and Pen/Strep. Plates were incubated for 4 days at 37°C in 5% CO2. Overlays were removed, cells were fixed with formalin and stained with 2.5% crystal violet in 30% ethanol, and plaque-forming units per ml (PFU/ml) were counted. For assaying temperature sensitivity, infections and post-infection incubations were performed at 32°C in 5% CO2, and plaques were counted 7 days p.i.. For quantitative RT-PCR (qRT-PCR), 18 μl-reactions were set up with 2 μl RNA, DENV-3 specific primers (Johnson et al., 2005), 0.1 μM DENV-3 probe (Johnson et al., 2005), and reagents from the Verso 1-Step qRT-PCR kit (Thermo Scientific). Reaction conditions were as follows: 1 cycle of reverse transcription (30 minutes at 50°C), 1 cycle of Thermo-Start activation (12.5 minutes at 95°C), and 40 cycles of denaturation (15 seconds at 95°C) and annealing/extension (1 minute at 60°C). A standard curve was used to calculate DENV-3 genome equivalents per ml (GE/ml) for each test sample.
Relative Quantitation of Viruses by Sequencing
Viral RNA was amplified using reagents from the SuperScript III One-Step RT-PCR System, with primers (5’- GACTCGGAAGCTTGCTTAAC-3’ and 5’-TATTGACAGGCTCCTCCTTCTTAG-3’) designed to capture the prM100,101 and E315 hotspot loci. Amplicons were gel-purified and sequenced by the Sanger method using primers 5’-TCAATATGCTGAAACGCGTG-3’ and 5’-CGGACAGGTTTGGATTTCAATG-3’ for capturing chromatogram peak heights at the prM100,101 and E315 loci, respectively. Relative levels of WT and mutant viruses were calculated from quantitation of chromatogram peak heights at the hotspot loci corresponding to WT and mutant nucleotides using the PolySNP program (Hall and Little, 2007).
Replication in C6/36 Cells
For each infection, we used six-well plates with 2 X 105 C6/36 cells per well, and a virus mix containing 1000 GE/cell of WT and 1000 GE/cell of prM100,101 or E315 viruses (as determined by qRT-PCR) diluted in Hank’s Balanced Salt Solution (HBSS; Gibco). One hundred μl of the virus mix was stored for assessing input ratios. C6/36 cells were infected with each virus mix for 2 h at 32°C in 5% CO2, washed with PBS, and incubated in MEM containing 2% FBS, 1% Pen/Strep, 1% NEAA and 1% L-Glutamine. Supernatants were collected from independent wells at 3, 4 and 5 d.p.i.. Viral RNA was extracted from supernatants and the input virus mix using the QIAamp Viral RNA kit (Qiagen). All experiments were performed in duplicate. Relative levels of WT and mutant viruses were estimated using Sanger sequencing and the polySNP program (Hall and Little, 2007).
Replication in U937-DC-SIGN Cells
Infections were set up in 96-well plates with 5 x 104 U937-DC-SIGN cells per well. Infections were performed with 625 GE/cell of WT, prM100,101 or E315 viruses, diluted in HBSS. Cells were infected with each virus mix for 2 h at 37°C in 5% CO2, washed and incubated in medium containing RPMI 1640, 2% FBS, 1% Pen/Strep and 1% HEPES for 24, 48 or 72 h at 37°C in 5% CO2. Cells were stained at room temperature with a 1:500 dilution of Zombie Aqua stain (BioLegend), fixed, permeabilized, and stained with a 1:250 dilution of an anti-E (5D4) (Henchal et al., 1982) monoclonal antibody conjugated to Texas Red and a 1:500 dilution of an anti-NS3 (E1D8) (Balsitis et al., 2009) monoclonal antibody conjugated to APC. All experiments were performed in duplicate. The percent of infected cells was measured via flow cytometry on an LSR Fortessa cell analyzer (BD Biosciences), and the data were analyzed using FlowJo 8.8.7 software (TreeStar).
RT-PCR from Serum
Reverse primers specific for WT or E315 mutant virus were designed as follows: primer WT (DV3Ewt_r: CGACCTTAATGAGTATTGTCCCAT); primer E315 (DV3E∗_r: CGACCTTAATGAGTATTGTCCCAa). In combination with the forward primer PP49_DENV3_32_F (GACTCGGAAGCTTGCTTAAC), both primer sets generated an RT-PCR amplicon of 1,865bp. RT-PCR was carried out using SuperScript® III One-Step RT-PCR System with Platinum® Taq High Fidelity (Invitrogen). The RT-PCR products were visualized using an agarose gel (1%).
Entry Assays
The entry assay was performed as previously described (Diamond and Harris, 2001) with slight modifications. Specifically, 10μl of U937-DC-SIGN cells (1x107 cells/ml, total of 1x105 cells) was aliquoted into microcentrifuge tubes in triplicate, mixed with WT virus (multiplicity of infection of 0.5), E315 variant virus or prM100,101 variant virus diluted in 240μl of U937-DC-SIGN culture medium, and incubated at 37°C for 2 hours with occasional agitation to avoid pelleting. All samples were then iced and washed three times with 500μl of U937-DC-SIGN culture medium at 4°C, and washed twice with 500μl PBS at 4°C. Then 350μl of lysis buffer was added to cell pellets, and RNA extraction was performed using RNeasy Mini Kit (QIAGEN). qRT-PCR was performed as described above.
Quantification and Statistical Analysis
Different statistical analyses were performed and are specified in the corresponding results in the main text or in figure legends when describing the analysis, such as Fisher’s exact test (Figure 2C, main text), Wilcoxon test for unpaired samples (Figure 1B, figure legend), Wilcoxon signed-rank test (Figures 1A, 1D, and 1E, figure legend), Wilcoxon signed-rank test for paired samples (Figure 1F, figure legend). Significance was generally defined as ∗P≤0.01, ∗∗P≤0.001, while P < 0.0001 was also indicated where applicable. The number of samples (n) used for all comparisons is indicated in the figure legends as well as in the main text. Briefly, samples from 53 individuals with 1° and 46 individuals with 2° dengue were used for comparisons presented in Figures 1A–1C. Samples from 22 paired PBMC and plasma samples were used in Figures 1D and 1E. Samples from 12 paired PBMC and plasma samples were used in Figure 1F. Samples from 68 PBMC and 31 plasma samples were used in Figures 2A–2D. In Figure 2E, 690 full-length DENV-3 genomes were analyzed. Data from 8 PBMC/plasma samples (prM/M hotspot) and 39 PBMC/plasma samples (E hotspot) are shown in Figure 3. Data from 2 independent experiments were used in Figure 4.
Data and Software Availability
Fastq files for all the sequences generated in this study have been deposited into NCBI's Sequence Read Archive, and can be accessed using BioProject ID PRJNA394021.
Author Contributions
Design of Experiments, P.P., C.W., and E.H.; Performed Experiments, S.B.T., C.W., M.E., P.P., and M.M.; Data Analysis, P.P., C.W., S.B.T., M.E., M.M., and E.H.; Resources, A.B. and E.H.; Writing – Review & Editing, P.P., C.W., and E.H.; Funding Acquisition, P.P., A.B., and E.H.
Acknowledgments
This work was funded by the following grants from NIAID, NIH: R01 GM087405 to E.H., U54 AI065359 to A.B., U54 AI065359 Development Award to P.P. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors would like to thank Shayna M. Cave, Sanjana Shah, Edwina B. Tran, and Alejandro Ramirez for technical assistance with experiments, and Dr. Andrew Fire for scientific advice. We thank members of the study team based at the Hospital Infantil Manuel de Jesús Rivera, the National Virology Laboratory in the Centro Nacional de Diagnóstico y Referencia, and the Sustainable Sciences Institute in Nicaragua for their dedication and high-quality work, as well as the children who participated in the studies and their families.
Published: September 13, 2017
Footnotes
Supplemental Information includes five figures and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.chom.2017.08.003.
Supplemental Information
References
- Aye K.S., Charngkaew K., Win N., Wai K.Z., Moe K., Punyadee N., Thiemmeca S., Suttitheptumrong A., Sukpanichnant S., Prida M. Pathologic highlights of dengue hemorrhagic fever in 13 autopsy cases from Myanmar. Hum. Pathol. 2014;45:1221–1233. doi: 10.1016/j.humpath.2014.01.022. [DOI] [PubMed] [Google Scholar]
- Balsitis S.J., Coloma J., Castro G., Alava A., Flores D., McKerrow J.H., Beatty P.R., Harris E. Tropism of dengue virus in mice and humans defined by viral nonstructural protein 3-specific immunostaining. Am. J. Trop. Med. Hyg. 2009;80:416–424. [PubMed] [Google Scholar]
- Beltramello M., Williams K.L., Simmons C.P., Macagno A., Simonelli L., Quyen N.T., Sukupolvi-Petty S., Navarro-Sanchez E., Young P.R., de Silva A.M. The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe. 2010;8:271–283. doi: 10.1016/j.chom.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S., Gething P.W., Brady O.J., Messina J.P., Farlow A.W., Moyes C.L., Drake J.M., Brownstein J.S., Hoen A.G., Sankoh O. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull R.A., Eden J.S., Luciani F., McElroy K., Rawlinson W.D., White P.A. Contribution of intra- and interhost dynamics to norovirus evolution. J. Virol. 2012;86:3219–3229. doi: 10.1128/JVI.06712-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byk L.A., Gamarnik A.V. Properties and functions of the dengue virus capsid protein. Annu. Rev. Virol. 2016;3:263–281. doi: 10.1146/annurev-virology-110615-042334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyde K., Barrera J., Harris E. The capsid-coding region hairpin element (cHP) is a critical determinant of dengue virus and West Nile virus RNA synthesis. Virology. 2008;379:314–323. doi: 10.1016/j.virol.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alwis R., Williams K.L., Schmid M.A., Lai C.Y., Patel B., Smith S.A., Crowe J.E., Wang W.K., Harris E., de Silva A.M. Dengue viruses are enhanced by distinct populations of serotype cross-reactive antibodies in human immune sera. PLoS Pathog. 2014;10:e1004386. doi: 10.1371/journal.ppat.1004386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Borba L., Villordo S.M., Iglesias N.G., Filomatori C.V., Gebhard L.G., Gamarnik A.V. Overlapping local and long-range RNA-RNA interactions modulate dengue virus genome cyclization and replication. J. Virol. 2015;89:3430–3437. doi: 10.1128/JVI.02677-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debbink K., Lindesmith L.C., Ferris M.T., Swanstrom J., Beltramello M., Corti D., Lanzavecchia A., Baric R.S. Within-host evolution results in antigenically distinct GII.4 noroviruses. J. Virol. 2014;88:7244–7255. doi: 10.1128/JVI.00203-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejnirattisai W., Jumnainsong A., Onsirisakul N., Fitton P., Vasanawathana S., Limpitikul W., Puttikhunt C., Edwards C., Duangchinda T., Supasa S. Cross-reacting antibodies enhance dengue virus infection in humans. Science. 2010;328:745–748. doi: 10.1126/science.1185181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descloux E., Cao-Lormeau V.M., Roche C., De Lamballerie X. Dengue 1 diversity and microevolution, French Polynesia 2001-2006: connection with epidemiology and clinics. PLoS Negl. Trop. Dis. 2009;3:e493. doi: 10.1371/journal.pntd.0000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond M.S., Harris E. Interferon inhibits dengue virus infection by preventing translation of viral RNA through a PKR-independent mechanism. Virology. 2001;289:297–311. doi: 10.1006/viro.2001.1114. [DOI] [PubMed] [Google Scholar]
- Domingo E., Holland J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997;51:151–178. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- Farci P., Strazzera R., Alter H.J., Farci S., Degioannis D., Coiana A., Peddis G., Usai F., Serra G., Chessa L. Early changes in hepatitis C viral quasispecies during interferon therapy predict the therapeutic outcome. Proc. Natl. Acad. Sci. USA. 2002;99:3081–3086. doi: 10.1073/pnas.052712599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe P., Shi P.Y., Harris E. The 5' and 3' downstream AUG region elements are required for mosquito-borne flavivirus RNA replication. J. Virol. 2011;85:1900–1905. doi: 10.1128/JVI.02037-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gire S.K., Goba A., Andersen K.G., Sealfon R.S., Park D.J., Kanneh L., Jalloh S., Momoh M., Fullah M., Dudas G. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345:1369–1372. doi: 10.1126/science.1259657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalvez A.P., Purcell R.H., Lai C.-J. Epitope determinants of a chimpanzee Fab antibody that efficiently cross-neutralizes dengue type 1 and type 2 viruses map to inside and in close proximity to fusion loop of the dengue type 2 virus envelope protein. J. Virol. 2004;78:12919–12928. doi: 10.1128/JVI.78.23.12919-12928.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez G., Gresh L., Perez M.A., Elizondo D., Aviles W., Kuan G., Balmaseda A., Harris E. Evaluation of the diagnostic utility of the traditional and revised WHO dengue case definitions. PLoS Negl. Trop. Dis. 2013;7:e2385. doi: 10.1371/journal.pntd.0002385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall G.S., Little D.P. Relative quantitation of virus population size in mixed genotype infections using sequencing chromatograms. J. Virol. Methods. 2007;146:22–28. doi: 10.1016/j.jviromet.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead S.B. Antibodies determine virulence in dengue. Ann. N. Y Acad. Sci. 2009;1171(Suppl 1):E48–E56. doi: 10.1111/j.1749-6632.2009.05052.x. [DOI] [PubMed] [Google Scholar]
- Halstead S.B., Yamarat C. Recent epidemics of hemorrhagic fever in Thailand. observations related to pathogenesis of a “new” dengue disease. Am. J. Public Health Nations Health. 1965;55:1386–1395. doi: 10.2105/ajph.55.9.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond S.N., Balmaseda A., Perez L., Tellez Y., Saborio S.I., Mercado J.C., Videa E., Rodriguez Y., Perez M.A., Cuadra R. Differences in dengue severity in infants, children, and adults in a 3-year hospital-based study in Nicaragua. Am. J. Trop. Med. Hyg. 2005;73:1063–1070. [PubMed] [Google Scholar]
- Henchal E.A., Gentry M.K., McCown J.M., Brandt W.E. Dengue virus-specific and flavivirus group determinants identified with monoclonal antibodies by indirect immunofluorescence. Am. J. Trop. Med. Hyg. 1982;31:830–836. doi: 10.4269/ajtmh.1982.31.830. [DOI] [PubMed] [Google Scholar]
- Jessie K., Fong M.Y., Devi S., Lam S.K., Wong K.T. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J. Infect. Dis. 2004;189:1411–1418. doi: 10.1086/383043. [DOI] [PubMed] [Google Scholar]
- Johnson B.W., Russell B.J., Lanciotti R.S. Serotype-specific detection of dengue viruses in a fourplex real-time reverse transcriptase PCR assay. J. Clin. Microbiol. 2005;43:4977–4983. doi: 10.1128/JCM.43.10.4977-4983.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joos B., Trkola A., Fischer M., Kuster H., Rusert P., Leemann C., Boni J., Oxenius A., Price D.A., Phillips R.E. Low human immunodeficiency virus envelope diversity correlates with low in vitro replication capacity and predicts spontaneous control of plasma viremia after treatment interruptions. J. Virol. 2005;79:9026–9037. doi: 10.1128/JVI.79.14.9026-9037.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C.Y., Williams K.L., Wu Y.C., Knight S., Balmaseda A., Harris E., Wang W.K. Analysis of cross-reactive antibodies recognizing the fusion loop of envelope protein and correlation with neutralizing antibody titers in Nicaraguan dengue cases. PLoS Negl. Trop. Dis. 2013;7:e2451. doi: 10.1371/journal.pntd.0002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.Y., Perelson A.S., Park S.C., Leitner T. Dynamic correlation between intrahost HIV-1 quasispecies evolution and disease progression. PLoS Comput. Biol. 2008;4:e1000240. doi: 10.1371/journal.pcbi.1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing S. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.R., Hsieh S.C., Yueh Y.Y., Lin T.H., Chao D.Y., Chen W.J., King C.C., Wang W.K. Study of sequence variation of dengue type 3 virus in naturally infected mosquitoes and human hosts: implications for transmission and evolution. J. Virol. 2004;78:12717–12721. doi: 10.1128/JVI.78.22.12717-12721.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer W.B., Gubler D.J., Harris E., Sivananthan K., de Silva A.M. Emergence and global spread of a dengue serotype 3, subtype III virus. Emerg. Infect. Dis. 2003;9:800–809. doi: 10.3201/eid0907.030038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer W.B., Yount B., Hacker K.E., Donaldson E.F., Huynh J.P., de Silva A.M., Baric R.S. Development and characterization of a reverse genetic system for studying dengue virus serotype 3 strain variation and neutralization. PLoS Negl. Trop. Dis. 2012;6:e1486. doi: 10.1371/journal.pntd.0001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau I., Levis J., Crosbie O., Kenny-Walsh E., Fanning L.J. Correlation between pre-treatment quasispecies complexity and treatment outcome in chronic HCV genotype 3a. Virol. J. 2008;5:78. doi: 10.1186/1743-422X-5-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narvaez F., Gutierrez G., Perez M.A., Elizondo D., Nunez A., Balmaseda A., Harris E. Evaluation of the traditional and revised WHO classifications of Dengue disease severity. PLoS Negl. Trop. Dis. 2011;5:e1397. doi: 10.1371/journal.pntd.0001397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S., Poddar S., Lin T.Y., Pierson T.C. Protonation of individual histidine residues is not required for the pH-dependent entry of West Nile virus: evaluation of the “histidine switch” hypothesis. J. Virol. 2009;83:12631–12635. doi: 10.1128/JVI.01072-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T.P., Kikuchi M., Vu T.Q., Do Q.H., Tran T.T., Vo D.T., Ha M.T., Vo V.T., Cao T.P., Tran V.D. Protective and enhancing HLA alleles, HLA-DRB1∗0901 and HLA-A∗24, for severe forms of dengue virus infection, dengue hemorrhagic fever and dengue shock syndrome. PLoS Negl. Trop. Dis. 2008;2:e304. doi: 10.1371/journal.pntd.0000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni M., Chen C., Qian J., Xiao H.X., Shi W.F., Luo Y., Wang H.Y., Li Z., Wu J., Xu P.S. Intra-host dynamics of Ebola virus during 2014. Nat. Microbiol. 2016;1:16151. doi: 10.1038/nmicrobiol.2016.151. [DOI] [PubMed] [Google Scholar]
- OhAinle M., Balmaseda A., Macalalad A.R., Tellez Y., Zody M.C., Saborio S., Nunez A., Lennon N.J., Birren B.W., Gordon A. Dynamics of dengue disease severity determined by the interplay between viral genetics and serotype-specific immunity. Sci. Transl. Med. 2011;3:114ra128. doi: 10.1126/scitranslmed.3003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran P., Charlebois P., Tellez Y., Nunez A., Ryan E.M., Malboeuf C.M., Levin J.Z., Lennon N.J., Balmaseda A., Harris E. Genome-wide patterns of intrahuman dengue virus diversity reveal associations with viral phylogenetic clade and interhost diversity. J. Virol. 2012;86:8546–8558. doi: 10.1128/JVI.00736-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D., Huh H.J., Kim Y.J., Son D.S., Jeon H.J., Im E.H., Kim J.W., Lee N.Y., Kang E.S., Kang C.I. Analysis of intrapatient heterogeneity uncovers the microevolution of Middle East respiratory syndrome coronavirus. Cold Spring Harb. Mol. Case Stud. 2016;2:a001214. doi: 10.1101/mcs.a001214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Porterfield J.S. Antibody-mediated enhancement of Flavivirus replication in macrophage-like cell lines. Nature. 1979;282:509–511. doi: 10.1038/282509a0. [DOI] [PubMed] [Google Scholar]
- Quiner C.A., Parameswaran P., Ciota A.T., Ehrbar D.J., Dodson B.L., Schlesinger S., Kramer L.D., Harris E. Increased replicative fitness of a dengue virus 2 clade in native mosquitoes: potential contribution to a clade replacement event in Nicaragua. J. Virol. 2014;88:13125–13134. doi: 10.1128/JVI.01822-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico-Hesse R., Harrison L.M., Salas R.A., Tovar D., Nisalak A., Ramos C., Boshell J., de Mesa M.T., Nogueira R.M., da Rosa A.T. Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology. 1997;230:244–251. doi: 10.1006/viro.1997.8504. [DOI] [PubMed] [Google Scholar]
- Rivino L., Kumaran E.A., Jovanovic V., Nadua K., Teo E.W., Pang S.W., Teo G.H., Gan V.C., Lye D.C., Leo Y.S. Differential targeting of viral components by CD4+ versus CD8+ T lymphocytes in dengue virus infection. J. Virol. 2013;87:2693–2706. doi: 10.1128/JVI.02675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Roche R., Blanc H., Borderia A.V., Diaz G., Henningsson R., Gonzalez D., Santana E., Alvarez M., Castro O., Fontes M. Increasing clinical severity during a dengue virus type 3 Cuban epidemic: deep sequencing of evolving viral populations. J. Virol. 2016;90:4320–4333. doi: 10.1128/JVI.02647-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers M.B., Song T., Sebra R., Greenbaum B.D., Hamelin M.E., Fitch A., Twaddle A., Cui L., Holmes E.C., Boivin G. Intrahost dynamics of antiviral resistance in influenza a virus reflect complex patterns of segment linkage, reassortment, and natural selection. MBio. 2015;6 doi: 10.1128/mBio.02464-14. e02464-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman A.L., Ennis F.A. Immunopathogenesis of Dengue hemorrhagic fever. Virology. 1999;257:1–6. doi: 10.1006/viro.1999.9656. [DOI] [PubMed] [Google Scholar]
- Selisko B., Wang C., Harris E., Canard B. Regulation of flavivirus RNA synthesis and replication. Curr. Opin. Virol. 2014;9:74–83. doi: 10.1016/j.coviro.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessions O.M., Wilm A., Kamaraj U.S., Choy M.M., Chow A., Chong Y., Ong X.M., Nagarajan N., Cook A.R., Ooi E.E. Analysis of dengue virus genetic diversity during human and mosquito infection reveals genetic constraints. PLoS Negl. Trop. Dis. 2015;9:e0004044. doi: 10.1371/journal.pntd.0004044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim S., Aw P.P., Wilm A., Teoh G., Hue K.D., Nguyen N.M., Nagarajan N., Simmons C.P., Hibberd M.L. Tracking dengue virus intra-host genetic diversity during human-to-mosquito transmission. PLoS Negl. Trop. Dis. 2015;9:e0004052. doi: 10.1371/journal.pntd.0004052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons C.P., Dong T., Chau N.V., Dung N.T., Chau T.N., Thao le T.T., Dung N.T., Hien T.T., Rowland-Jones S., Farrar J. Early T-cell responses to dengue virus epitopes in Vietnamese adults with secondary dengue virus infections. J. Virol. 2005;79:5665–5675. doi: 10.1128/JVI.79.9.5665-5675.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel Leonard A., McClain M.T., Smith G.J., Wentworth D.E., Halpin R.A., Lin X., Ransier A., Stockwell T.B., Das S.R., Gilbert A.S. Deep sequencing of influenza a virus from a human challenge study reveals a selective bottleneck and only limited intrahost genetic diversification. J. Virol. 2016;90:11247–11258. doi: 10.1128/JVI.01657-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleford K.A., Moratorio G., Henningsson R., Chen R., Matheus S., Enfissi A., Weissglas-Volkov D., Isakov O., Blanc H., Mounce B.C. Whole-genome sequencing analysis from the chikungunya virus Caribbean outbreak reveals novel evolutionary genomic elements. PLoS Negl. Trop. Dis. 2016;10:e0004402. doi: 10.1371/journal.pntd.0004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan D.G., Bruden D., Deubner H., McArdle S., Chung M., Christensen C., Hennessy T., Homan C., Williams J., McMahon B.J. Hepatitis C virus dynamics during natural infection are associated with long-term histological outcome of chronic hepatitis C disease. J. Infect. Dis. 2007;196:239–248. doi: 10.1086/518895. [DOI] [PubMed] [Google Scholar]
- Tassaneetrithep B., Burgess T.H., Granelli-Piperno A., Trumpfheller C., Finke J., Sun W., Eller M.A., Pattanapanyasat K., Sarasombath S., Birx D.L. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 2003;197:823–829. doi: 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai K.T., Henn M.R., Zody M.C., Tricou V., Nguyet N.M., Charlebois P., Lennon N.J., Green L., de Vries P.J., Hien T.T. High-resolution analysis of intrahost genetic diversity in dengue virus serotype 1 infection identifies mixed infections. J. Virol. 2012;86:835–843. doi: 10.1128/JVI.05985-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkenburg S.A., Quinones-Parra S., Gras S., Komadina N., McVernon J., Wang Z., Halim H., Iannello P., Cole C., Laurie K. Acute emergence and reversion of influenza A virus quasispecies within CD8+ T cell antigenic peptides. Nat. Commun. 2013;4:2663. doi: 10.1038/ncomms3663. [DOI] [PubMed] [Google Scholar]
- Wang W.K., Lin S.R., Lee C.M., King C.C., Chang S.C. Dengue type 3 virus in plasma is a population of closely related genomes: quasispecies. J. Virol. 2002;76:4662–4665. doi: 10.1128/JVI.76.9.4662-4665.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.K., Sung T.L., Lee C.N., Lin T.Y., King C.C. Sequence diversity of the capsid gene and the nonstructural gene NS2B of dengue-3 virus in vivo. Virology. 2002;303:181–191. doi: 10.1006/viro.2002.1635. [DOI] [PubMed] [Google Scholar]
- Weiskopf D., Angelo M.A., de Azeredo E.L., Sidney J., Greenbaum J.A., Fernando A.N., Broadwater A., Kolla R.V., De Silva A.D., de Silva A.M. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc. Natl. Acad. Sci. USA. 2013;110:E2046–E2053. doi: 10.1073/pnas.1305227110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . Second Edition. WHO; 1997. Dengue Haemorrhagic Fever: Diagnosis, Treatment, Prevention and Control. [Google Scholar]
- Zagordi O., Bhattacharya A., Eriksson N., Beerenwinkel N. ShoRAH: estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinformatics. 2011;12:119. doi: 10.1186/1471-2105-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng A., Yuan F., Kleinfelter L.M., Kielian M. A toggle switch controls the low pH-triggered rearrangement and maturation of the dengue virus envelope proteins. Nat. Commun. 2014;5:3877. doi: 10.1038/ncomms4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Fastq files for all the sequences generated in this study have been deposited into NCBI's Sequence Read Archive, and can be accessed using BioProject ID PRJNA394021.