

Abstract
Four studies in this issue of Nature Genetics report new mechanisms underlying SWI/SNF complex's function in controlling gene expression and suppressing tumor development, providing valuable insights into treating cancers harboring mutations in SWI/SNF complex subunits.
SWI/SNF complex (also known as BAF complex) was originally described in yeast as the complex critical for cellular responses to mating type switching (SWI) or sucrose fermentation (SNF)1,2. A large (∼2MDa) complex containing more than 15 subunits, SWI/SNF complex activates gene expression, a long-held function to result from its activity to remodel and evict nucleosomes at gene promoters3. Recently, SWI/SNF complex has been linked to a number of human diseases, most notably cancer. Mutations, translocations and deletions involving various subunits of SWI/SNF complex were found in ∼20% of human tumors, making the complex as one of the most commonly affected target in cancer4. In this issue, four research articles offer novel insights into the mechanisms by which SWI/SNF complex alterations lead to defective complex assembly and recruitment, abnormal gene silencing and tumor development5-8. Importantly, these mechanisms help explain the preclinical efficacies of several therapeutic approaches developed to target SWI/SNF-mutated tumors.
‘Remodeling’ the enhancers
Loss of SMARCB1 (also known as SNF5) is the defining molecular feature of childhood malignant rhabdoid tumors. To investigate the impact of SMARCB1 inactivation on SWI/SNF complex assembly and targeting, Roberts, Bernstein, Park and colleagues5 examined the genome-wide localization of SWI/SNF complex in SMARCB1-wildtype and deficient rhabdoid tumors and cell lines. In addition to gene promoters, they found that a substantial portion of SWI/SNF complexes were recruited to enhancers and so-called ‘super-enhancers’ (clusters of active enhancers) in SMARCB1-wildtype cells. Interestingly, loss of SMARCB1 caused disassembly of most SWI/SNF complexes at promoters and typical enhancers, but only minimally affected super-enhancer-associated complexes. The authors propose that since typical enhancers regulate the expression of genes involved in lineage specification while super-enhancers are linked to genes maintaining cell identity, the redistribution of SWI/SNF complexes may impair SMARCB1-deficient cells' potential to differentiate while maintain their self-renewal, survival and proliferation capacity, thus promoting tumorigenesis.
Another study from the Roberts laboratory focused on cancer-associated ARID1A inactivation. They found that genetic knockout of Arid1a in mouse colon tissues resulted in development of invasive colon adenocarcinomas resembling human colorectal cancers. The tumor growth was independent of APC inactivation and represents a new mouse model for the study of colon cancer. Similarly, the authors observed that the majority of SWI/SNF complexes, particularly those at enhancers, were compromised upon ARID1A loss, which in turn caused marked deregulation of developmental gene expression. The remaining SWI/SNF complexes were maintained by the ARID1B isoform and seemed to be critical for tumor survival, as depletion of ARID1B was selectively toxic to ARID1A-deficient cells, consistent with previous findings9.
Taken together, these two studies identified a previously underappreciated role for SWI/SNF complex at gene enhancers/super-enhancers. They also provide potential explanations for why SWI/SNF mutations in cancer, particularly those affecting the regulatory subunits such as ARID1A/B and SMARCB1, exhibit a high degree of tissue specificity and why SWI/SNF-mutated tumors are hypersensitive to the inhibition of residual complex activity9,10.
‘SWIng’ polycomb complex off chromatin
What is the molecular connection between abnormal SWI/SNF activity and deregulated gene expression? Studies in Drosophila have pointed to a genetic antagonism between SWI/SNF complex and the transcriptionally silencing Polycomb Repressive Complex 1 and 2 (PRC1/2)11. Consistently, tumors carrying SWI/SNF inactivating mutations are particularly vulnerable to genetic and pharmacological inhibition of PRC212,13. The biochemical mechanism underlying such opposition, however, has remained elusive. Two studies from the Crabtree and Kadoch laboratories have provided compelling evidence in support of an unexpected function of SWI/SNF complex in direct eviction of PRC1 from chromatin7,8. Stanton and Hodges et al. found that upon deletion of the catalytic subunit Smarca4 in mouse embryonic stem cells, there was a genome-wide increase in the localization of PRC1 and PRC2, as well as enrichment of H3K27me3. The enhanced polycomb silencing effects could be reversed by re-expressing the wildtype but not cancer-associated ATPase mutant Smarca4. Importantly, co-immunoprecipitation experiments identified a chromatin-independent interaction between SWI/SNF complex and PRC1 complex components, which could be disrupted upon stimulation of ATP hydrolysis, consistent with a model whereby ATPase activity of SWI/SNF directly releases PRC1.
To study the dynamic mechanisms underlying SWI/SNF and polycomb complex opposition in vivo, Kadoch et al. developed a novel system to chemically induce local recruitment of SWI/SNF complex in a rapid and reversible manner8. They found that loss of PRC1 from chromatin occurred within minutes after SWI/SNF complex recruitment. Furthermore, SWI/SNF-mediated PRC1 eviction is reversible, dependent on its ATPase activity, and independent of transcription, replication, nucleosome turnover and changes in chromatin accessibility. Removal of the tumor suppressor SMARCB1 from SWI/SNF complex resulted in a reduced ability to oppose PRC1, whereas the oncogenic SWI/SNF complex containing the SS18-SSX1 fusion protein, which is found in 100% of synovial sarcomas, displayed enhanced chromatin occupancy and PRC1 eviction. Collectively, these results suggest that, outside of the commonly held belief, polycomb complexes are critical non-nucleosomal substrates of SWI/SNF. They also provide strong rationale for targeting polycomb-mediated gene silencing, such as pharmacological inhibition of PRC1, as a therapeutic strategy for SWI/SNF-inactivated tumors.
While these studies reveal exciting new aspects of SWI/SNF complex biology (Figure 1), they also raise several interesting concepts begging future studies. In addition to the well-characterized inter-cellular heterogeneity of SWI/SNF complex, it now seems plausible that intra-cellular heterogeneity of this complex also exists and may be functionally important. Proteomic and epigenomic analyses are required to determine whether variants of SWI/SNF complex, such as SMARCB1-null or ARID1B-containing complexes, localize to distinct genomic regions (eg. typical vs. super-enhancers) and differ in their abilities to remodel nucleosomes and/or oppose polycomb-mediated silencing. Notably, several subunits of SWI/SNF complex possess putative DNA-binding domains and “reader” domains for chromatin modifications, which may underline the potential variant-specific chromatin recruitment mechanism. Further characterization of the SWI/SNF-mediated PRC1 chromatin eviction using purified proteins and customized nucleosome arrays will be critical to understand the biochemical mechanisms, especially in relation to the well-known nucleosome-remodeling activity of this complex. Lastly, given the conserved genetic antagonism, whether inhibiting SWI/SNF complex could effectively treat tumors carrying inactivating mutations/deletions of polycomb complex subunits remains to be explored14. Because of their high prevalence in cancer, a better understanding of the interplay between SWI/SNF and polycomb complexes will have far-reaching implications for clinical strategies moving forward.
Figure 1. New ‘remodeling’ mechanisms underline SWI/SNF complex's tumor suppressive function.
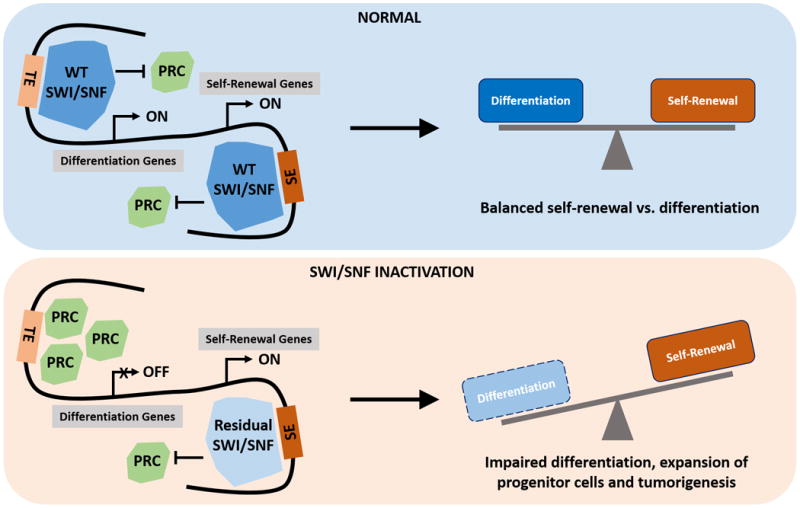
Inactivating mutations/deletions of SWI/SNF subunits cause defective complex assembly and failure to oppose PRC1/2 at promoters and typical enhancers (TE) of genes involved in differentiation. The residual functional SWI/SNF complexes are preferentially localized to super-enhancers (SE) of genes maintaining cell identity. The resulting imbalance between differentiation vs. self-renewal promotes tumorigenesis.
References
- 1.Stern M, Jensen R, Herskowitz I. Five SWI genes are required for expression of the HO gene in yeast. J Mol Biol. 1984;178:853–868. doi: 10.1016/0022-2836(84)90315-2. [DOI] [PubMed] [Google Scholar]
- 2.Neigeborn L, Carlson M. Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genetics. 1984;108:845–858. doi: 10.1093/genetics/108.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwon H, Imbalzano AN, Khavari PA, Kingston RE, Green MR. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature. 1994;370:477–481. doi: 10.1038/370477a0. [DOI] [PubMed] [Google Scholar]
- 4.Kadoch C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet. 2016 doi: 10.1038/ng.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathur R, et al. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat Genet. 2016 doi: 10.1038/ng.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stanton BZ, et al. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat Genet. 2016 doi: 10.1038/ng.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadoch C, et al. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat Genet. 2016 doi: 10.1038/ng.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helming KC, et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med. 2014;20:251–254. doi: 10.1038/nm.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, et al. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009;69:8094–8101. doi: 10.1158/0008-5472.CAN-09-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tamkun JW, et al. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell. 1992;68:561–572. doi: 10.1016/0092-8674(92)90191-e. [DOI] [PubMed] [Google Scholar]
- 12.Wilson BG, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18:316–328. doi: 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bitler BG, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21:231–238. doi: 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Comet I, Riising EM, Leblanc B, Helin K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat Rev Cancer. 2016 doi: 10.1038/nrc.2016.83. advance online publication. [DOI] [PubMed] [Google Scholar]